

Abstract
In previous studies from our laboratory, chronic noncontingent morphine administration decreased μ opioid receptor-activated G-proteins in specific brainstem nuclei. In the present study, μ opioid receptor binding and receptor-activated G-proteins were examined after chronic heroin self-administration. Rats were trained to self-administer intravenous heroin for up to 39 d, achieving heroin intake up to 366 mg · kg−1 · d−1. μ opioid-stimulated [35S]GTPγS and [3H]naloxone autoradiography were performed in adjacent brain sections. Agonist-stimulated [35S]GTPγS autoradiography also examined other G-protein-coupled receptors, including δ opioid, ORL-1, GABAB, adenosine A1, cannabinoid, and 5-HT1A. In brains from heroin self-administering rats, decreased μ opioid-stimulated [35S]GTPγS binding was observed in periaqueductal gray, locus coeruleus, lateral parabrachial nucleus, and commissural nucleus tractus solitarius, as previously observed in chronic morphine-treated animals. In addition, decreased μ opioid-stimulated [35S]GTPγS binding was found in thalamus and amygdala after heroin self-administration. Despite this decrease in μ-activated G-proteins, [3H]naloxone binding demonstrated increased μ opioid receptor binding in several brain regions after heroin self-administration, and there was a significant decrease in μ receptor G-protein efficiency as expressed as a ratio between agonist-activated G-proteins and μ receptor binding. No effects on agonist-stimulated [35S]GTPγS binding were found for any other receptor examined. The effect of chronic heroin self-administration to decrease μ-stimulated [35S]GTPγS binding varied between regions and was highest in brainstem and lowest in the cortex and striatum. These results not only provide potential neuronal mechanisms that may contribute to opioid tolerance and dependence, but also may explain why various chronic effects of opioids develop to different degrees.
Keywords: μ opioid receptor, heroin, G-protein, desensitization, δ opioid receptor, nociceptin/orphanin FQ receptor
Chronic abuse of heroin remains a major problem: the ability of heroin to produce high levels of tolerance and physical dependence, combined with its reinforcing properties, create a chronic relapsing disease with a significant fatality rate (Goldstein and Herrera, 1995). The acute CNS effects of heroin (3,6-diacetylmorphine) are primarily mediated by the binding of its metabolites, 6-monoacetylmorphine (6-MAM) and morphine, to μ opioid receptors (Umans and Inturrisi, 1981; Inturrisi et al., 1983), although recent evidence also suggests the potential existence of specific receptors for heroin or its metabolites (Rossi et al., 1997;Schuller et al., 1999). μ opioid receptors are located in brain regions known to mediate the acute actions of opiates (Young and Kuhar, 1979; Herkenham and Pert, 1982), which include reinforcement, analgesia, sympathetic nervous system effects, and respiratory depression. Chronic opiate administration leads to the development of tolerance and physical dependence on most opiate-mediated effects, although the rate and magnitude of the development of these chronic effects vary among different symptoms (Way et al., 1969).
Despite numerous investigations, the neuronal basis of opiate tolerance and dependence remains unclear. Opioid receptors belong to the family of inhibitory G-protein-coupled receptors (Evans et al., 1992; Kieffer et al., 1992; Chen et al., 1993; Thompson et al., 1993), and chronic opiate exposure in cultured cell lines results in desensitization followed by receptor downregulation (Law et al., 1983; Puttfarcken and Cox, 1989; Breivogel et al., 1997). However, previous studies in animals have produced conflicting results regarding the effects of chronic opiate administration on opioid receptor number (Klee and Streaty, 1974; Tao et al., 1987; Brady et al., 1989; Yoburn et al., 1993), and it has been suggested that many of the cellular adaptations underlying tolerance and dependence occur at the level of the signal transducing G-protein (Blasig et al., 1979; Tao et al., 1993) or by compensatory mechanisms involving downstream effectors (Nestler, 1992;Noble and Cox, 1996, 1997).
The development of agonist-stimulated [35S]GTPγS binding for opioid receptors (Traynor and Nahorski, 1995) has provided an opportunity not only to determine acute mechanisms of agonist efficacy (Clark et al., 1997; Selley et al., 1997b; Alt et al., 1998; Selley et al., 1998) but also to examine effects of chronic agonist treatment on coupling of receptors to G-proteins (Breivogel et al., 1997; Selley et al., 1997a). In brain, this technique has been extended by the development of [35S]GTPγS autoradiography (Sim et al., 1995), which allows the visualization of receptor-activated G-proteins in brain sections. Using this technique, our laboratory (Sim et al., 1996a) showed previously that chronic morphine administration decreased μ opioid-activated G-proteins in specific nuclei in brainstem, but not in forebrain structures thought to contribute to the reinforcing effects of opiates (Bozarth and Wise, 1984; Vaccarino et al., 1985). These results suggested that although μ opioid receptor desensitization occurs after chronic morphine administration, cellular adaptations may depend on the brain region examined. The present study was designed to extend those results by determining the effects of heroin self-administration on opioid receptors and receptor-activated G-proteins in brain.
MATERIALS AND METHODS
Materials. Male Fischer 344 rats (250–300 gm) were purchased from Harlan Laboratories (Indianapolis, IN). [35S]GTPγS (1250 Ci/mmol) and [3H]naloxone (48 Ci/mmol) were purchased from New England Nuclear (Boston, MA). [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) was obtained from Peninsula Laboratories (Belmont, CA). R(+)-baclofen HCl, R(+)-WIN 55,212–2 mesylate, 5-carboxamidotryptamine maleate (5-CT), and R(−)N6-(2-phenylisopropyl)adenosine (PIA) were purchased from Research Biochemicals International (Natick, MA). Adenosine deaminase,p-Cl-[d-penicillamine2,5,p-Cl-Phe4]enkephalin (p-Cl-DPDPE), and GDP were obtained from Sigma (St. Louis, MO). The nociceptin/orphanin FQ (N/OFQ) peptide was synthesized in the Protein Core Laboratory of the Cancer Center at Wake Forest University. Reflections autoradiography film was purchased from New England Nuclear. Heroin hydrochloride was provided by Research Triangle Institute (Research Triangle Park, NC) through the Drug Supply Program of the National Institute on Drug Abuse. All doses are reported in terms of the free base of heroin. Pentobarbital (Nembutal) was purchased from Abbott Laboratories (North Chicago, IL) in a vehicle of 10:40:50 ethanol/propylene glycol/water at a concentration of 50 mg/ml. Atropine sulfate was purchased from Sigma, and heparin was purchased from Elkins-Sinn (Cherry Hill, NJ). All other reagent grade chemicals were obtained from Sigma or Fisher Scientific (Pittsburgh, PA).
Heroin self-administration. Male Fischer 344 rats were implanted with chronic indwelling catheters for intravenous administration of drugs as described previously (Martin et al., 1995). Control rats were subjected to the same surgery and catheters as drug-treated rats. After recovery from surgery, animals were trained to self-administer intravenous infusions of 0.06 mg/kg heroin during daily 4 hr sessions on a fixed-ratio 10 schedule of reinforcement (Martin et al., 1995). Once drug intake was stable (∼1 mg · kg−1 · d−1), this dose of heroin was made available for self-administration 24 hr/d. The schedule for increasing heroin doses, determined from the rate of drug self-administration until the final infusion rate (6 mg/kg) produced intakes of up to 366 mg · kg−1 · d−1, is presented in Results. The rats were maintained on this schedule of increasing heroin doses for 29–39 d. Animals were killed after 5 d of self-administration of 6 mg/kg of heroin, during which time total heroin intake did not vary by >10% of the mean for each animal.
Rats were decapitated, and brains were removed and frozen in isopentane at −35°. Coronal sections (20 μ m) were cut throughout the rostral-caudal extent of the brain on a cryostat maintained at −20°, mounted on gelatin-subbed slides, and stored at −80° until processed as described below.
Agonist-stimulated [35S]GTPγS autoradiography in brain sections. Agonist-stimulated [35S]GTPγS autoradiography was performed as described previously (Sim et al., 1995). Sections were rinsed in TME buffer (50 mm Tris-HCl, 3 mmMgCl2, 0.2 mm EGTA, 100 mm NaCl, pH 7.4) at 25° for 10 min, followed by a 15 min preincubation in TME buffer containing 2 mm GDP and adenosine deaminase (9.5 mU/ml) at 25°. Sections were then incubated in TME buffer with GDP, adenosine deaminase, 0.04 nm[35S]GTPγS, and appropriate agonist at 25° for 2 hr. The following agonists were used: 10 μ mDAMGO (μ opioid), 10 μ mp-Cl-DPDPE (δ opioid), 3 μ m N/OFQ, 10 μm WIN 55212–2 (cannabinoid), 300 μm baclofen (GABAB), 1 μm PIA (adenosine A1), or 2 μ m 5-CT (5HT1A). Sections incubated with N/OFQ contained protease inhibitors, and those incubated with WIN 55,212–2 contained bovine serum albumin, as described previously (Sim et al., 1995, 1996c). Slides were rinsed twice for 2 min each in cold Tris buffer (50 mmTris-HCl, pH 7.4) and once in deionized H2O. Slides were dried overnight and exposed to film for 48 hr in film cassettes containing [14C] microscales (Amersham, Arlington Heights, IL) for densitometric analysis.
[3H]Naloxone autoradiography. Sections were equilibrated in TME buffer for 15 min at 25°. Slides were then incubated in 2 nm[3H]naloxone in TME buffer containing 1 μ mp-Cl-DPDPE (to block δ receptors) for 1 hr at 25°. Slides were rinsed three times for 2 min each in 50 mm Tris buffer at 4°C, then briefly in deionized H2O at 4°. Nonspecific binding was assessed in the presence of 10 μ mlevallorphan. Slides were dried under a cool stream of air and exposed to Hyperfilm-3H for ∼8 weeks. All film cassettes included a [3H] microscale (Amersham) for calibration of results.
Analysis of data. Films were digitized with a Sony XC-77 video camera and analyzed densitometrically using the NIH IMAGE program for Macintosh computers. For [35S]GTPγS autoradiography, resulting values were expressed as nanocuries of [35S]per gram of tissue and were corrected for [35S] from [14C] standards based on incorporation of [35S] into sections of frozen brain paste, as described previously (Sim et al., 1997). Net agonist-stimulated [35S]GTPγS binding was calculated by subtracting basal binding (obtained in the absence of agonist) from agonist-stimulated binding. For [3H]naloxone binding, nonspecific binding was densitometrically subtracted from total binding before analysis, and resulting values represent specific nanocuries per gram of [3H]naloxone binding. Data are reported as mean values ± SE of triplicate sections of brains from eight treated and seven control animals. Statistical comparison between control and heroin self-administering rats was performed by ANOVA followed by post hoc analysis using the two-tailed non-paired Student's t test.
RESULTS
Heroin self-administration
To produce opiate tolerance and dependence using a procedure that parallels the behavior of chronic opiate use in humans, the current study established a paradigm of heroin self-administration in rats. The results of this procedure are illustrated in Figure1, which shows daily heroin intake for self-administering animals over the course of the study. In daily 4 hr sessions, animals were trained to self-administer intravenous infusions of 0.06 mg/kg heroin on a fixed-ratio 10 schedule of reinforcement (Martin et al., 1995). The dose of heroin was increased as drug intake escalated across several days, thereby allowing the animals to maintain high levels of daily heroin intake and still have time for feeding and sleeping. Beginning with a dose of 0.06 mg/kg per infusion, heroin intake increased from an average of 1.8 ± 0.33 mg · kg−1 · d−1to 5.1 ± 1.6 mg · kg−1 · d−1 over the next 7.6 ± 1.1 d. The dose of heroin was subsequently increased to 0.3 mg/kg per infusion. Animals compensated for the dose increase by taking fewer infusions; however, daily heroin intake again increased from 5.6 ± 0.93 mg · kg−1 · d−1 to 16.9 ± 3.7 mg · kg−1 · d−1 over an average of 6.9 ± 1.1 d. The dose of heroin was then increased to 1.2 mg/kg per infusion, and daily drug intake subsequently increased from 16.2 ± 2.8 mg ·kg−1 · d−1 to 81.4 ± 17.0 mg · kg−1 · d−1 over 8.8 ± 1.8 d. The dose of heroin was finally increased to 6 mg/kg per infusion, and daily heroin intake reached a maximum of 366 mg · kg−1 · d−1, with animals maintaining self-administration for 29–39 d. The tolerance produced by this paradigm is illustrated by the fact that this daily heroin intake was lethal in drug-naive animals. Moreover, these animals were also highly dependent, because interruption of drug intake produced significant withdrawal symptoms (ptosis, diarrhea, vocalization) soon after drug cessation in five animals that were not included in the autoradiographic studies (data not shown).
Fig. 1.

Heroin intake in self-administering animals. Rats were initially trained to self-administer intravenous infusions of 0.06 mg/kg heroin during daily 4 hr sessions on a fixed-ratio 10 schedule of reinforcement. As drug intake became stable and began to increase, the dose of heroin was gradually increased to 6 mg · kg−1 · d−1 as described in Results. Data are mean values ± SEM of daily heroin intake in eight rats.
Autoradiography of μ opioid-stimulated [35S]GTPγS binding and [3H]naloxone binding
Coronal sections from several brain levels were processed for DAMGO-stimulated [35S]GTPγS binding and [3H]naloxone binding to examine μ opioid-activated G-proteins and μ receptors, respectively. The use of [3H]naloxone along with DAMGO-stimulated [35S]GTPγS autoradiography allows both parameters to be determined under similar assay conditions and provides direct comparison between receptor binding and activation of G-proteins. Representative sections of DAMGO-stimulated [35S]GTPγS binding from control and chronic heroin self-administering rats are shown in Figure2. Figure 2, A andB, shows autoradiograms at the level of locus coeruleus (A) and periaqueductal gray/interpeduncular nucleus (B), regions in which decreased DAMGO-stimulated [35S]GTPγS binding was evident in our previous study of chronic morphine-treated rats (Sim et al., 1996a). In sections from control rats, high levels of μ receptor-activated G-proteins were observed in these brainstem nuclei. However, in sections from chronic heroin self-administering rats, μ receptor-stimulated G-protein activity was visibly reduced in these regions. Figure 2C shows autoradiograms of thalamus, amygdala, and hypothalamus, where decreases in DAMGO-stimulated [35S]GTPγS binding in sections from heroin self-administering rats were particularly evident in the medial thalamus and amygdala, with a magnitude of decrease similar to that observed in brainstem. These data are in contrast to the previous study using chronic morphine-treated rats, where none of these regions displayed any decrease in μ-stimulated [35S]GTPγS binding after chronic morphine treatment (Sim et al., 1996a). On the other hand, only a small decrease was seen in caudate putamen, cingulate cortex, and nucleus accumbens from heroin self-administering rats (Fig.2D).
Fig. 2.

Representative autoradiograms showing μ opioid-stimulated [35S]GTPγS binding in coronal brain sections from control and heroin self-administering rats. Decreases in DAMGO-stimulated [35S]GTPγS binding are evident in nuclei in sections from heroin-treated rats, including locus coeruleus (A), periaqueductal gray/interpeduncular nucleus (B), and thalamus/amygdala (C), but not caudate putamen/nucleus accumbens/cingulate cortex (D).
The anatomical distribution of [3H]naloxone binding was the same as that seen for DAMGO-stimulated [35S]GTPγS binding. Representative sections in Figure 3 (which show adjacent sections from the same animals presented in Fig. 2) show an increase in [3H]naloxone binding in several regions of heroin self-administration versus control brains; these regions include locus coeruleus (Fig. 3A), periaqueductal gray/interpeduncular nucleus (Fig. 3B), and hypothalamus (Fig. 3C). Little visible change was apparent was in the amygdala, thalamus (Fig. 3C), caudate putamen, or cingulate cortex (Fig. 3D) from heroin self-administering rats.
Fig. 3.

Representative autoradiograms showing [3H]naloxone binding in control and heroin self-administering rats. Sections are at the same levels and from the same animals as those in Figure 2. Increases in [3H]naloxone binding are evident in sections from heroin-treated rats in regions including locus coeruleus (A), periaqueductal gray/interpeduncular nucleus (B), and hypothalamus (C) but not thalamus/amygdala (C) or caudate putamen (D).
Quantification of basal and agonist-stimulated [35S]GTPγS binding, and specific [3H]naloxone binding, was obtained by measuring optical density in autoradiograms of regions of interest. Basal [35S]GTPγS binding was generally unaffected by chronic heroin self-administration (data not shown), except in the interpeduncular nucleus and locus coeruleus, where heroin self-administration significantly decreased basal [35S]GTPγS binding by 18 and 20%, respectively (p < 0.05). This finding confirms previous observations of decreased basal [35S]GTPγS binding in the locus coeruleus of chronic morphine-treated rats (Sim et al., 1996a; Selley et al., 1997a).
Table 1 shows the analysis of net DAMGO-stimulated [35S]GTPγS binding and specific [3H]naloxone binding from both groups of animals. Results showed that the effect of chronic heroin self-administration in decreasing μ receptor-stimulated [35S]GTPγS binding varied across brain regions. The largest decreases were observed in brainstem, with 25–39% reductions in heroin self-administering rats compared with controls. Decreases in μ-stimulated [35S]GTPγS binding were also observed in medial thalamus (28%) and amygdala (32%). DAMGO-stimulated [35S]GTPγS binding also appeared to be reduced in hypothalamus (17% decrease), but this effect did not reach the level of statistical significance (p > 0.05). Although trends of decreased DAMGO-stimulated [35S]GTPγS binding were observed in other regions, there were no significant decreases observed in caudate putamen, nucleus accumbens, cingulate cortex, and prefrontal cortex in brains from heroin self-administering rats, with the exception of the rostral pole of the nucleus accumbens (p < 0.005). Similar results were obtained whether the nucleus accumbens was analyzed as a whole or separated into shell and core regions.
Table 1.
Effect of chronic heroin self-administration on μ opioid-stimulated [35S]GTPγS binding and [3H] naloxone binding in rat brain
| Region | Net μ-stimulated [35S]GTPγS binding (nCi/gm) | [3H]naloxone binding (nCi/gm) | ||
|---|---|---|---|---|
| Control | Heroin (% change) | Control | Heroin (% change) | |
| Prefrontal cortex | 250 ± 22 | 268 ± 12 | 363 ± 6 | 401 ± 7*(+10) |
| Cingulate cortex | 332 ± 18 | 293 ± 18 | 570 ± 14 | 620 ± 10**(+9) |
| Rostral nucleus accumbens | 620 ± 16 | 509 ± 12*(−18) | 923 ± 73 | 1004 ± 83 (+10) |
| Nucleus accumbens | 417 ± 19 | 370 ± 13 | 730 ± 16 | 804 ± 17**(+10) |
| Caudate putamen | 407 ± 20 | 372 ± 12 | 642 ± 14 | 680 ± 12 |
| Amygdala | 438 ± 25 | 299 ± 22*(−32) | 386 ± 11 | 413 ± 7 |
| Hippocampus | 76 ± 11 | 61 ± 10 | 229 ± 5 | 243 ± 3***(+6) |
| Medial thalamus | 603 ± 33 | 435 ± 30*(−28) | 488 ± 15 | 507 ± 13 |
| Mediodorsal thalamus | 531 ± 34 | 386 ± 25*(−27) | 389 ± 8 | 383 ± 10 |
| Hypothalamus | 226 ± 14 | 188 ± 22 | 281 ± 7 | 356 ± 13*(+27) |
| Ventral tegmental area | 123 ± 21 | 113 ± 15 | ND | |
| Interpeduncular nucleus | 618 ± 19 | 463 ± 13*(−25) | 889 ± 69 | 1180 ± 87***(+33) |
| Periaqueductal gray | 244 ± 17 | 166 ± 13*(−32) | 431 ± 6 | 560 ± 26*(+30) |
| Dorsal raphe nucleus | 269 ± 27 | 195 ± 21**(−28) | ND | |
| Locus coeruleus | 370 ± 23 | 279 ± **(−25) | 463 ± 13 | 549 ± 28***(+19) |
| Lateral parabrachial nucleus | 357 ± 19 | 219 ± 17*(−39) | 644 ± 11 | 640 ± 28 |
| Nucleus tractus solitarius | 530 ± 21 | 400 ± 33**(−25) | 686 ± 13 | 1048 ± 69*(+35) |
Brain sections were assayed for DAMGO-stimulated [35S]GTPγS and [3H]naloxone binding as described in Materials and Methods. Data represent nanocuries per gram of [35S] or [3H] from densitometric analysis of autoradiograms and are mean values ± SE of triplicate sections of brains from eight treated and seven control animals. Numbers in parentheses represent percentage change in heroin self-administering animals compared with control. Significantly different from control by Student's t test: *p ≤ 0.005; **p ≤ 0.01; ***p ≤ 0.05. ND, Not determined.
In contrast to the inhibitory effects of chronic heroin self-administration on μ opioid receptor-activated G-proteins, increased [3H]naloxone binding was observed in several brain regions after chronic heroin self-administration (Table 1). Although the increase in [3H]naloxone binding in forebrain was generally small, in some of these areas this increase (∼10%) was significant (i.e., prefrontal and cingulate cortices, nucleus accumbens, hippocampus). A greater increase (27%) was seen in hypothalamus, an effect comparable to the increases measured in brainstem nuclei. With the exception of the lateral parabrachial nucleus, increased [3H]naloxone binding was found in all of the brainstem nuclei analyzed. In general, the magnitude of the increases in [3H]naloxone binding in brainstem (20–35%) was greater than those measured in the forebrain.
Table 1 shows that eight of the nine regions with significant decreased μ-stimulated [35S]GTPγS binding after chronic heroin self-administration also demonstrated increased [3H]naloxone binding. This finding suggests that decreased μ receptor activation of G-proteins after chronic heroin self-administration occurred despite a significant increase of μ receptors, indicating a loss in receptor–G-protein coupling efficiency after chronic heroin exposure. To further explore this concept, data from Table 1 were expressed as the ratios between μ-stimulated [35S]GTPγS binding and [3H]naloxone binding (G/R ratios) in each region, in both control and chronic heroin animals. Results (Fig.4) show that the decrease in μ receptor activation of G-proteins after chronic heroin self-administration was even more evident when expressed in this manner, with significant decreases in G/R ratios in every region except prefrontal cortex and hippocampus. For example, chronic heroin self-administration produced 35% decreases in the G/R ratio in medial thalamus and amygdala and 40–50% decreases in brainstem nuclei. These results indicate a significant decline in the efficiency of μ receptor–G-protein coupling in these regions after chronic heroin self-administration.
Fig. 4.
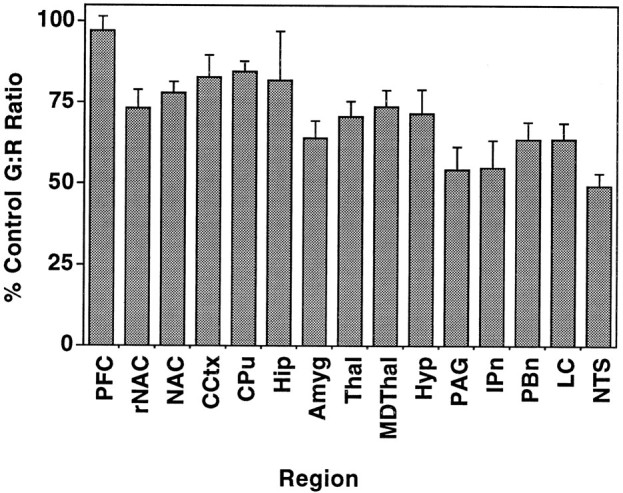
Ratios of DAMGO-stimulated [35S]GTPγS binding to [3H]naloxone binding (G/R ratios) in regions from control and heroin self-administering rats, expressed as percentage of values in control animals. Data from both autoradiographic assays (nanocuries per gram for [35S] and [3H]) were obtained from Table 1, and ratios of [35S] to [3H] were calculated in both control and heroin-treated animals. Ratios (which varied from 0.25 to 1.4) were separately obtained in sections from each animal, and data are expressed as mean values ± SEM. PFC, Prefrontal cortex; rNAC, nucleus accumbens rostral pole;NAC, nucleus accumbens; CCtx, cingulate cortex; CPu, caudate putamen; Hip, hippocampus; Amyg, amygdala; Thal, medial thalamus; MDT, medial dorsal thalamus;Hyp, hypothalamus; PAG, periaqueductal gray; IPn, interpeduncular nucleus; PBn, lateral parabrachial nucleus; LC, locus coeruleus;NTS, commissural nucleus tractus solitarius.
Agonist-stimulated [35S]GTPγS autoradiography for other receptors
Receptor-coupled G-protein activity for several other G-protein-coupled receptors was examined in control and heroin self-administration brains to determine whether other Gi/Go-coupled receptors were affected by heroin treatment. δ opioid-stimulated [35S]GTPγS binding was of particular interest, because at relatively high concentrations heroin metabolites also bind to δ opioid receptors. However, no significant differences in δ opioid-stimulated [35S]GTPγS binding were found in forebrain regions, including amygdala, nucleus accumbens, caudate putamen, cingulate cortex, and prefrontal cortex (Table 2). There was a trend toward a decrease in δ-stimulated [35S]GTPγS binding observed in amygdala, but the variability precluded any significant differences. Nevertheless, the possibility of concerted changes in μ and δ receptors in amygdala represents an intriguing possibility, especially considering the recent evidence of opioid receptor heterodimers (Jordan and Devi, 1999). Data from brainstem nuclei were not included in the analysis because the level ofp-Cl-DPDPE-stimulated [35S]GTPγS binding was too low in these regions to be accurately quantified. Several other G-protein-coupled receptors were also examined using agonist-stimulated [35S]GTPγS binding to determine whether the activity of other receptor types was affected by chronic heroin self-administration. However, none of these receptors, including ORL-1, GABAB, adenosine A1, or cannabinoid receptors, showed any changes in G-protein activation (Table 2). In addition, 5-HT1A-stimulated [35S]GTPγS binding was examined in dorsal raphe nucleus and hippocampus, but no significant changes were found (data not shown). These data suggest that the desensitization produced by uncoupling μ opioid receptors from G-proteins in brain is homologous in nature.
Table 2.
Effect of chronic heroin self-administration on agonist-stimulated [35S]GTPγS binding in rat brain
| % Net [35S]GTPγS binding in control animals | |||||
|---|---|---|---|---|---|
| Delta | ORL-1 | GABAB | A1 | CB | |
| Prefrontal cortex | 116 ± 15 | 108 ± 6 | 88 ± 9 | 99 ± 4 | 91 ± 3 |
| Cingulate cortex | 96 ± 5 | 91 ± 7 | 115 ± 4 | 92 ± 5 | 99 ± 5 |
| Caudate putamen | 98 ± 4 | ND | ND | 91 ± 5 | 96 ± 5 |
| Nucleus accumbens | 96 ± 5 | ND | ND | 91 ± 6 | ND |
| Amygdala | 84 ± 13 | 90 ± 12 | ND | 84 ± 6 | 102 ± 6 |
| Hippocampus | ND | 95 ± 7 | ND | 95 ± 3 | 102 ± 3 |
| Thalamus | 96 ± 8 | 92 ± 16 | 99 ± 5 | 101 ± 8 | ND |
Brain sections from control and heroin self-administering rats were assayed for agonist-stimulated [35S]GTPγS binding as described in Materials and Methods. Receptors (and agonists) included delta opioid (p-CI-DPDPE), ORL-1 (nociceptin/orphanin-F/Q), GABAB (baclofen), adenosine A1 (phenylisopropyladenosine), and cannabinoid (CB) (WIN 55212-2). Data were calculated from nanocuries per gram of [35S] from densitometric analysis of autoradiograms and are expressed as percentage net agonist-stimulated [35S]GTPγS binding of control animals using triplicate sections of brains from eight treated and seven control animals. ND, Not determined.
DISCUSSION
The current study used heroin self-administration for 29–39 d to assess the effect of chronic opiate exposure on μ receptors. The effects of chronic opiate administration have typically been studied using scheduled injections or subcutaneously implanted morphine pellets. The choice of dose, dosing frequency, and route of administration is usually made for practical considerations, rather than behavioral or pharmacological measures made during the dosing regimen. Although these paradigms efficiently produce opiate tolerance and physical dependence, they may not necessarily mimic the gradual development of tolerance and physical dependence that occurs with illicit heroin use in humans, where doses are escalated for subjective effects. In the present study, a paradigm that incorporates this behavior-driven dose escalation has been applied to heroin self-administration in rats.
Desensitization of μ receptor-activated G-proteins in heroin self-administering rats was observed in brain areas crucial in mediating many of the acute and chronic actions of opiates. These include analgesia (periaqueductal gray, thalamus), respiratory depression and cardiovascular effects [parabrachial nucleus, commissural nucleus tractus solitarius (cNTS)], sympathetic symptoms of withdrawal (locus coeruleus), and emotional responses (amygdala). In contrast, nucleus accumbens, which mediates reinforcing effects of several psychoactive drugs including opiates, displayed less desensitization in heroin self-administering rats compared with areas like thalamus, amygdala, and brainstem nuclei. The effect of chronic heroin self-administration in caudate putamen, which mediates some of the motor effects of opiates, was not significant. The decreases found in agonist-stimulated [35S]GTPγS binding after heroin self-administration could be caused by a decrease in either agonist potency or efficacy; preliminary results from amygdala membranes suggest that the primary effect of chronic heroin treatment is to decrease agonist efficacy (C. Maher, T. Martin, and S. Childers, unpublished observations).
An important consideration in any study of chronic agonist treatment is whether residual drug remains bound to tissue and affects results in an artifactual manner. This possibility is unlikely for several reasons. First, brain sections were incubated in buffers containing sodium and GDP, which increase agonist dissociation from receptors (Childers and Snyder, 1980) and remove endogenous and exogenous agonists from membranes. Second, residual agonist in sections would increase basal [35S]GTPγS binding. In fact, heroin self-administration had no effect on basal [35S]GTPγS binding except in two regions, locus coeruleus and interpeduncular nucleus, where basal binding was decreased. Finally, no decreases were seen in [3H]naloxone binding in any region. These results, coupled with the fact that the effect of chronic heroin self-administration varied across brain regions, indicate that these effects were not caused by residual heroin in brain sections.
These studies in heroin self-administering rats confirm previous results from rats treated noncontingently with morphine for 12 d (Sim et al., 1996a). In that study, desensitization of DAMGO-stimulated [35S]GTPγS binding was observed only in specific brainstem nuclei, including periaqueductal gray, dorsal raphe nucleus, parabrachial nucleus, locus coeruleus, and cNTS. The present study confirms these effects and shows that heroin self-administration produces the highest level of μ opioid desensitization in these areas. This confirmation is important, because it was accomplished using a different drug, dose, and administration paradigm. Therefore, these effects on μ opioid activation of G-proteins appear to be a fundamental response of neurons in these regions to chronic opioid agonist exposure. Interestingly, in the previous study, no effects of chronic morphine treatment were observed in forebrain regions. The finding that chronic heroin self-administration produced μ receptor desensitization in thalamus and amygdala may be the result of several differences between the two studies, including administration of different opiates (heroin vs morphine) and a longer treatment duration (39 d of heroin vs 12 d of morphine). Indeed, the brain levels of total opioid are significantly higher after intravenous administration of heroin compared with morphine, and peak levels are reached more rapidly (Way et al., 1965). There may also be a difference of drug self-administration versus noncontingent administration (Smith et al., 1980).
Another goal of this study was to use [3H]naloxone autoradiography under the same assay conditions as [35S]GTPγS autoradiography to determine whether changes in μ receptors accompanied the decrease in receptor–G-protein coupling. A radiolabeled antagonist was used to avoid any changes in agonist high-affinity states that might accompany receptor–G-protein uncoupling. Most regions with decreased DAMGO-stimulated [35S]GTPγS binding demonstrated significant increases in [3H]naloxone binding. This change in [3H]naloxone binding may be the result of changes in binding kinetics; however, this would produce changes in [3H]naloxoneKD values, which has not been observed in cell culture studies with chronic agonist treatment (Breivogel et al., 1997).
The G/R ratio between agonist-activated G-proteins and receptors showed a greater decrease after chronic heroin self-administration than μ-stimulated [35S]GTPγS binding by itself. Although these results suggest that efficiency of μ receptor–G-protein coupling was reduced after chronic heroin self-administration, the G/R ratio should be interpreted with caution: because it does not determine Bmax of receptors or agonist-activated G-proteins, it is not equivalent to the amplification factor (Sim et al., 1996b). Indeed, without a precise measure of Bmax values, true receptor efficiency cannot be calculated, and the apparent increase in binding observed in the present studies may be produced by changes in receptor affinity. On the other hand, this G/R ratio is a relevant measure of efficiency between μ receptors and activated G-proteins because (1) [3H]naloxone, as an antagonist, does not vary in its KD value at μ receptors across brain regions (Maher et al., 2000), and (2) DAMGO-stimulated [35S]GTPγS binding was determined with saturating concentrations of agonist. The fact that almost all regions with decreased μ-stimulated G-proteins also displayed increased μ receptor binding may indicate an important response of this system to chronic agonist exposure. It is possible that the initial event of receptor–G-protein uncoupling could be accompanied by later increases in receptor synthesis to compensate for receptor desensitization, as described previously for β-adrenergic receptors (Lefkowitz et al., 1992).
The relationship between changes in μ receptor binding and development of tolerance/dependence is not clear. Previous studies provided conflicting results regarding the effect of chronic opioid treatment on opioid receptor number, and the data are difficult to interpret because different paradigms were used. Moreover, previous findings of decreased receptor binding in brain after chronic agonist treatment (Yoburn et al., 1993) may in fact be consistent with the present findings: because most previous studies were accomplished with [3H]agonist binding, desensitization caused by receptor–G-protein uncoupling would result in a decrease in high-affinity agonist binding with no actual decrease in total receptor number. Interestingly, a previous study using chronic morphine administration with [3H]DAMGO autoradiography also reported an increase in μ receptor binding (Brady et al., 1989). Although data from cell lines indicate that chronic opioid treatment decreases receptor number along with G-protein uncoupling (Puttfarcken et al., 1988; Breivogel et al., 1997), other studies have shown that different opiate agonists have differential effects on receptor phosphorylation (Chakrabarti et al., 1997; Keith et al., 1998) and that morphine (a major metabolite of heroin) does not promote μ receptor internalization (Keith et al., 1998).
The desensitization in μ-activated G-proteins after chronic heroin self-administration was homologous, because no changes in other receptors coupling to G-proteins were observed. Moreover, less μ opioid receptor desensitization was found in forebrain regions that may mediate reinforcing behaviors, particularly the nucleus accumbens (Vaccarino et al., 1985). The small magnitude of the effect on μ opioid-activated G-proteins in these areas may explain why although it is clear that tolerance develops to effects of opiates such as analgesia and respiratory depression, there is controversy regarding the degree of tolerance that develops to the reinforcing and discriminative stimulus effects of opiates (Colpaert, 1995; Contarino et al., 1997).
Chronic heroin self-administration is a complex interplay of a number of effects of opioid agonists, including tolerance, physical dependence, and reinforcement. The large increase in the daily intake of heroin in self-administering animals reflects the dramatic tolerance that develops to this drug, as well as its potent reinforcing properties. Opiate addiction results from both the positive reinforcing effects of opiates as well as avoidance of the negative effects of physical dependence (Schulteis and Koob, 1996). Certainly, the animals in the current study were highly physically dependent, because they experienced withdrawal symptoms if not allowed free access to heroin. Therefore, an important question is which of these facets of chronic opiate effects (tolerance or physical dependence) are mediated by μ opioid receptor desensitization in specific brain nuclei. The issue of tolerance cannot be addressed at the current time without a careful a time course study. However, the present results may have important implications regarding the development of physical dependence. A number of studies (Kogan et al., 1992) suggest that withdrawal may be associated with increased excitatory tone in specific brain regions, and μ opioid receptors are predominantly inhibitory in nature (Christie et al., 1987). If μ receptors are desensitized during chronic agonist exposure, the resulting loss in inhibitory tone may increase excitatory neuronal function in specific brain nuclei and thus contribute to the development of physical dependence. These results suggest that regionally specific adaptations in inhibitory signal transduction may underlie the differential development of opiate dependence.
Footnotes
This work was supported by Public Health Service Grants DA-00287 (L.J.S.), DA-10770 (D.E.S.), DA-06634 (S.R.C.), and DA-00247 (T.J.M.) from the National Institute on Drug Abuse. C. Todd Hairston and Ruoyu Xiao provided technical assistance.
Correspondence should be addressed to Dr. Steven R. Childers, Center for Investigative Neuroscience, Wake Forest University School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157. E-mail:childers@wfubmc.edu.
REFERENCES
- 1.Alt A, Mansour A, Akil H, Medzihradsky F, Traynor JR, Woods JH. Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding by endogenous opioids acting at a cloned mu receptor. J Pharmacol Exp Ther. 1998;286:282–288. [PubMed] [Google Scholar]
- 2.Blasig J, Hollt V, Hengstenberg J, Dum J, Herz A. Non-competitive nature of the antagonistic mechanism responsible for tolerance to opiate-induced analgesia. Neuropharmacology. 1979;18:473–481. doi: 10.1016/0028-3908(79)90073-x. [DOI] [PubMed] [Google Scholar]
- 3.Bozarth MA, Wise RA. Anatomically distinct opiate receptor fields mediate reward and physical dependence. Science. 1984;224:516–517. doi: 10.1126/science.6324347. [DOI] [PubMed] [Google Scholar]
- 4.Brady LS, Herkenham M, Long JB, Rothman RB. Chronic morphine increases μ-opiate receptor binding in rat brain: a quantitative autoradiographic study. Brain Res. 1989;477:382–386. doi: 10.1016/0006-8993(89)91432-7. [DOI] [PubMed] [Google Scholar]
- 5.Breivogel CS, Selley DE, Childers SR. Acute and chronic effects of opioids on delta and mu receptor activation of G-proteins in NG108–15 and SK-N-SH cell membranes. J Neurochem. 1997;68:1462–1472. doi: 10.1046/j.1471-4159.1997.68041462.x. [DOI] [PubMed] [Google Scholar]
- 6.Chakrabarti S, Yang W, Law PY, Loh HH. The mu-opioid receptor down-regulates differently from the delta-opioid receptor: requirement of a high affinity receptor/G protein complex formation. Mol Pharmacol. 1997;52:105–113. [PubMed] [Google Scholar]
- 7.Chen Y, Mestek A, Liu J, Hurley JA, Yu L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- 8.Childers SR, Snyder SH. Differential regulation by guanine nucleotides of opiate agonist and antagonist receptor interactions. J Neurochem. 1980;34:583–593. doi: 10.1111/j.1471-4159.1980.tb11184.x. [DOI] [PubMed] [Google Scholar]
- 9.Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- 10.Clark MJ, Emmerson PJ, Mansour A, Akil H, Woods JH, Portoghese PS, Remmers AE, Medzihradsky F. Opioid efficacy in a C6 glioma cell line stably expressing the delta opioid receptor. J Pharmacol Exp Ther. 1997;283:501–510. [PubMed] [Google Scholar]
- 11.Colpaert FC. Drug discrimination: no evidence for tolerance to opiates. Pharmacol Rev. 1995;47:605–629. [PubMed] [Google Scholar]
- 12.Contarino A, Zanotti A, Drago F, Natolino F, Lipartiti M, Giusti P. Conditioned place preference: no tolerance to the rewarding properties of morphine. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:589–594. doi: 10.1007/pl00004988. [DOI] [PubMed] [Google Scholar]
- 13.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein A, Herrera J. Heroin addicts and methadone treatment in Albuquerque: a 22-year follow-up. Drug Alcohol Dep. 1995;40:139–150. doi: 10.1016/0376-8716(95)01205-2. [DOI] [PubMed] [Google Scholar]
- 15.Herkenham M, Pert CB. Light microscopic localization of brain opiate receptors: a general autoradiographic method which preserves tissue quality. J Neurosci. 1982;2:1129–1149. doi: 10.1523/JNEUROSCI.02-08-01129.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inturrisi CE, Schultz M, Shin S, Umans JG, Angel L, Simon EJ. Evidence from opiate binding studies that heroin acts through its metabolites. Life Sci. 1983;33:773–776. doi: 10.1016/0024-3205(83)90616-1. [DOI] [PubMed] [Google Scholar]
- 17.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- 19.Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The δ-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci USA. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klee WA, Streaty RA. Narcotic receptor sites in morphine-dependent rats. Nature. 1974;248:61–63. doi: 10.1038/248061a0. [DOI] [PubMed] [Google Scholar]
- 21.Kogan JH, Nestler EJ, Aghajanian GK. Elevated basal firing rates and enhanced responses to 8-Br-cAMP in locus coeruleus in brain slices from opiate-dependent rats. Eur J Pharmacol. 1992;211:47–53. doi: 10.1016/0014-2999(92)90261-2. [DOI] [PubMed] [Google Scholar]
- 22.Law PY, Hom DS, Loh HH. Opiate receptor down-regulation and desensitization in neuroblastoma x glioma NG108–15 hybrid cells are two separate cellular adaptation processes. Mol Pharmacol. 1983;24:413–424. [PubMed] [Google Scholar]
- 23.Lefkowitz RJ, Inglese J, Koch WJ, Pitcher J, Attramadal H, Caron MG. G-protein-coupled receptors: regulatory role of receptor kinases and arrestin proteins. Cold Spring Harbor Symp Quant Biol. 1992;57:127–133. doi: 10.1101/sqb.1992.057.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Maher C, Selley D, Childers S. Relationship of mu opioid receptor binding to activation of G-proteins in specific rat brain regions. Biochem Pharmacol. 2000;59:1395–1401. doi: 10.1016/s0006-2952(00)00272-0. [DOI] [PubMed] [Google Scholar]
- 25.Martin TJ, Dworkin SI, Smith JE. Alkylation of mu opioid receptors by β-funaltrexamine in vivo: comparison of the effects on in situ binding and heroin self-administration in rats. J Pharmacol Exp Ther. 1995;272:1135–1140. [PubMed] [Google Scholar]
- 26.Nestler EJ. Molecular mechanisms of drug addiction. J Neurosci. 1992;12:2439–2450. doi: 10.1523/JNEUROSCI.12-07-02439.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noble F, Cox BM. Differential desensitization of mu- and delta-opioid receptors in selected neural pathways following chronic morphine treatment. Br J Pharmacol. 1996;117:161–169. doi: 10.1111/j.1476-5381.1996.tb15169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noble F, Cox BM. The role of dopaminergic systems in opioid receptor desensitization in nucleus accumbens and caudate putamen of rat after chronic morphine treatment. J Pharmacol Exp Ther. 1997;283:557–565. [PubMed] [Google Scholar]
- 29.Puttfarcken PS, Cox BM. Morphine-induced desensitization and downregulation at mu-receptors in 7315C pituitary tumor cells. Life Sci. 1989;45:1937–1942. doi: 10.1016/0024-3205(89)90548-1. [DOI] [PubMed] [Google Scholar]
- 30.Puttfarcken PS, Werling LL, Cox BM. Effects of chronic morphine exposure on opioid inhibition of adenylyl cylcase in 7315c cell membranes: a useful model for the study of tolerance at mu opioid receptors. Mol Pharmacol. 1988;33:520–527. [PubMed] [Google Scholar]
- 31.Rossi CG, Leventhal L, Pan YX, Cole J, Su W, Bodnar RJ, Pasternak GW. Antisense mapping of MOR-1 in rats: distinguishing between morphine and morphine-6β-glucuronide antinociception. J Pharmacol Exp Ther. 1997;281:109–114. [PubMed] [Google Scholar]
- 32.Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, Pintar JE. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci. 1999;2:151–156. doi: 10.1038/5706. [DOI] [PubMed] [Google Scholar]
- 33.Schulteis G, Koob GF. Reinforcement processes in opiate addiction: a homeostatic model. Neurochem Res. 1996;21:1437–1454. doi: 10.1007/BF02532385. [DOI] [PubMed] [Google Scholar]
- 34.Selley DE, Nestler EJ, Breivogel CS, Childers SR. Opioid receptor-coupled G-proteins in rat locus coeruleus membranes: decrease in activity after chronic morphine treatment. Brain Res. 1997a;746:10–18. doi: 10.1016/s0006-8993(96)01125-0. [DOI] [PubMed] [Google Scholar]
- 35.Selley DE, Sim LJ, Xiao R, Liu Q, Childers SR. Mu opioid receptor-stimulated [35S]GTPγS binding in rat thalamus and cultured cell lines: signal transduction mechanisms underlying agonist efficacy. Mol Pharmacol. 1997b;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- 36.Selley DE, Liu Q, Childers SR. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTPγS binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- 37.Sim LJ, Selley DE, Childers SR. In vitro autoradiography of receptor-activated G-proteins in rat brain by agonist-stimulated guanylyl 5′-[γ-[35S]thio]-triphosphate binding. Proc Natl Acad Sci USA. 1995;92:7242–7246. doi: 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sim LJ, Selley DE, Dworkin SI, Childers SR. Effects of chronic morphine administration on mu opioid receptor-stimulated [35S]GTPγS autoradiography in rat brain. J Neurosci. 1996a;16:2684–2692. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sim LJ, Selley DE, Xiao R, Childers SR. Differences in G-protein activation by mu and delta opioid, and cannabinoid, receptors in rat striatum. Eur J Pharmacol. 1996b;307:95–107. doi: 10.1016/0014-2999(96)00211-7. [DOI] [PubMed] [Google Scholar]
- 40.Sim LJ, Xiao R, Childers SR. Identification of opioid receptor-like (ORL1) peptide-stimulated [35S]GTPγS binding in rat brain. NeuroReport. 1996c;7:729–733. doi: 10.1097/00001756-199602290-00012. [DOI] [PubMed] [Google Scholar]
- 41.Sim LJ, Selley DE, Childers SR. Autoradiographic visualization in brain of receptor-G-protein coupling using [35S]GTPγS binding. In: Challiss RS, editor. Methods in molecular biology: receptor signal transduction protocols. Humana; Totowa, NJ: 1997. pp. 117–132. [DOI] [PubMed] [Google Scholar]
- 42.Smith J, Co C, Freeman M, Sands M, Lane J. Neurotransmitter turnover in rat striatum is correlated with morphine self-administration. Nature. 1980;287:152–154. doi: 10.1038/287152a0. [DOI] [PubMed] [Google Scholar]
- 43.Tao P-L, Law P-Y, Loh HH. Decrease in delta and mu opioid receptor binding capacity in rat brain after chronic etorphine treatment. J Pharmacol Exp Ther. 1987;240:809–816. [PubMed] [Google Scholar]
- 44.Tao P-L, Chang L-R, Chou Y-P, Law P-Y, Loh HH. Chronic opioid treatment may uncouple opioid receptors and G-proteins: evidence from irradiation inactivation analysis. Eur J Pharmacol. 1993;246:233–238. doi: 10.1016/0922-4106(93)90036-9. [DOI] [PubMed] [Google Scholar]
- 45.Thompson RC, Mansour A, Akil H, Watson SJ. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- 46.Traynor JR, Nahorski SR. Modulation by μ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1995;47:848–854. [PubMed] [Google Scholar]
- 47.Umans JG, Inturrisi CE. Pharmacodynamics of subcutaneously administered diacetylmorphine, 6-acetylmorphine and morphine in mice. J Pharmacol Exp Ther. 1981;218:409–415. [PubMed] [Google Scholar]
- 48.Vaccarino FJ, Bloom FE, Koob GF. Blockade of nucleus accumbens opiate receptors attenuates intravenous heroin reward in the rat. Psychopharmacology. 1985;86:37–42. doi: 10.1007/BF00431681. [DOI] [PubMed] [Google Scholar]
- 49.Way EL, Young JM, Kemp JW. Metabolism of heroin and its pharmacologic implications. Bull Narcotics. 1965;17:25–33. [Google Scholar]
- 50.Way EL, Loh HH, Shen FH. Simultaneous quantitative assessment of morphine tolerance and physical dependence. J Pharmacol Exp Ther. 1969;167:1–8. [PubMed] [Google Scholar]
- 51.Yoburn BC, Billings B, Duttaroy A. Opioid receptor regulation in mice. J Pharmacol Exp Ther. 1993;265:314–320. [PubMed] [Google Scholar]
- 52.Young WS, Kuhar MJ. A new method for receptor autoradiography: [3H]opioid receptors in rat brain. Brain Res. 1979;179:255–270. doi: 10.1016/0006-8993(79)90442-6. [DOI] [PubMed] [Google Scholar]