

Abstract
Broad spectrum caspase inhibitors have been found to reduce neurodegeneration caused by cerebral ischemia. We studied whether blockade of group I caspases, mainly caspase-1, using the inhibitor Ac-YVAD.cmk reduced infarct volume and produced prolonged neuroprotection. Ac-YVAD.cmk (300 ng/rat) was injected intracerebroventricularly 10 min after permanent middle cerebral artery occlusion in the rat. Drug treatment induced a significant reduction of infarct volume not only 24 hr after ischemia (total damage, percentage of hemisphere volume: control, 41.1 ± 2.3%; treated, 26.5 ± 2.1%; p < 0.05) but also 6 d later (total damage: control, 30.6 ± 2.2%; treated, 23.0 ± 2.2%; p < 0.05). Ac-YVAD.cmk treatment resulted in a reduction not only of caspase-1 (control, 100 ± 20.3%; treated, 3.4 ± 10.4%; p< 0.01) but also of caspase-3 (control, 100 ± 30.3%; treated, 13.2 ± 9.5%; p < 0.05) activity at 24 hr and led to a parallel decrease of apoptosis as measured by nucleosome quantitation (control, 100 ± 11.8%; treated, 47 ± 5.9%;p < 0.05). Six days after treatment no differences in these parameters could be detected between control and treated animals. Likewise, brain levels of the proinflammatory cytokines IL-1β and TNF-α were reduced at 24 hr (39.5 ± 23.7 and 51.9 ± 10.3% of control, respectively) but not at 6 d. Other cytokines, IL-10, MCP-1, MIP-2, and the gaseous mediator nitric oxide, were not modified by the treatment. These findings indicate that blockade of caspase-1-like activity induces a long-lasting neuroprotective effect that, in our experimental conditions, takes place in the early stages of damage progression. Finally, this effect is achieved by interfering with both apoptotic and inflammatory mechanisms.
Keywords: permanent focal cerebral ischemia, caspase inhibition, Ac-YVAD.cmk, neuroprotection, apoptosis, TNF-α, IL-1β
In rodents the ischemic area induced by middle cerebral artery occlusion (MCAo) can be divided into an ischemic core and the so called penumbra area. In the core there is a rapid necrotic cell death, whereas in the penumbra neuronal damage develops more slowly (Furlan et al., 1996). Among the factors contributing to delayed injury progression in the penumbra, apoptosis and inflammation have a key role (Linnik et al., 1993; Du et al., 1996;MacManus and Linnik, 1997; Becker, 1998). Caspases, a family of cysteinyl-aspartate proteases (Namura et al., 1998) that include at least 14 members divided into three groups (I, II, and III), are essential players in apoptotic death. Prompted by this background research, groups have studied the effects of caspase inhibition on cerebral ischemia-induced neurodegeneration by using the broad spectrum caspase inhibitor z-VAD, either in the fluoromethylketone (fmk) or dichlorobenzoyloxopentanoic acid (dcb) form and z-DVED.fmk. Both inhibitors induced a significant neuroprotection in mouse models of transient cerebral ischemia (Hara et al., 1997b; Endres et al. 1998; Ma et al., 1998), and z-VAD was neuroprotective also in transient and permanent models in the rat (Loddick et al., 1996; Hara et al., 1997b). Ac-YVAD.cmk (Ac-Tyr-Val-Ala-Asp-cmk), a caspase group I (caspase-1-like) inhibitor mainly active on caspase-1 (Ki 0.8 nm) compared to caspase-4 and caspase-5 (Ki 362 and 163 nm, respectively; Garcia-Calvo et al., 1998), was shown to be neuroprotective in a mouse transient model of cerebral ischemia (Hara et al., 1997b).
An inflammatory reaction, involving cytokines (IL-1β, TNF-α, and MCP-1) production and release, astroglia and microglia activation, and inflammatory cell infiltration, occurs in permanent cerebral ischemia and contributes to subsequent damage (Liu et al., 1993, 1994; Garcia et al., 1994; Schroeter et al., 1994, Stroemer and Rothwell, 1998).
Interplay between inflammation and apoptosis is likely to be a key event in ischemia-induced neurodegeneration. Furthermore, the two processes have certain, preliminary steps in common. For example, IL-1β exhibits both proapoptotic and proinflammatory activity and originates from cleavage of its immature form by caspase-1 (Rothwell and Relton, 1993). On the other hand, caspase-1 is responsible for the activation of executioner caspases, directly involved in apoptosis progression (Denner, 1999). Further evidence that caspase-1 activation could play a pivotal role in the ischemic neurodegeneration comes from caspase-1 −/− mice and mice expressing a dominant-negative mutant caspase-1 gene. Both of these genetically modified mouse strains are more resistant to ischemic insult than wild-type littermates (Hara et al., 1997a; Schielke et al., 1998). However, there is no evidence that pharmacological caspase-1 inhibition induces long-lasting neuroprotection in relevant stroke models, and the mechanisms through which neuroprotection is achieved have not been elucidated.
The present study provides the first evidence of a long-lasting neuroprotection after caspase-1-like activity inhibition. We used the group I caspase inhibitor Ac-YVAD.cmk and administered it after having produced focal ischemia in the rat. Moreover, we show that caspase-1-like activity blockade leads to neuroprotective effects by inhibiting both cell death via apoptosis and release of proinflammatory mediators.
MATERIALS AND METHODS
Surgical procedure and treatments
Studies were performed in male Sprague Dawley rats (Charles River, Calco, Como, Italy), weighing 250–275 gm. Procedures involving animals and their care were conducted in conformity with the institutional guidelines, in compliance with the European Community Council, Directive 86/609 (OJ L 358, 1, December 12, 1987). The animals were caged for at least 3 d before surgery, with ad libitum access to food (until 12 hr before surgery) and water, and maintained on a 12 hr light/dark schedule (lights on at 7:00 A.M.). Focal cerebral ischemia was induced by permanent, unilateral occlusion of the left MCA in rats anesthetized with chloral hydrate (400 mg/kg, i.p.). The pMCAo was performed according to the method described byShigeno et al. (1985) with minor changes. Briefly, all rats underwent subtemporal subperiosteal craniotomy (with intact zygoma) and exposure of the main trunk of MCA under 16× magnification of an operating stereomicroscope (model M351; Leica, Heerbrugg, Switzerland). The exposed artery was electrocoagulated close to its origin at the junction with the olfactory branch and resected. Each rat was allowed to breathe spontaneously, and rectal temperature was maintained at 37°C (36.5–37.5°C) with a homeothermic heating blanket. All necessary care was taken to perform surgery under sterile conditions.
Rats were given either Ac-YVAD.cmk (300 ng/rat in 3 μl) or vehicle (0.6% DMSO in saline, 3 μl) by intracerebroventricular injection into the right lateral ventricle (1.5 mm lateral and 0.8 mm posterior to bregma and 4.0 mm below bone surface), 10 min after the pMCAo. In a separate group of animals, blood pressure, blood gases, blood glucose concentration, and heart rate were monitored before and after drug treatment. All such parameters did not show any significant variation with respect to vehicle-injected animals.
Infarct size analysis
Rats were killed by decapitation 24 hr or 6 d after pMCAo, and the brains were rapidly removed and fixed in Carnoy. Infarct volume was determined on paraffin-embedded coronal slices stained with cresyl violet to assess cortical and subcortical damage. Sections (10 μm) were collected at 1 mm intervals in correspondence of eight different levels (from +3.2 to −3.8 from bregma). Before quantitation, each set of sections was inspected for qualitative evaluation of infarct magnitude. Animals in which the ischemic area was present in less then four sections were excluded from analysis. Total brain and infarct areas were measured by using an image analyzer (Image-Pro Plus; Media Cybernetics). The volume of infarction was computed with Cavalieri's estimator of morphometric volume and corrected for edema. Operators blind to the experimental treatment performed all analyses.
Caspase activity assay
Male Sprague Dawley rats were injected intracerebroventricularly with Ac-YVAD.cmk (300 ng/rat in 3 μl) or vehicle (0.6% DMSO in saline, 3 μl) 10 min after the pMCAo and killed 24 hr or 6 d later. The cerebral cortex of each hemisphere was separately dissected and homogenized in ice-cold lysis buffer containing HEPES 25 mm, pH 7.4, 3-[(3-cholamidopropyl)dimethyl-ammonio]1-propanesulfonate 0.1%, MgCl2 5 mm, EDTA 1.3 mm, EGTA 1 mm, 10 μg/ml pepstatin, aprotinin, and leupeptin, and 1 mm PMSF. The homogenates were centrifuged (15 min at 50,000 rpm) and stored at −80°C. Protein content was assayed by the bicinchoninic acid (BCA) procedure (Pierce, Rockford, IL). Homogenates (200 μg of protein) were incubated at 37°C in a buffer containing 25 mm HEPES, pH 7.4, and 2 mm dithiothreitol supplemented with Ac-YVAD.aminomethylcoumarin (amc; 50 μm; Calbiochem, San Diego, CA) or Ac-DEVD.aminotrifluoromethylcoumarin (afc; 12.5 μm;Calbiochem) to assess, respectively, caspase-1-like and caspase-3 activity. The increase of fluorescence following the cleavage of the fluorogenic amc or afc moiety was monitored for 20 min in a LS50 Perkin-Elmer fluorimeter (excitation, 380 nm; emission, 460 nm for amc moiety; excitation, 400 nm, emission, 505 nm, for afc moiety). For quantitation, standard curves using increasing concentrations of amc or afc moiety (2–30 pmol; Sigma, Steinheim, Germany) were performed in parallel. Activity was expressed as the difference (Δ picomoles of substrate per minute per milligram of protein) between caspase activity in the ischemic cortex and in the contralateral cortex.
The possibility of a direct caspase-3 activity inhibition by Ac-YVAD.cmk was checked by incubating brain homogenate from ischemic hemisphere of vehicle-treated animals killed 24 hr after pMCAo with Ac-YVAD.cmk (500 nm). Caspase-1-like and caspase-3 activity were measured as described above.
Apoptosis detection
Free nucleosome assay. Male Sprague Dawley rats were injected intracerebroventricularly with Ac-YVAD.cmk (300 ng/rat in 3 μl) or vehicle (0.6% DMSO in saline, 3 μl) 10 min after the pMCAo and killed 24 hr and 6 d later. Cerebral cortex homogenates were prepared as described above. The amount of apoptosis was assessed in homogenates by monitoring internucleosomal fragmentation of genomic DNA (Cell Death Detection Elisa Plus kit; Roche Molecular Biochemicals, Mannheim, Germany). This immunoassay is based on recognition of released nucleosomes by mouse monoclonal antibodies directed against DNA and histones. The assay was performed following the manufacturer's instruction.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling technique. The terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining was performed according to the method of Gavrieli et al. (1992) using the ApopTag in situ apoptosis detection kit (Intergen, Purchase, NY). Brains were prepared as described above (see section: infarct size analysis). After deparaffinizing brain sections, proteins were digested with proteinase K, and endogenous peroxidase activity was quenched with 2% H2O2 in PBS. Slices were placed in the equilibration buffer before addition of terminal deoxynucleotidyl transferase (TdT) enzyme. Incubation with TdT in the presence of digoxigenin-labeled nucleotides lasted for 60 min, and the reaction was terminated by rinse in stop/wash buffer. A peroxidase-coupled anti-digoxigenin Fab fragment was used to identify labeled cells and visualized using 3,3′-diaminobenzidine tetrahydrochloride (Sigma, Steinheim, Germany; 150 μg/ml in 50 mm Tris-HCl, pH 7.6) as a chromogen in the presence of 0.0025% hydrogen peroxide. Negative controls were performed by replacing TdT enzyme with distilled water. Coronal sections stained with the TUNEL method were counterstained with cresyl violet.
Biochemical analysis
Cytokine immunoassay. Brain levels of cytokines (IL-1β, TNF-α, IL-10, MCP-1, and MIP-2) were determined using commercially available ELISAs (Biosource International, Camarillo, CA; Endogen, Woburn, MA). Tissue homogenates were prepared by Dounce homogenization of brain cortices as described above and processed in duplicate. The assay was performed following the manufacturer's instruction. For quantitation, reference curves, obtained using increasing concentration of recombinant rat cytokines, were done in parallel. Protein content was estimated by BCA. Cytokine levels were measured as the difference (Δ picograms per milliliter per milligram) between levels in the ischemic cortex and that in the contralateral cortex.
Nitric oxide generation measurement. Nitric oxide production was evaluated measuring the nitrite content of the homogenates with the Griess reaction (Green et al., 1982). Briefly, samples, processed in duplicate, were transferred into a 96 well plate and mixed with freshly prepared solution containing 1% sulfanilamide in orthophosphoric acid and 0.1% naphtyl-ethylendiamide (1:1, v/v). The plate was then read on a multiscan plate reader using a 550 nm filter. A standard curve with increasing concentrations of sodium nitrite was done in parallel and used for quantitation. NO levels were expressed as the difference (Δ micromolar concentration per milligram of protein) between levels in the ischemic cortex and that in the contralateral cortex.
Morphofunctional analysis
Histopathological changes occurring in the ischemic area were analyzed by histological and immunohistochemical stainings. A conventional hematoxylin-eosin stain was used for general morphological analysis. Cells of the microglial/macrophage lineage were identified with a biotinylated Griffonia simplicifolia lectin I isolectin B4 (25 μg/ml diluted in PBS and 0.1% Triton X-100; Vector Laboratories, Burlingame, CA). Lectin incubation was performed overnight, and biotinylated reagents were visualized through a conventional avidin–biotin–HRP technique (Elite kit; Vector Laboratories) using 3,3′-diaminobenzidine tetrahydrochloride (Sigma; 150 μg/ml in 50 mm Tris-HCl, pH 7.6) as a chromogen in the presence of 0.0025% hydrogen peroxide. Nuclei were counterstained with hematoxylin, and after dehydration, sections were coverslipped with DPX.
Statistical analysis
Data are presented as mean ± SEM. Statistical comparisons were made with one-way ANOVA for infarct volumes, whereas for biochemical dosages we applied an analysis of covariance (ANCOVA) model for factorial design, with right hemisphere as covariate, or the Kruskall–Wallis ANOVA and Dunn multiple comparison tests. Software used was SAS System for Windows, version 6.12, and Instat, version 2.03. p < 0.05 was considered statistically significant.
RESULTS
Treatment with Ac-YVAD.cmk induces a long-lasting reduction of ischemic infarct volume
Representative images of ischemic area of control and Ac-YVAD.cmk-treated animals are shown in Figure1. Twenty-four hour after pMCAo, in control animals, the ischemic area encompassed cortical and subcortical (including caudate putamen, globus pallidus, and, more rarely, also nucleus accumbens and amygdaloid complex) structures. Treatment clearly reduced the ischemic damage, as indicated by the decreased size of the area of pallor in the section (Fig. 1A,B). At 6 d, tissue morphology was dramatically changed: the area of pallor had been substituted by a cavitation surrounded by infiltrated and activated cells. There was still reduction of the damaged area after Ac-YVAD.cmk treatment (Fig. 1C,D). Quantitation of these results indicates that Ac-YVAD.cmk significantly reduced infarct volumes 24 hr after occlusion (damage vs hemisphere volume: vehicle, total 41.1 ± 2.3%, cortical 30.9 ± 2.1%, and subcortical 10.2 ± 0.6%; Ac-YVAD.cmk, total 26.5 ± 2.1%, cortical 21.3 ± 1.9%, and subcortical 5.2 ± 0.5%) (Fig.2). At 6 d, the volume of total and cortical infarcts was significantly reduced (damage vs hemisphere volume: vehicle, total 30.6 ± 2.2%, cortical 23.2 ± 1.7%; Ac-YVAD.cmk, total 23.0 ± 2.2%, cortical 17.9 ± 1.7%), but that in subcortical region was not (vehicle, 7.4 ± 0.8%; Ac-YVAD.cmk, 5.9 ± 0.8%) (Fig. 2). Difference in terms of absolute ischemic volume between 24 hr and 6 d could be ascribed to slight differences in the protocol used to perform dehydration before paraffin embedding.
Fig. 1.

Morphological evidence of infarct reduction after Ac-YVAD.cmk treatment. Ischemic brain coronal sections of representative animals injected with DMSO 0.6% (left) or with Ac-YVAD.cmk (right), analyzed 24 hr or 6 d after pMCA occlusion. Scale bar, 5 mm.
Fig. 2.

Ac-YVAD.cmk treatment reduces cerebral infarct volume. Total, cortical, and subcortical infarct volumes were measured on cresyl violet-stained coronal paraffin sections 24 hr and 6 d after pMCA occlusion in animals injected intracerebroventricularly 10 min after the ischemic insult with Ac-YVAD.cmk (white bar) or vehicle only (black bar). Data are expressed as mean ± SEM; *p < 0.05. Absolute values in cubic millimeters at 24 hr; total infarct volume, 111.1 ± 6.6 versus 80 ± 6.4; cortical infarct volume, 83.5 ± 5.7 versus 64.1 ± 5.5; striatal infarct volume, 27.6 ± 1.6 versus 15.9 ± 1.6. Values at 6 d (in cubic millimeters); total infarct volume, 77.6 ± 6.4 versus 55.3 ± 6; cortical infarct volume, 58.7 ± 4.8 versus 43.3 ± 4.5; striatal infarct volume, 18.9 ± 2.2 versus 13.7 ± 2.
Ac-YVAD.cmk treatment reduces caspase-1-like and caspase-3 activity 24 hr but not 6 d after ischemia
To evaluate whether caspase-1-like activity was inhibited, we assessed enzyme activity in cortical homogenates by measuring the cleavage of the fluorogenic substrate Ac-YVAD.amc at 24 hr and 6 d, after a single postischemic administration of the inhibitor. Caspase-1-like activity was almost completely inhibited at 24 hr (Fig.3A). At 6 d, caspase-1-like activity was decreased uniformly in both treated and control animals (Fig. 3A). We also analyzed the activity of caspase-3 after Ac-YVAD.cmk administration. Twenty-four hours after ischemia caspase-3 activity, as evaluated by the fluorogenic substrate Ac-DEVD.afc, was significantly reduced (p < 0.05) compared to the control, whereas at 6 d no difference was observed between control and treated animals (Fig. 3B).
Fig. 3.

Effects of Ac-YVAD.cmk on caspase-1 and -3 activity. Caspase-1 activity was determined by measuring cleavage of the fluorogenic substrate Ac-YVAD.amc in cortical homogenates 24 hr and 6 d after ischemic lesion in rats injected intracerebroventricularly with Ac-YVAD.cmk (white) or vehicle (black) 10 min after occlusion (A). Caspase-3 activity measured as described for caspase-1 activity using the fluorogenic substrate Ac-DEVD.afc (B). Data are expressed as mean ± SEM; **p < 0.01; *p < 0.05. Statistical analysis was performed by ANCOVA using right hemisphere as a covariate (n = 7–9 for evaluation of caspase-1 activity at 24 hr; n = 17 for evaluation at 6 d; n = 7–9 for evaluation of caspase-3 activity at 24 hr; n = 8–9 for evaluation at 6 d).
Separate experiments on brain homogenates indicate that 500 nm Ac-YVAD.cmk inhibited caspase-1-like activity by 91%, but had almost no effect on caspase-3 activity.
Ac-YVAD.cmk treatment reduces free nucleosome formation but has no effect on TUNEL labeling
Analysis of brain homogenates by ELISA of the histone-conjugated DNA fragments originating during apoptotic cell death showed that in ischemic cortices there was an increased level of nucleosomes compared with the contralateral cortices (Fig. 4). Treatment with Ac-YVAD.cmk induced a significant reduction of histone-conjugated DNA fragments at 24 hr (Fig. 4). At 6 d, the amount of free nucleosomes was already reduced in control animals, and drug treatment did not further decrease their level (Fig. 4).
Fig. 4.

Evaluation of apoptosis after Ac-YVAD.cmk. Biochemical quantification of apoptotic cells assessed by monitoring histone-associated DNA fragments in cortical homogenates obtained 24 hr and 6 d after pMCA occlusion from animals treated with Ac-YVAD.cmk (white) or with vehicle (black). Data shown are expressed as mean ± SEM; **p < 0.01. Statistical analysis was performed by ANCOVA using right hemisphere as a covariate (n = 17–19 for evaluation at 24 hr; n = 7–9 for evaluation at 6 d).
Hallmarks of caspase-induced DNA fragmentation were evaluated in situ with both TUNEL technique and in hematoxylin–eosin-stained sections. TUNEL-positive cells, which appeared as early as 6 hr after the occlusion of the MCA, were abundant between 24 and 48 hr within the infarct area (Fig. 5A; data not shown). At 6 d, TUNEL-labeled nuclei persisted within the ischemic tissue predominantly in areas invaded by inflammatory cells, as evidenced by nuclear counterstaining (Fig. 5C). These results suggest that TUNEL staining is likely localized in degenerating neurons initially and in inflammatory cells (macrophages, neutrophils, and lymphocytes at 6 d). Morphological analysis failed to reveal major qualitative differences in the TUNEL-staining pattern after Ac-YVAD.cmk treatment at both time points considered (Fig.5B,D). No TUNEL staining was observed in any area of the contralateral hemispheres in the different experimental groups (data not shown).
Fig. 5.
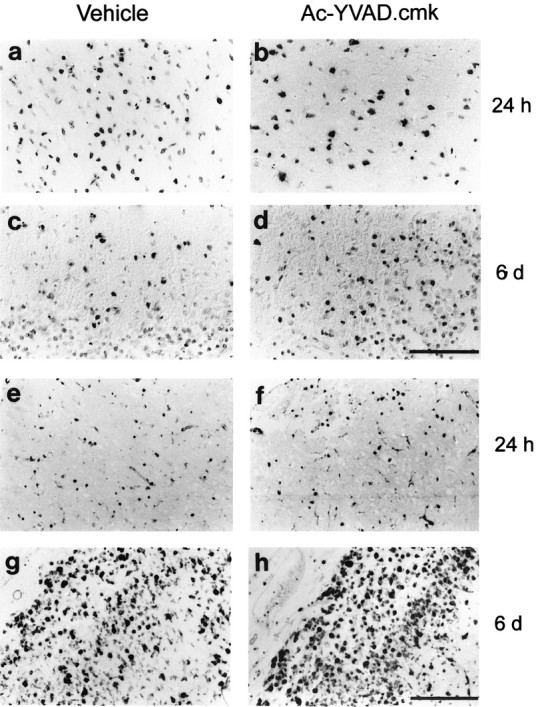
Morphofunctional evaluation of apoptosis and microglia activation. In situ detection of necrotic and apoptotic cell by the TUNEL method (A–D, top panels) and identification of macrophage/activated microglia by lectin histochemistry using Griffonia simplicifolia B4 isolectin staining (E–H, bottom panels) 24 hr and 6 d after left MCA occlusion. Scale bars: A–D, 100 μm; E–H, 200 μm.
IL-1β and TNF-α are reduced after Ac-YVAD.cmk treatment, whereas other inflammatory markers are not
To evaluate microglial activation and peripheral macrophage infiltration, we performed lectin histochemistry on tissue sections obtained from ischemic brains. At 24 hr few rounded macrophage-like cells were scattered throughout the ischemic tissue (mostly in cortex, Fig. 5E), whereas the edges of the lesion were outlined by a rim of activated microglia. Intensely labeled lectin-positive macrophages were also present within cortical blood vessels or in their close proximity within the brain parenchyma. At 6 d both cortical and subcortical areas of the infarct were invaded by phagocytic microglia and infiltrated macrophages (Fig. 5G), mostly localized in areas characterized by a remarkable degree of hypercellularity in hematoxylin–eosin-stained sections. No differences were observed in the distribution of microglial/macrophages cells in the ischemic territory between Ac-YVAD.cmk and vehicle-treated animals at either 24 hr or 6 d after MCAo (Fig. 5F,H). Very weak labeling of resting microglia could be observed on the contralateral hemisphere in all experimental groups (data not shown).
Caspase-1 activation leads to processing and release of mature IL-1β. To assess whether Ac-YVAD.cmk inhibition of caspase-1 activity induced also a reduction of IL-1β production, we measured IL-1β levels in the cortex. They resulted to be high at 24 hr after ischemia and were significantly reduced by Ac-YVAD.cmk treatment (p < 0.05) (Fig.6A). Six days after ischemia there was no difference between treated and control animals (Fig. 6A). It has to be noted that the ELISA kit used for IL-1β could not be tested for cross-reactivity with pro-IL1β because of the lack of commercially available precursor form of this cytokine. However, even in the presence of cross-reactivity, ELISA results are a good proxy for IL-1β because the cytokine can only be generated from its precursor form. To further investigate the effect of Ac-YVAD.cmk on the inflammatory reaction elicited by pMCAo we also measured the level of selected cytokines (one proinflammatory, TNF-α, and one anti-inflammatory, IL-10), chemokines (MCP-1 and MIP-2), and of nitric oxide (NO). Cortical levels of TNF-α in ischemic cortices, as measured by immunoassay on homogenates, were elevated at 24 hr but subsided by 6 d after ischemia (Fig. 6B). Treatment with Ac-YVAD.cmk induced a significant reduction of TNF-α levels in ipsilateral cortex at 24 hr (p < 0.05), whereas the effect disappeared at 6 d (Fig.6B). In control rats, IL-10 levels were similar at 24 hr and 6 d after ischemia and were not affected by Ac-YVAD.cmk treatment (Table 1). Conversely, MCP-1 and MIP-2 levels were elevated at 24 hr but sharply declined at 6 d (Table 1). As for IL-10, treatment with Ac-YVAD.cmk did not influence the amount of these chemokines in the cortex (Table 1). On the other hand, NO was the only mediator more abundant at 6 d than at 24 hr (Table 1). A similar trend was observed also in the treated animals (Table 1). Even though the increase at 6 d was less evident after treatment, no statistically significant difference could be noticed between treated and control animals (Table 1).
Fig. 6.

Effect of Ac-YVAD.cmk treatment on IL-1β and TNF-α level. A, Cerebral cortex IL-1β levels in animals treated with Ac-YVAD.cmk (white) or with vehicle (black) and killed 24 hr and 6 d after pMCA occlusion measured by ELISA assay and expressed as difference between left and right hemisphere. Data shown are expressed as mean ± SEM. Statistical analysis was performed by Mann–WhitneyU test (n = 6–10); *p < 0.05. B, Cerebral cortex TNF-α levels in animals treated as described above. Statistical analysis was performed by ANCOVA using right hemisphere as a covariate (n = 17–19 for evaluation at 24 hr;n = 7–9 for evaluation at 6 d); *p < 0.05.
Table 1.
Production of inflammatory mediators after permanent middle cerebral artery occlusion after Ac-YVAD.cmk or vehicle (DMSO 0.6%) treatment
| 24 hr | 6 d | |||
|---|---|---|---|---|
| Vehicle | Ac-YVAD.cmk | Vehicle | Ac-YVAD.cmk | |
| MCP-11-a | 141.6 ± 8.5 | 153.6 ± 20.2 | 19.7 ± 3.7 | 15.8 ± 3.4 |
| MIP-21-a | 568.9 ± 54.6 | 508.1 ± 63.7 | 77.6 ± 15.3 | 72.7 ± 17.2 |
| IL-101-a | 7.5 ± 4.3 | 5.5 ± 1.7 | 6.7 ± 1.9 | 2.7 ± 1.7 |
| NO1-b | 12.1 ± 2.1 | 10.9 ± 4.7 | 25.1 ± 3.5 | 15.9 ± 3.6 |
Data are expressed in picograms per milliliter per milligram of protein as difference between ipsilateral and contralateral hemisphere.
Data are expressed in micromolar concentration per milligram of protein as difference between ipsilateral and contralateral hemisphere.
DISCUSSION
Administration of broad spectrum caspase inhibitors to ischemic rodents induces neuroprotection. For the elucidation of the underlying processes it is crucial to identify which caspase or caspases are involved. Some caspases have a major role in apoptosis induction either as initiators (caspase-8, -9, and -6) or executioners (caspase-3 and -7), whereas others (caspase-4 and -5) seem to be mainly involved in promoting inflammation (Denner, 1999). Caspase-1 has the peculiarity of being involved in the activation of both apoptosis and inflammation, through the intermediate of the pro-inflammatory cytokine IL-1β (Kuida et al., 1995; Rothwell et al., 1997). Evidence from studies performed with knock-out mice (Friedlander and Yuan, 1998) indicate that caspase-1 is important in the development of cerebral ischemia damage. However, evaluation of neuroprotective effects in knock-out and transgenic mice should be appraised with caution. To circumvent these problems, we addressed the question of caspase-1 involvement in the formation of postischemia infarct, by using a pharmacological-based approach combined with biochemical and morphofunctional analysis.
Ac-YVAD.cmk is an irreversible caspase-1-like activity inhibitor. A previous report on administration of Ac-YVAD.cmk to ischemic mice demonstrated neuroprotective effects of this compound (Hara et al., 1997b). However, ischemia was performed with the transient MCAo model in mouse, compared with the pMCAo model in rat that we used in our study. The compound was administered twice, 45 min before ischemia onset and immediately after reperfusion, whereas our experimental setting consisted of a single drug administration 10 min after ischemia onset. Finally, in the study by Hara et al. (1997b) the ischemic volume was assessed only at 18 hr after reperfusion. Nevertheless, in our studies Ac-YVAD.cmk induced a significant reduction of ischemic volume at 24 hr. More importantly neuroprotection persisted at least up to 6 d after ischemia.
Three considerations are of particular interest. Primarily, administration of Ac-YVAD.cmk resulted in neuroprotection even though the treatment started after cerebral ischemia onset and the drug was delivered by a single bolus injection, aspects that are relevant to possible therapeutic exploitation (Jonas et al., 1997). Second, neuroprotection induced by the inhibition of caspase-1-like activity is long-lasting. This suggests that blockade of caspase-1-like activity does not simply delay cellular death, but it interferes with key events in the initiation of neurodegeneration and blocks this process in a significant manner in the ischemic tissue. Third, the majority of these were performed with the transient model and mainly in the mouse (Hara et al., 1997b; Endres et al., 1998, Fink et al., 1998; Ma et al., 1998). Our results obtained with the permanent model in the rat clearly establish that caspase activation is a general process in the formation of neuronal damage after an ischemic insult that depends neither on the type of ischemia nor on the species studied.
Treatment of ischemic rats with Ac-YVAD.cmk clearly reduced caspase-1-like activity in cortical homogenates at 24 hr, but the effect was lost 6 d after treatment. Particularly intriguing was the observation that caspase-3 was also significantly inhibited by Ac-YVAD.cmk treatment at 24 hr. The possibility that Ac-YVAD.cmkin vivo may directly inhibit caspase-3 cannot be ruled out. However, two observations argue strongly against this possibility. First, in vitro results indicate that theKi of Ac-YVAD.cmk for caspase-1 and -3 are extremely different (Ki: caspase-1, 0.8 nm; caspase-3, >10.000 nm; Garcia-Calvo et al., 1998). Second, we observed that relatively high concentration of Ac-YVAD.cmk used for anin vitro assay on ischemic cortical homogenate did not affect caspase-3 activity. Therefore, it seems more likely that, because of the activator role of caspase-1, reduction in caspase-3 activity could be a downstream effect of caspase-1 inhibition, as supported by the observation that caspase-1 is able to directly process pro-caspase-3 to its active form (Tewari et al., 1995).
Data obtained by analysis of DNA nucleosomal fragmentation using a quantitative biochemical assay revealed a clear decrease in free nucleosomes 24 hr after treatment (but not at 6 d), confirming that reduction of apoptotic cell death is involved in this model of neuroprotection. Taken together, these results suggest that a decrease in apoptotic events induced by inhibition of caspase-1-like activity is taking place in the early phase of ischemic damage development, but it does not occur at later stages. On the other hand, we were unable to see any significant difference in apoptosis between Ac-YVAD.cmk-treated and control animals by using the TUNEL technique. This result is not completely surprising when considering that the specificity of the TUNEL technique has been questioned in in vivo models, in which both apoptotic and necrotic cells have been shown to be labeled with this method (de Torres et al., 1997).
As mentioned above, caspase-1 is also involved in the generation of the proinflammatory cytokine IL-1β. Our results clearly indicate that inhibition of caspase-1-like activity dramatically reduces the level of IL-1β, although, with the available antibodies, it is impossible to discriminate between pro- and mature IL-1β. Studies on the role of IL-1β in neurodegeneration support the hypothesis that IL-1β exacerbates tissue damage induced by either ischemia or excitotoxicity (Rothwell, 1997; Rothwell et al., 1997), even though it does not seem to have a direct toxic action per se (Rothwell et al., 1997). In agreement with these results, it is conceivable that a decrease in IL-1β levels could be an important cofactor in the reduction of brain tissue degeneration observed in our experiments. Recently, another cytokine, IL-18, has been identified. IL-18 is cleaved and activated by caspase-1. It induces IFN-γ, which can enhance caspase-1 expression, and a positive feedback loop has been hypothesized (Culhane et al., 1998). Inhibition of caspase-1 by Ac-YVAD.cmk, could reduce IL-18 levels, interrupt this loop, and thus potentiate enzyme inhibition. However, various other mechanisms could participate to this neuroprotective effect. Release of IL-1β induces several effects, among them stimulation of TNF-α production (Benveniste, 1995). In turn, TNF-α may directly trigger apoptosis through the activation of the TNF-α receptor. Release of IL-1β has also been linked to the induction of nitric oxide production (Liu et al., 1996; Romero et al., 1996), a molecule that has both neuroprotective and neurotoxic effects (Iadecola, 1997). On the other hand, the neuroprotective effect of caspase-1-like activity inhibition could also be related to a reduction of chemokines such as MCP-1 and MIP-2 that are involved in the recruitment of inflammatory cells. However, the levels of IL-10, MCP-1, MIP-2 and nitric oxide were not modified by the treatment either at 24 hr or at 6 d, suggesting that these mediators are not involved in the neuroprotection we observed. Conversely, TNF-α levels at 24 hr were reduced by almost half by the treatment, but no effect was observed at 6 d. The role of TNF-α in the neurodegeneration is still a matter of discussion. The knock-out technology has made it possible to study the effect of ischemia in mice lacking TNF-α receptors, either the p75, the p55 or both of them (Bruce et al., 1996;Gary et al., 1998). The picture emerging from these studies suggests that TNF-α is neuroprotective, because the knock-out mice showed an increase in infarct volume. On the contrary, several other studies using different techniques indicate that TNF-α is neurotoxic and capable of inducing both apoptosis, through activation of the caspase cascade, or inflammatory reaction by enhancing the synthesis and release of IL-1β, IL-6, colony-stimulating factors, TNF-α itself, and by increasing leukocyte adhesion and infiltration (Liu et al., 1994; Benveniste, 1995; Barone et al., 1997; Pan et al., 1997). Our data support the hypothesis that an elevated level of TNF-α is associated with an exacerbation of the tissue damage induced by cerebral ischemia (Bertorelli et al., 1998). However, it is possible that TNF-α may have opposite actions at different time points inducing damage in the early ischemic stages, and neuroprotection through the stimulation of neurotrophic factor production at later stages (Pan et al., 1997; Munoz-Fernandez and Fresno, 1998). Thus, further studies are necessary to thoroughly address this problem.
Our results together with the data present in the literature suggest that the neuroprotective effect of caspase-1-like activity inhibition could be achieved by both prevention of apoptosis, taking place mainly in neuronal cells, and reduction of inflammatory cytokine production principally involving activated astrocytes and microglial cells. Thus, direct prevention of neuronal death by apoptosis blockade and indirect protection by reduction of proinflammatory cytokine production and release are probably acting in concert to prevent neurodegeneration.
To summarize, our data indicate that inhibition of caspase-1 and, more generally caspase-1-like activity, results in a long-term neuroprotection from ischemic insult. This neuroprotective effect is achieved not only by partially blocking the apoptotic pathway, but also by decreasing the level of IL-1β. Conversely, additional studies are necessary to fully establish a causative link between neuroprotection and the reduction of TNF-α level we observed. Taken together, these results indicate that caspase-1-like activity inhibition could be a useful way to interfere simultaneously with two of the major mechanisms involved in neurodegeneration, apoptosis, and inflammation, and thus represent a promising therapeutic approach to reduce neuronal damage after cerebral ischemia.
Footnotes
We thank Dr. E. Ongini for critical revision of this manuscript and Drs. S. Bortolazzi and M. Campanella for assistance with experiments.
M.R. and C.S. contributed equally to this work.
Correspondence should be addressed to Massimiliano Beltramo, Schering-Plough Research Institute, San Raffaele Biomedical Science Park, Milan, Italy. E-mail: massimiliano.beltramo@spcorp.com.
REFERENCES
- 1.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-α a mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 2.Benveniste EN. Cytokine production. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 700–713. [Google Scholar]
- 3.Bertorelli R, Adami M, Di Santo E, Ghezzi P. MK 801 and dexamethasone reduce both tumor necrosis factor levels and infarct volume after focal cerebral ischemia in the rat brain. Neurosci Lett. 1998;246:41–44. doi: 10.1016/s0304-3940(98)00221-3. [DOI] [PubMed] [Google Scholar]
- 4.Becker KJ. Inflammation and acute stroke. Curr Opin Neurol. 1998;11:45–49. doi: 10.1097/00019052-199802000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:2788–2794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 6.Culhane AC, Hall MD, Rothwell NJ, Luheshi GN. Cloning of rat interleukin-18 cDNA. Mol Psychiatry. 1998;3:362–366. doi: 10.1038/sj.mp.4000389. [DOI] [PubMed] [Google Scholar]
- 7.Denner L. Caspases in apoptotic death. Exp Opin Invest Drugs. 1999;8:37–50. doi: 10.1517/13543784.8.1.37. [DOI] [PubMed] [Google Scholar]
- 8.de Torres C, Munell F, Ferrer I, Reventós J, Macaya A. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci Lett. 1997;230:1–4. doi: 10.1016/s0304-3940(97)00445-x. [DOI] [PubMed] [Google Scholar]
- 9.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis ? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Fink K, Zhu J, Namura S, Shimizu-Sasamata M, Endres M, Ma J, Dalkara T, Yuan J, Moskowitz MA. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J Cereb Blood Flow Metab. 1998;18:1071–1076. doi: 10.1097/00004647-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Friedlander RM, Yuan J. ICE, neuronal apoptosis and neurodegeneration. Cell Death Diff. 1998;5:823–831. doi: 10.1038/sj.cdd.4400433. [DOI] [PubMed] [Google Scholar]
- 13.Furlan M, Marchal G, Viader F, Derlon JM, Baron JC. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann Neurol. 1996;40:216–226. doi: 10.1002/ana.410400213. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;49:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 15.Garcia GH, Liu KF, Yoshida Y, Zhang ZG, Lian J, Chen S, del Zoppo GJ. Influx of leucokytes and platelets in an evolving brain infarct (Wistar rat). Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- 16.Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green LC, Wagner DA, Glogouski J, Skipper PL, Wishnok JS, Tarmenbeum SR. Analysis of nitrate, nitrite and [15]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 19.Hara H, Fink K, Endres M, Friedlander RM, Gaglardini V, Yuan J, Moskowitz MA. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997a;17:370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1 ? converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA. 1997b;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 22.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 23.Jonas S, Tran AQ, Eisenberg E, Azam M, Viera D, Grumet S. Does effect of a neuroprotective agent on volume of experimental animal cerebral infarct predict effect of the agent on clinical outcome in human stroke? Ann NY Acad Sci. 1997;825:281–287. doi: 10.1111/j.1749-6632.1997.tb48439.x. [DOI] [PubMed] [Google Scholar]
- 24.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2009. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Zhao ML, Brosnan CF, Lee SC. Expression of type II nitric oxide synthase in primary human astrocytes and microglia: role of IL-1β and IL-1 receptor antagonist. J Immunol. 1996;157:3569–3576. [PubMed] [Google Scholar]
- 26.Liu T, Mc Donnell PC, Young PR, White RF, Siren AL, Hallenbeck JM, Barone FC, Feuerstein GZ. Interleukin 1-β mRNA expression in ischemic rat cortex. Stroke. 1993;24:1746–1751. doi: 10.1161/01.str.24.11.1746. [DOI] [PubMed] [Google Scholar]
- 27.Liu T, Clark RK, Mc Donnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-α expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 28.Loddick SA, MacKenzie A, Rothwell NJ. An ICE inhibitor, z-VAD-dbc attenuates ischaemic brain damage in the rat. NeuroReport. 1996;7:1465–1468. doi: 10.1097/00001756-199606170-00004. [DOI] [PubMed] [Google Scholar]
- 29.Ma J, Endres M, Moskowitz MA. Synergistic effects of caspase inhibitors and MK-801 in brain injury after transient focal cerebral ischaemia in mice. Br J Pharmacol. 1998;124:756–762. doi: 10.1038/sj.bjp.0701871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacManus JP, Linnik MD. Gene expression induced by cerebral ischemia: an apoptotic perspective. J Cereb Blood Flow Metab. 1997;17:815–832. doi: 10.1038/aj.jcbfm.9590266. [DOI] [PubMed] [Google Scholar]
- 31.Munoz-Fernandez MA, Fresno M. The role of tumor necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog Neurobiol. 1998;56:307–340. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 32.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan W, Zadina JE, Harlan RE, Weber JT, Banks WA, Kastin AJ. Tumor necrosis factor-α: a neuromodulator in the CNS. Neurosci Biobehav Rev. 1997;21:603–613. doi: 10.1016/s0149-7634(96)00047-4. [DOI] [PubMed] [Google Scholar]
- 34.Romero LI, Tatro JB, Field JA, Reichlin S. Roles of IL-1β and TNFα in the endotoxin-induced activation of nitric oxide synthase in cultured rat brain. Am J Physiol. 1996;270:R326–332. doi: 10.1152/ajpregu.1996.270.2.R326. [DOI] [PubMed] [Google Scholar]
- 35.Rothwell NJ. Neuroimmune interactions: the role of cytokines. Br J Pharmacol. 1997;121:841–847. doi: 10.1038/sj.bjp.0701248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothwell NJ, Relton JK. Involvement of interleukin-1 and lipocortin-1 in ischaemic brain damage. Cerebrovasc Brain Metab Rev. 1993;5:178–198. [PubMed] [Google Scholar]
- 37.Rothwell NJ, Allan S, Toulmond S. The role of interleukin-1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. 1997;100:2648–2652. doi: 10.1172/JCI119808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schielke GP, Yang GY, Shivers BD, Lorris Betz A. Reduced ischemic brain injury in interleukin-1β converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- 39.Schroeter M, Jander S, Witte OW, Stoll G. Local immunoresponses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 40.Shigeno T, McCulloch J, Graham DI, Mendelow AD, Teasdale GM. Pure cortical ischemia versus striatal ischemia. Circulatory, metabolic, and neuropathologic consequences. Surg Neurol. 1985;24:47–51. doi: 10.1016/0090-3019(85)90063-1. [DOI] [PubMed] [Google Scholar]
- 41.Stroemer RP, Rothwell NJ. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1β in the rat. J Cereb Blood Flow Metab. 1998;18:833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]