

Abstract
Monoamines modulate neuronal differentiation, and alteration of monoamine neurotransmission during development produces specific changes in neuronal structure, function, and pattern formation. We have previously observed that prenatal exposure to cocaine in a clinically relevant animal model produces increased length of pyramidal neuron dendrites in the anterior cingulate cortex (ACC) postnatally. We now report that cocaine administered intravenously to pregnant rabbits at gestational stages preceding and during cortical histogenesis results in the early onset of hypertrophic dendritic outgrowth in the embryonic ACC. Confocal microscopy of DiI-labeled neurons revealed that the atypical, tortuous dendritic profiles seen postnatally in ACC-cocaine neurons already are apparent in utero. No defects in neuronal growth were observed in visual cortex (VC), a region lacking prominent dopamine innervation. In striking correlation with ourin vivo results, in vitro experiments revealed a significant enhancement of spontaneous process outgrowth of ACC neurons isolated from cocaine-exposed fetuses but no changes in neurons derived from visual cortex. The onset of modified growthin vivo is paralleled by reduced D1Areceptor coupling to its G-protein. These data suggest that the dynamic growth of neurons can be regulated by early neurotransmitter signaling in a selective fashion. Prenatal onset of defects in dopamine receptor signaling contributes to abnormal circuit formation and may underlie specific cognitive and behavioral dysfunction.
Keywords: prenatal cocaine, dendritic outgrowth, anterior cingulate cortex, development, dopamine, DiI, neurite, D1 receptor, Gs
Dysfunctions in neurotransmitter signaling contribute to the pathophysiology of numerous neurological disorders, which may in turn have origins in defective morphogenetic processes during brain development (Bloom, 1993; Weinberger, 1995;Levitt et al., 1997). The growth and development of the CNS is a prolonged process, commencing in utero and extending beyond birth, and alterations of neurotransmitter balances in the fetus can affect critical phases of brain histogenesis (Levitt et al., 1997). Monoamines, in particular, appear early in the developing mammalian CNS and act as morphogens (Molliver, 1982; Chubakov et al., 1986; Lauder, 1988; Mattson, 1988; Reinoso et al., 1996). An important role for monoamines in pattern formation has been further elucidated by the characterization of mice lacking monoamine oxidase A (Cases et al., 1995, 1996).
At least 1% of women use cocaine during pregnancy (National Institute on Drug Abuse, 1996), and some studies suggest prevalence rates of 5–17% in urban regions (Day et al., 1993). Monoamine uptake-storage mechanisms form early in development (Coyle, 1977; Moody et al., 1993), and cocaine readily penetrates the placental barrier (Wiggins, 1992). Cocaine can thus interfere with fetal monoamine uptake mechanisms (House, 1990; Meyer and Dupont, 1993; Shearman et al., 1996) and produce CNS deficits in offspring (Kosofsky et al., 1994; Lidow, 1995).
An animal model of in utero intravenous cocaine administration has been characterized previously (Levitt et al., 1997). Injection of low doses (2–4 mg/kg) of cocaine twice a day to pregnant rabbits produces long-lasting and specific effects on the structure and function of cortical neurons receiving dopaminergic innervation in the offspring, without altering general developmental parameters (Murphy et al., 1995, 1997). For example, prenatal exposure to cocaine produces aberrant layer III/V dendritic outgrowth postnatally (Jones et al., 1996), an increase in GABA-immunoreactive neurons (Wang et al., 1995a), and long-term uncoupling of the D1 dopamine (DA) receptor from its G-protein (Friedman et al., 1996) in the anterior cingulate cortex (ACC) of offspring. These changes are not induced in the visual cortex (VC). Functional outcomes of prenatal cocaine exposure in this model include anomalous behavior on motor and discriminative tasks (Romano et al., 1995; Romano and Harvey, 1996;Simansky and Kachelries, 1996) and altered regulation of DA release (Wang et al., 1995c; Du et al., 1999).
In the present study, we administered cocaine before and during cerebral cortical histogenesis (Stensaas, 1967a,b) and analyzed the dendritic growth patterns of embryonic rabbit neurons to determine (1) how early in cortical development cocaine alters dendritic growth, (2) whether neurons exposed to cocaine in utero, when isolatedin vitro, can recover and develop normally in culture in the absence of the drug, and (3) whether these alterations in dendritic growth are induced by a loss of D1 receptor coupling. The results indicate that prenatal exposure to cocaine induces defects in the mechanisms that control neuronal differentiation and D1 receptor signaling. These morphological alterations impressively persist even in dissociated cell culture conditions, providing a unique model system in which to study the cellular and molecular control of neuronal differentiation.
MATERIALS AND METHODS
Animals. Proven breeder Dutch-belted rabbits from Myrtle's Rabbitry (Thompson Station, TN) were used for these studies. They were housed individually in a 12 hr light/dark cycle with free access to food and water, and female rabbits were mated with a male rabbit ∼1 week after arrival. The day of breeding was designated as embryonic day 0 (E0), and injections were initiated after implantation on E8. The injections were given intravenously (through the marginal ear vein) twice daily (8 A.M., 3 P.M.) from E8 until the day before animals were killed for studies of prenatal ages and until E29 for postnatal analyses. The dams received either 3 mg/kg of cocaine or an equal volume of saline as a control injection. This dose, administered from E8 through E29, induces abnormal ACC dendritic morphology postnatally, concomitant with D1receptor uncoupling and behavioral abnormalities (for review, seeLevitt et al., 1997; Levitt, 1998). This dose did not cause grand mal seizure activity or weight changes in pregnant dams, and gross parameters such as birth weight and litter size were normal as reported previously (Murphy et al., 1995, 1997). Dams were given an overdose of sodium pentobarbitol (50 mg/kg), and embryos were removed at E21 or E24 for morphological analysis. Crown-rump lengths and external morphological features of ear pinnae and digits were monitored to assure the appropriate developmental stage (Stensaas, 1967b,c). Embryos were harvested, and brains were isolated for use in DiI labeling of cortical pyramidal neurons, D1 receptor coupling assays, or cell culture experiments. For simplicity, we refer to neurons analyzed from the saline-exposed fetuses as ACC- or VC-saline neurons and those from cocaine-exposed animals as ACC- or VC-cocaine neurons.
In vivo dendritic analysis using DiI. Fetal rabbit brains were removed from the skull and immersed in 4% formalin for 1–2 weeks at 4°C. Brains were then blocked to reveal the ACC and VC. Labeling of projection neurons was achieved by placing DiI-coated glass spears in the white matter underlying the area of interest (either ACC or VC). The tissue slabs were returned back to fixative, and containers were wrapped in aluminum foil and incubated at 37°C for 3–5 d (Barbe and Levitt, 1992). Longer incubation periods did not result in more labeling of cortical neurons. The slabs were embedded in 3% agar and sectioned in the coronal plane at 200 μm using a vibratome and collected in PBS. The sections were mounted in glycerol-PBS and viewed on a Leica or Nikon microscope equipped with rhodamine fluorescence optics. Dendritic length was measured by collecting digitized images using the Bioquant True Color image analysis system (Memphis, TN). Dendritic trajectories were analyzed using a Leica confocal microscope system. Dendritic profiles of rabbit neurons at ages E21 and E24 (the majority of the DiI-labeled neurons at these ages reside in layers V and VI) were analyzed. There were no differences in the laminar distribution of DiI-labeled neurons as a function of drug treatment. For all digitized images, a landmark was placed at the center of the cell body, and the length of individual apical dendrites was traced and measured. For each animal, an average length was calculated from at least seven neurons in ACC and VC of each animal, and the mean length between control and experimental groups was determined (five at each fetal age for each group). A population analysis of dendritic length distribution was performed such that lengths for all neurons were grouped into 25 μm intervals, and the percentage of dendritic profiles belonging within these different intervals was plotted. A χ2 test was performed to determine statistical significance of changes in length distribution.
In vitro analysis of neurite outgrowth. Primary neuronal cultures were prepared from ACC and VC areas of saline controls and cocaine-exposed rabbit embryos using slight modifications of previously published methods (Reinoso et al., 1996). Embryos were removed for analysis at E21, corresponding to the latter period of neurogenesis in the rabbit (Stensaas, 1967a,b). Using the rostrum, genu, and splenium of the corpus callosum as landmarks, the VC and medial frontal cortex that includes the ACC were removed from each embryo and cut into small pieces (∼2 mm2) in Earle's balanced salt solution. Tissue was then incubated in 0.35% collagenase-dispase for 1 hr at room temperature, and a cell suspension was prepared by repeated trituration using a fire-polished Pasteur pipette. Cells were then plated at low density (5 × 104/cm2) onto poly-l-lysine-coated coverslips in 16-mm-diameter tissue culture wells. The cells initially were incubated in fetal bovine serum-supplemented media for 3 hr to enhance attachment, then switched to N2-supplemented glia conditioned media (GCM) (Bottenstein, 1985) and maintained for 2–4 d. The GCM was harvested from neonatal rat cerebral cortical glial cultures (Morrison and de Vellis, 1984) and was found to provide the best condition for neuronal survival and baseline growth of the rabbit cell cultures (data not shown). The procedure for harvesting GCM has been described previously (Reinoso et al., 1996). Cells were incubated in the chemically defined neuron–glial medium, modified fromBottenstein (1985) and Morrison and de Vellis (1984). Cells were maintained in culture for 48–96 hr, fixed, and stained as described previously (Reinoso et al., 1996) at 1:100 with a polyclonal antibody against the somatodendritic marker MAP2 (Crandall and Fischer, 1989) (generous gift of Dr. Itzhak Fischer, Medical College of Pennsylvania-Hahnemann) and at 1:50 with a monoclonal antibody against the early phosphorylated form of neurofilament protein (NFp-H) (Pennypacker et al., 1991). The coverslips were washed and then incubated in secondary anti-rabbit IgG-FITC and anti-mouse Ig M-Texas Red (Jackson Immunoresearch).
The coverslips containing immunocytochemically labeled neurons were analyzed using Leica fluorescence optics (20× objective). A horizontal or vertical sweep of each coverslip was performed to analyze 12 individual fields, and the fraction of each field (0.15 mm2) occupied by MAP2 or NFp-H staining was measured (area fraction analysis). Soma number and size were determined (MAP2-positive cells) and used to obtain a net area fraction, based on neurites per field. This method of analysis was performed, rather than measuring lengths of individual neurites, because outgrowth in the cultures harvested from the cocaine-exposed embryos, even by 48 hr, was too extensive to allow consistent tracing of individual processes. Plating at lower densities permitted direct measurement of neurite length in cocaine-treated cells, but under these very low density conditions, neurons from saline-exposed embryos failed to thrive in culture, precluding direct comparisons (data not shown). The data were collected from five coverslips in each of four to five experiments using different litters. The values are expressed as mean net area fraction, and statistical significance was determined by the Mann–Whitney U test.
Coimmunoprecipitation of D1A dopamine receptor with Gαs protein. Determination of the linkage between receptor and G-protein was performed as described previously (Wang et al., 1995b;Friedman et al., 1996). Medial frontal cortex including the ACC was dissected from E15, E22, E25, postnatal day 1 (P1), and P20 rabbits and homogenized in 10 vol of buffer containing 25 mm HEPES, pH 7.5, 2 mm MgCl2, 1 mmEDTA, 0.2% 2-mercaptomethanol, 50 μg/ml leupeptin, 25 μg/ml pepstatin A, 5 μg/ml aprotinin, 0.01 U/ml soybean trypsin inhibitor, and 0.04 mm PMSF. The homogenate was centrifuged at 800 × g for 5 min, and the supernatant was centrifuged for 10 min at 49,000 × g. The resulting pellet was washed and resuspended in immunoprecipitation buffer containing 100 mm Tris-HCl, pH 7.5, 200 mmNaCl, 2 mm MgCl2, 1 mm EDTA, 0.2% 2-mercaptomethanol, 50 μg/ml leupeptin, 25 μg/ml pepstatin A, 0.01 U/ml soybean trypsin inhibitor, and 0.04 mm PMSF. Fifty micrograms of membrane proteins were solubilized in 1 ml of the immunoprecipitation buffer supplemented with 0.2% cholate and 0.5% digitonin. Solubilized tissues were precleared by the addition of normal rabbit serum (1:100 dilution) at 4°C for 60 min followed by 30 min incubation with 100 μl of a 10% suspension of protein A-bearing Staphylococcus aureus cells (Pansorbin cells). The suspension was centrifuged, and the supernatant was combined with antisera (1:1000 dilution) raised against a specific Gαs peptide and incubated for 3 hr followed by an additional 30 min with 100 μl of Pansorbin. After centrifugation, the pellet was suspended in 100 μl of sample preparation buffer and boiled for 5 min. The D1A dopamine receptors in the immunoprecipitates were assessed by immunoblotting using a monoclonal D1A dopamine receptor antibody (RBI; 1:1000 dilution).
Immunoblot analysis. Twenty-five micrograms of membrane proteins were solubilized in sample preparation buffer, and proteins were separated by SDS-PAGE (12%) and transferred electrophoretically to nitrocellulose membrane (NC). The completeness of the transfer was checked by Coomassie blue staining of the gel. The NCs were incubated at 4°C overnight with 10% nonfat dry milk in PBS containing 0.1% Tween 20 (0.1% TBS) to block nonspecific sites. The NC preparation was washed with 0.1% TBS and incubated for 2 hr with Gαs antiserum at 1:2000 dilution or with specific D1A dopamine receptor antibody at 1:1000 dilution. The unbound antibody was washed out with 0.1% TBS. The blot was incubated for 60 min with 1:10,000 dilution of HRP-conjugated anti-rabbit IgG (for Gαs protein blot) or anti-mouse IgG (for D1A dopamine receptor blot) followed by washing in 0.3% and 0.1% TBS. The immunoreactive proteins were detected by the ECL Western blot detection system and visualized by exposure to x-ray film. The blots were quantified using laser densitometry. Data are presented as mean ± SE. Two-tailed ANOVA was used to compare the data among the groups followed by the Newman–Keuls test. Significance was considered at p < 0.05.
RESULTS
DiI-labeled dendrites of cortical projection neurons
The morphology of the apical dendrites of early differentiating neurons was assessed by examining DiI-labeled neurons at E21 and E24 in the rabbit ACC and VC. ACC dendrites of layer V and VI neurons were visible in both the saline controls and the cocaine-exposed fetuses. Dendrites of the saline controls were very straight and thick, as described by Stensaas (1967c) using Golgi impregnation. The dendrites typically extended to the marginal zone and could be traced without altering microscope focus, indicative of outgrowth in a straight path. Most labeled dendrites of ACC-cocaine neurons also reached the marginal zone but could only be traced with changes in the focal plane (Fig. 1).
Fig. 1.
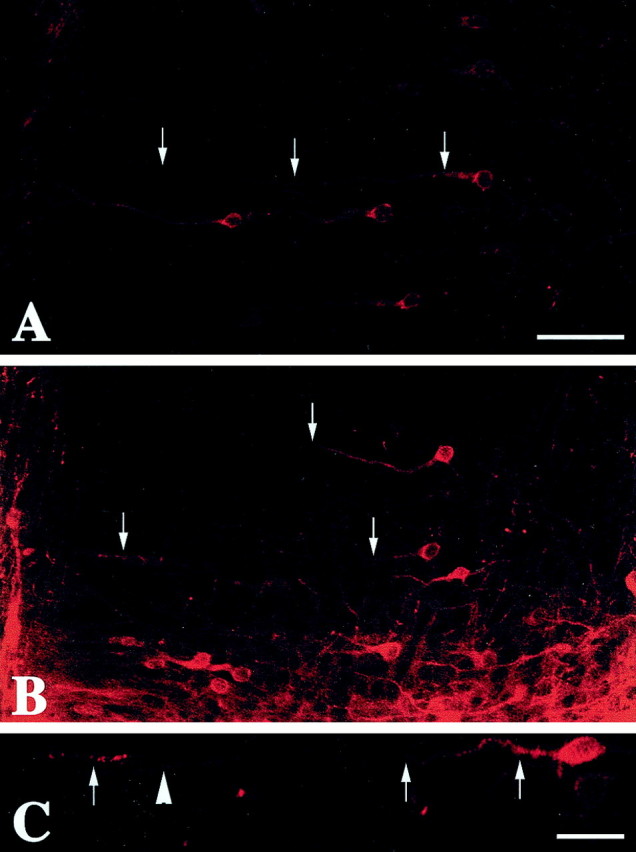
Photomicrographs display representative sections of anterior cingulate cortex neurons labeled with DiI in saline-exposed (A) and cocaine-exposed (B) E24 rabbit fetuses. In the saline sample, arrowsindicate a neuron the dendrites of which can be followed from its origin at the cell soma toward the pial surface. In the cocaine sample,arrows denote several apical dendrites that leave the plane of section. Scale bar, 50 μm. C, Higher-power photomicrograph of a neuron from a cocaine-treated E24 fetus the dendrite of which courses out of the plane of section but later returns near the marginal zone (arrows). This type of profile was regularly observed in cocaine-treated animals but rarely seen in saline controls. The arrowhead denotes the dendrite of a different cell the soma of which is out of the plane of the section. Scale bar, 25 μm.
Quantitative analysis of mean dendritic length in the E21 ACC-cocaine group revealed a 25% increase compared with controls (Fig.2A). Population analysis of the distribution of dendritic lengths revealed an even more striking effect. Approximately 30% of the ACC apical dendrites in the saline pups were longer than 75 μm, whereas more than twice as many of the total dendritic population in the cocaine-exposed fetuses (Fig.2B) were this category. These data indicate that as early as E21 the dendrites of neurons in the ACC-cocaine pups are considerably longer than their normal counterparts within the early developing cortical plate.
Fig. 2.
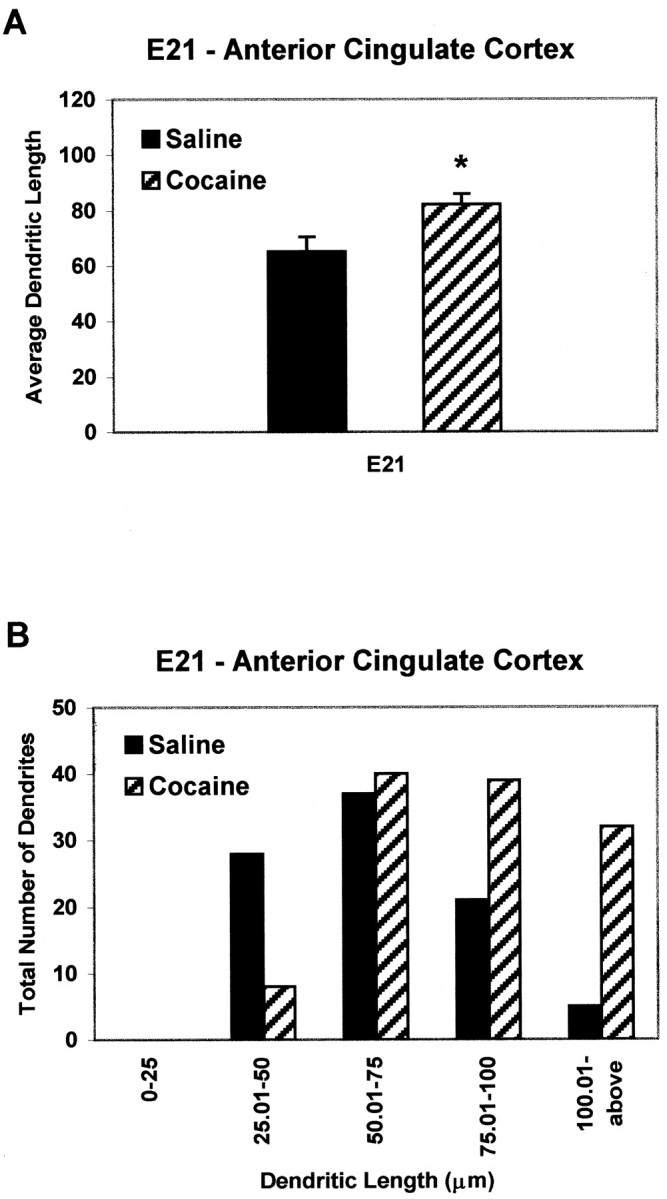
Quantitative analysis of apical dendrites from the anterior cingulate cortex of E21 saline and cocaine pups.A, The average apical dendritic length (in micrometers) at E21 is 25% longer in the cocaine-exposed pups compared with the saline controls. B, Note the sevenfold increase in the number of dendrites measuring >100 μm in cocaine-exposed embryos compared with saline controls. Statistical significance,p < 0.01.
By E24, nearly all cortical neurons have been generated, with neurons destined for layers VI–III located in their appropriate laminae. Morphological analysis of DiI-labeled ACC-cocaine dendrites in E24 ACC revealed a 73% increase in mean length (Fig.3A). Moreover, although only 5% of dendrites measured were >150 μm in the ACC-saline group, ∼70% of the dendrites in the ACC-cocaine fetuses reached or exceeded 150 μm (Fig. 3B). No such differences were apparent in the VC of cocaine-exposed fetuses, at either E21 or E24 (Fig.4), indicating regional specificity of the effects of cocaine exposure.
Fig. 3.
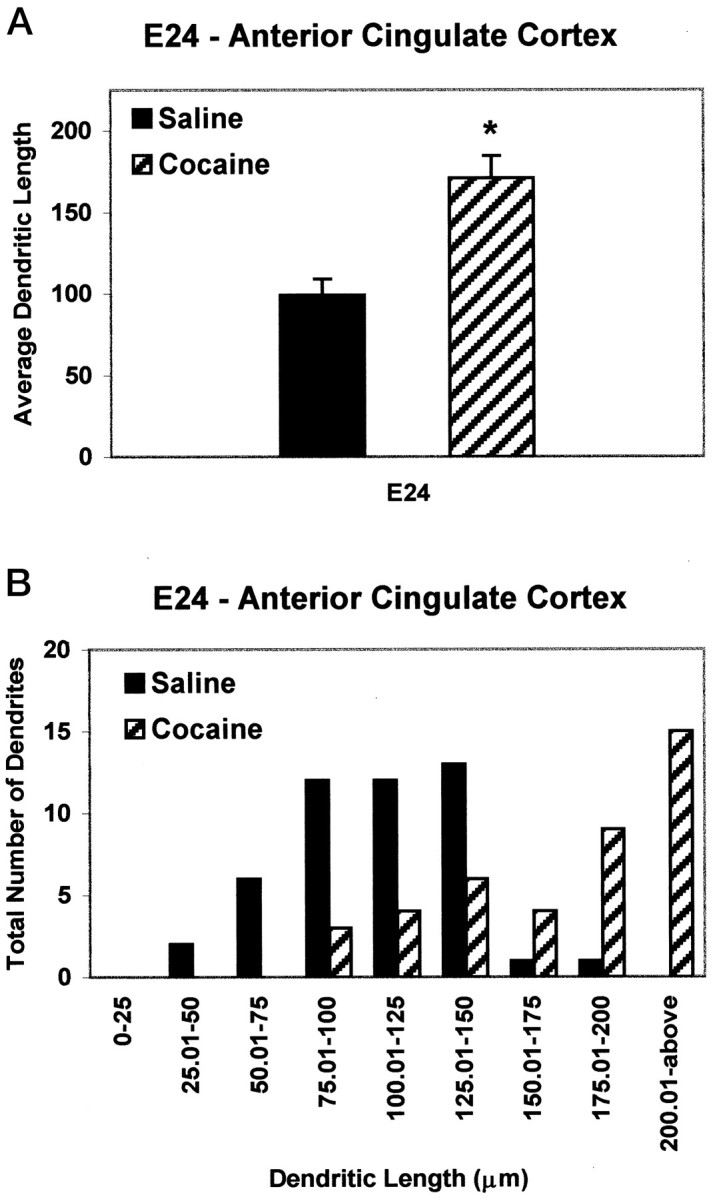
Quantitative analysis of apical dendritic lengths (in micrometers) from the anterior cingulate cortex of E24 saline and cocaine embryos. A, Average dendritic lengths of layer V cells are doubled in cocaine-exposed embryos compared with saline controls. B, The number of dendrites exceeding 150 μm in length increases by 75% in cocaine-exposed embryos compared with normal distribution patterns. Statistical significance,p < 0.01.
Fig. 4.
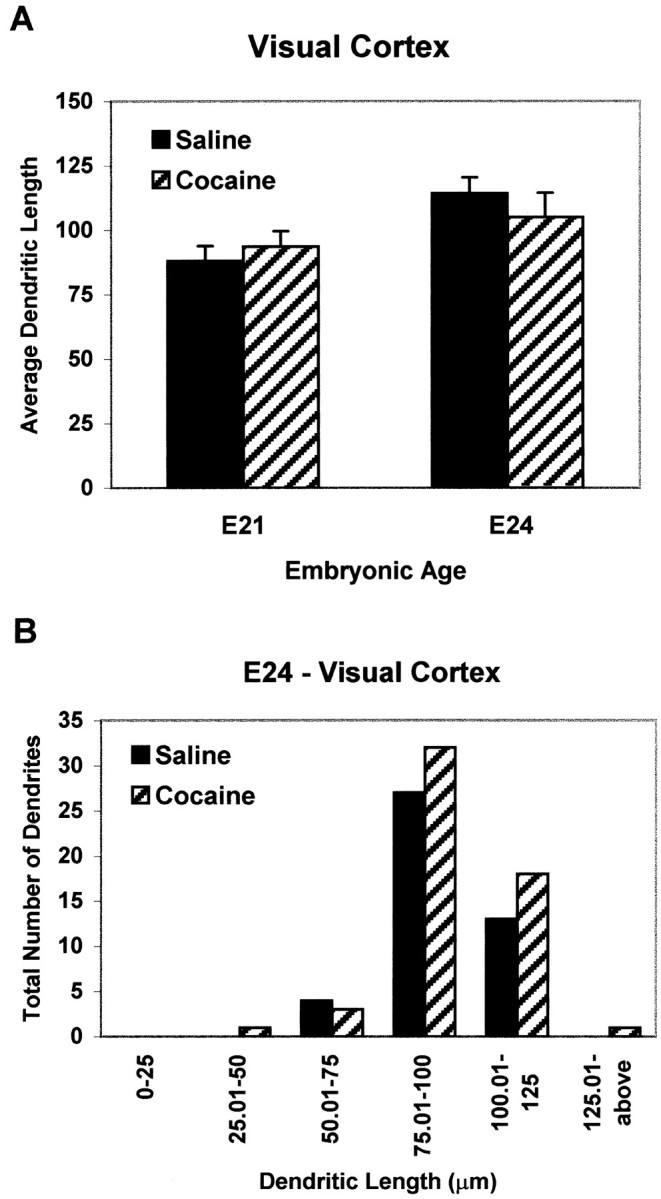
Quantitative analysis of visual cortical dendritic lengths (in micrometers) in saline or cocaine-exposed E21 and E24 embryos. Dendritic lengths did not vary between the saline and cocaine-exposed E21 and E24 embryos in this DA-poor brain region.
Dendritic growth patterns in vitro
We next sought to determine whether in utero cocaine exposure during corticogenesis causes defects in mechanisms that regulate growth of ACC neurons. Short-term monolayer cultures were prepared of neurons isolated at E21, the peak of neurogenesis in rabbit cerebral cortex, from saline controls and embryos exposed to cocainein utero beginning at E8. After 4 d in culture, neuronal processes of ACC-cocaine (Fig.5B) cells appeared longer and thicker compared with neurites of the same population of cells from saline controls (Fig. 5A). In contrast, no appreciable difference in neurite morphology was observed between VC-saline and VC-cocaine cells (Fig. 5C,D). The pattern of MAP2 staining of the E21 ACC cultures revealed marked differences in growth between groups. Neurons from saline-exposed embryos usually had two to four processes, with very few cells exhibiting long or branching neurites (Fig. 5A). In striking contrast, ACC-cocaine neurons exhibited longer, more complex fiber networks (Fig.5B) compared with the ACC-saline, VC-saline, and VC-cocaine neurons. In most ACC cultures of cocaine-exposed cells, the profiles of MAP2-positive neurites were difficult to trace because of the high process density, although soma density did not appear to differ (see below).
Fig. 5.

Photomicrographs of MAP2-stained E21 medial frontal cortical (including the anterior cingulate cortex) (A, B) and visual cortical (C, D) culture preparations from saline-exposed (A, C) and cocaine-exposed (B, D) embryos. ACC-cocaine neurons (B) have longer neurites than ACC-saline (A), VC-saline (C), and VC-cocaine (D). Scale bar, 50 μm.
Determination of net area fraction, the extent of neurites that occupy the culture substratum, revealed a 60% increase of MAP2-stained processes (Fig. 6A) in cocaine-exposed E21 embryos compared with saline controls. Importantly, soma density was not significantly different from cultures derived from saline-treated embryos (122 ± 11 MAP2-positive cells per field for saline-exposed cultures; 124 ± 16 MAP2-positive cells per field for cocaine-exposed cultures). Furthermore, the soma area of 50 cells per coverslip were measured in two separate culturing sessions (total of six coverslips per drug condition) and did not differ between saline- and cocaine-exposed cells (66.6 ± 3.1 μm2 for saline-exposed cells; 70.0 ± 5.3 μm2 for cocaine-exposed cells). This affirms that the increases in net area fraction in ACC-cocaine neurons are attributable to increases in neurite length and/or branching complexity.
Fig. 6.
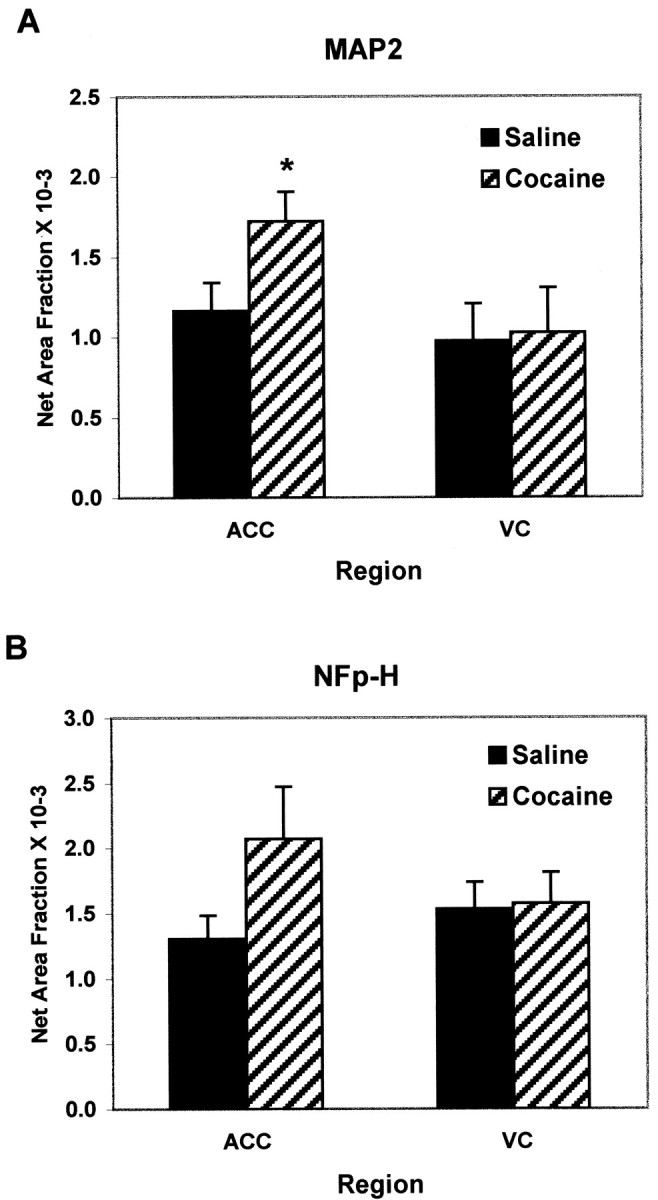
Quantitative analysis of MAP2-stained (A) and NF-H-stained (B) culture preparations from E21 medial frontal cortex including the anterior cingulate cortex (ACC) and visual cortex (VC). A, MAP2-stained E21 ACC cultures revealed a 50% increase in the net area fraction of the cocaine-exposed cultures as compared with the saline, suggestive of longer or a greater number of neurites; *p < 0.05. There was no effect in cultures derived from the VC. B, Likewise, E21 NF-H-stained cultures revealed a trend toward an increase in net area fraction of the cocaine-exposed as compared with the saline cultures, although this was not statistically significant. The effect was not present in VC cultures.
Expression of the early phosphorylated form of NFp-H may reflect maturation of axonal processes (Dotti et al., 1988; Pennypacker et al., 1991), and most embryonic neurons grown for 4 d in vitro only exhibited faint NFp-H staining, with very few labeled neurites. Most of the staining was nuclear, caused by cross-reactivity of the antibody with a common histone linker protein (Pennypacker et al., 1991). This pattern of staining, typical of cortical cultures in rodents (Pennypacker et al., 1991; Reinoso et al., 1996), was observed in the saline-exposed preparations from ACC and VC, but the NFp-H staining of ACC-cocaine neurons was more intense. However, area fraction analysis did not reveal a statistically significant change in NFp-H staining (Fig. 6B). This appears to be attributable to greater variability in the patterns of NFp-H staining between experiments.
D1 receptor uncoupling from Gαs
We have previously demonstrated that prenatal exposure to cocaine during E8–E29 leads to a pronounced reduction of D1 dopamine receptor-mediated activation of Gs proteins in the ACC and striatum of P10, P50, and P100 offspring (Wang et al., 1995b; Friedman et al., 1996). D1receptor activation inhibits neurite outgrowth of rodent cortical neurons in culture (Reinoso et al., 1996). We thus hypothesized that reduced D1 dopamine receptor coupling to Gαs produced by cocaine in utero may contribute to the aberrant, excessive outgrowth that we observed as early as E21. We therefore examined whether D1 receptor uncoupling is produced by in utero cocaine exposure at these embryonic ages. Coupling was assessed by coimmunoprecipitating D1A dopamine receptor protein with Gαs antiserum from solubilized cortical membrane preparations that were exposed to buffer (control) or dopamine. Although dopamine elicited an increase in coimmunoprecipitation of D1A receptor protein with Gs in frontal cortex of saline rabbits, in utero cocaine markedly reduced coupling (Fig.7). Incubation with 1 μm dopamine elicited four- to sevenfold increases in receptor-Gαs coupling in membranes obtained from saline-exposed offspring aged E22 through P20. This response was completely obviated in fetuses exposed to cocaine (E22 and E25) and was >80% reduced at the postnatal ages. These data are consistent with the hypothesis that a loss of D1 dopamine receptor-mediated inhibition of process elongation may underlie the aberrant growth of the apical dendrites of pyramidal neurons after fetal cocaine exposure. This state of reduced coupling is maintained throughout the postnatal life of offspring exposed to cocaine in utero (Fig. 7) (Wang et al., 1995b; Friedman et al., 1996).
Fig. 7.

Effects of prenatal cocaine exposure on dopamine-stimulated coupling of D1A dopamine receptor to Gαs in rabbit frontal cortex. Representative immunoblots of Gαs and of coimmunoprecipitated D1A receptors (A) and quantitative analyses of optical densities of the coimmunoprecipitated D1A receptor protein expressed as mean ± SEM (B) are shown; *p < 0.01. E, Embryonic age;P, postnatal age.
DISCUSSION
Effects of cocaine on cortical development
Cocaine is a potential teratogen with multiple mechanisms of inducing neurotoxicity (El-Bizri et al., 1991; Olsen, 1995). Descriptions of cocaine-induced modifications in behavior and function range from specific to global (Spear et al., 1989; Chen et al., 1993;Barron and Irvine, 1994; Kosofsky et al., 1994; Lidow, 1995;Vorhees et al., 1995; Wood et al., 1995; Levitt et al., 1997;Levitt, 1998). This widespread variability probably reflects the use of different species and administration paradigms (Dow-Edwards, 1996;Levitt, 1998). Ironically, the mode of cocaine administration in animal studies that most closely models the human pharmacokinetics is underrepresented, i.e., intravenous delivery (Dow-Edwards, 1996;Levitt, 1998). The rabbit model of intravenous cocaine use thus provides an opportunity to examine specific changes in ontogeny at low doses. There are no alterations in maternal weight, litter size, pup weight (at birth and throughout postnatal development), cortical thickness, laminar width, and cell number after doses up to 4 mg/kg per injection (Murphy et al., 1995; Wang et al., 1995a). Murphy et al. (1997) showed that 3 mg/kg induced identical morphological changes in the ACC of rabbits prenatally exposed to the 4 mg/kg dose of cocaine, and therefore we selected that dosage for our current study.
Animal models of drug exposure may be subject to caveats because of potential problems in maternal–infant care (Murphy et al., 1997;Levitt, 1998). Although our detailed analyses of the model has reduced the likelihood of such a mechanism to account for the observed developmental changes, our hypothesis of a specific cellular alteration in growth mechanisms is strongly supported by the current finding that cocaine-induced changes in neuronal growth are initiated around midgestation.
There was a threefold increase in the number of ACC-cocaine dendrites exceeding 75 μm as compared with saline control at E21, hypertrophic growth that was not apparent in the VC-cocaine group. The morphological alteration in the ACC-cocaine group was apparent within 1 week after the genesis of layer V neurons. Altered dendritic growth was even more pronounced in the ACC of E24 cocaine-exposed embryos, where the number of dendrites exceeding 150 μm was sevenfold greater than in the ACC-saline group. When administered intravenously, cocaine accumulates in DA-rich regions of the brain (Madras and Kaufman, 1994) and blocks DA reuptake (Meyer and Dupont, 1993). These observations are consistent with our findings of cocaine-induced alterations in the ACC, an area receiving a dense DA input, but not in the VC, a region receiving a sparse DA input (Fuxe et al., 1974; Reader et al., 1979; Goldman-Rakic and Brown, 1982; Levitt et al., 1984; Descarries et al., 1987). We have observed similar cocaine-induced morphological changes in the medial prefrontal cortex (a DA-rich area) but not in somatosensory cortex (DA-poor area) of rabbits exposed to cocaine in utero (our unpublished observations).
ACC-cocaine dendrites were longer and wavy, in marked contrast to the straight, single-plane path of VC-cocaine and ACC/VC-saline dendrites. Because the thickness of the cerebral cortex is not significantly different between saline- and cocaine-exposed pups (Wang et al., 1995a), the wavy dendritic profiles within the ACC-cocaine group may reflect a compensatory mechanism to counteract a more rapid rate of increase in the length of apical dendrites. Interestingly, mice with a null mutation in the L1 cell adhesion molecule gene also possess undulating apical dendrites of layer V pyramidal neurons, but in this case the altered processes are present in somatosensory, visual, and motor cortex (Demyanenko et al., 1999).
Our cell culture studies of neurons from fetal rabbits allowed us to examine the growth of neurites in an environment without additional cocaine, to establish whether prenatal cocaine exposure was sufficient to induce alterations in the basic properties of neuronal growth. It is important to note that cell survival, based on the number of MAP2-positive cells per coverslip, and soma area were the same, regardless of the origin (cocaine or saline) of the fetal tissue. The lack of significant differences in neuronal soma area and density among all groups affirms that the changes in net area fraction in ACC-cocaine neurons are attributable to changes in neurite length and/or branching complexity. Analyses of E21 ACC cultures revealed clear differences between cocaine and saline groups because cocaine-exposed neurons exhibited longer and more complex fiber networks than did VC-cocaine and ACC/VC-saline neurons. The advanced polarity and greater outgrowth by the same time in culture as the matched saline neurons indicates a faster maturation rate and outgrowth of neurites in the ACC-cocaine group.
Possible mechanisms involved in cocaine-induced abnormal neuronal growth
The present data suggest that prenatal exposure to cocaine induces an early desensitization of D1 receptor activation in the fetal ACC. The reduction in D1dopamine receptor–Gs coupling is not caused by changes in the amount of Gαs protein, as determined by immunoblot analysis. Furthermore, levels of D1 receptor protein do not appear to be altered by cocaine at the prenatal and postnatal day tested. These data are consistent with previous analyses of D1dopamine receptor densities at postnatal ages up to 100 d (Wang et al., 1995b; Friedman et al., 1996). These previous reports also showed selectivity of the effect because D2 dopamine receptor activation of Gαi was not altered by in uterococaine exposure. These results therefore indicate that D1 receptors are uncoupled from Gαs soon after the onset of normal differentiation in cocaine-exposed animals. Further confirmation that loss of D1 receptor signaling contributes to abnormal regulation of dendritic outgrowth after prenatal cocaine is suggested by our recent observation that morphological alterations may be present in the ACC of D1 receptor knockout mice (data not shown).
The observed increase in MAP2 staining in vitro is almost certainly attributable to morphological alterations and does not reflect an alteration in the intracellular distribution of MAP2 protein. In this regard, earlier studies of ACC dendritic organization in the rabbit model have confirmed that the anomalous MAP2 staining reflects an increase in dendritic length. Antibody staining against α-tubulin and Golgi silver impregnation showed a staining pattern identical to anti-MAP2, suggesting that these changes do not reflect an alteration in the expression or intracellular distribution of MAP2 protein (Jones et al., 1996). Second, our direct analysis of dendritic length after confocal analysis of DiI-filled neurons clearly shows that the apical dendrites of cocaine-exposed ACC neurons are significantly longer than those of saline-exposed controls. Last, phase-contrast photomicrographs of the cultured rabbit material reveal clear increases in neurite length in the cocaine condition (data not shown).
The in vitro pattern of neurite outgrowth in control and experimental groups, which parallels growth patterns seen in vivo, provides compelling evidence for a drug-induced alteration of neuronal responsiveness and intrinsic properties of neurons. We have previously observed that D1 receptor activation decreases neurite outgrowth in rodent cortical neurons in vitro, whereas D2 receptor stimulation increases process extension (Reinoso et al., 1996). Similar findings have been observed by other investigators (Lankford et al., 1987;Rodrigues and Dowling, 1990; Todd, 1992).
Moreover, synergistic interactions between D1 and D2 receptors have been documented, such that full expression of certain DA-mediated effects requires concurrent stimulation of both receptor subtypes (Clark and White, 1987; Seeman et al., 1989; Spealman et al., 1992). If such synergistic actions operate in the fetal cerebral cortex, then a lack of D1receptor and Gs coupling is likely to disrupt D1/D2 receptor synergism during development. When this is viewed from the perspective of DA receptor-mediated effects on neurite outgrowth (Reinoso et al., 1996), a mechanism to explain the cocaine-induced morphological changes involves the loss of the D1 receptor-mediated inhibitory control over a D2 receptor-mediated enhancement of neurite outgrowth. In this model, D1/Gαs uncoupling could potentiate D2 receptor-mediated neurite outgrowth. A potential caveat to this interpretation is the unknown proportion of ACC neurons during development that coexpress D1and D2 receptors. In the adult rat prefrontal cortex, most D1 and D2receptors may be localized on different neurons, with only 25% of neurons bearing colocalized D1 and D2 receptors (Vincent et al., 1995).
A second potential mechanism of the actions of cocaine on the developing CNS invokes a dopamine–glutamate interaction. Layer III and V pyramidal neurons are glutamatergic (Streit, 1984), and D1 receptors are localized on the dendritic spines of these neurons (Smiley et al., 1994) in specific regions of the cerebral cortex (Boyson et al., 1986; Fremeau et al., 1992). It is thus possible to disrupt signaling interactions between these neurotransmitters on the same neurons. Moreover, dopamine and glutamate influence each other's effects on neuronal responsiveness, primarily through D1 and NMDA receptors, respectively (Cepeda et al., 1993; Pralong and Jones, 1993), and experimental evidence supports the idea that dopamine–glutamate interactions are affected by cocaine (Karler et al., 1994; Cervo and Samanin, 1995). In some experimental systems, NMDA activation can induce neurite outgrowthin vitro (Lipton and Kater, 1989; Rashid and Cambray-Deakin, 1992). Thus a cocaine-induced D1 receptor uncoupling from Gαs could result in an imbalance in dopamine–glutamate signaling mechanisms (Halpain et al., 1990) and a disinhibition of NMDA receptor-mediated process elongation.
The early developmental onset, before birth, of changes in neuronal growth in DA-rich areas establishes long-term structural and biochemical defects. Prenatal exposure to cocaine produces a complex cascade of changes that result in specific cognitive and behavioral defects (Romano et al., 1995; Romano and Harvey, 1996; Simansky and Kachelries, 1996), many of which may be produced by the sustained disruption of D1 receptor signaling. The impact of in utero exposure to cocaine on structure–function relationships in DA-rich areas of the CNS will be the focus of future studies.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant DA 11165 to P.L., NIH Grant DA 11029 to E.F., a March of Dimes Birth Defects Foundation grant to H-Y.W., and a Pharmaceutical Research and Manufacturers of America Foundation fellowship to G.D.S. We thank Dr. Simon Watkins (Center for Biological Imaging, University of Pittsburgh), Dr. Herb Geller (Department of Pharmacology, University of Medicine and Dentistry of New Jersey/Robert Wood Johnson Medical School), and Dr. Elizabeth Powell for assistance in confocal image analysis.
L.B.J. and G.D.S. contributed equally to this work.
Correspondence should be addressed to Dr. Gregg D. Stanwood, Department of Neurobiology, University of Pittsburgh School of Medicine, E1414 Biomedical Science Tower, Pittsburgh PA 15261. E-mail:gstanwoo+@pitt.edu.
REFERENCES
- 1.Barbe MF, Levitt P. Attraction of specific thalamic input by cerebral grafts depends on the molecular identity of the implant. Proc Natl Acad Sci USA. 1992;89:3706–3710. doi: 10.1073/pnas.89.9.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barron S, Irvine J. Effects of neonatal cocaine exposure on two measures of balance and coordination. Neurotoxicol Teratol. 1994;16:89–94. doi: 10.1016/0892-0362(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 3.Bloom FE. Advancing a neurodevelopmental origin for schizophrenia. Arch Gen Psychiatry. 1993;50:224–227. doi: 10.1001/archpsyc.1993.01820150074008. [DOI] [PubMed] [Google Scholar]
- 4.Bottenstein JE. Growth and differentiation of neural cells in defined media. In Cell culture in the neurosciences (Bottenstein JE, and Sato G, eds), pp 3–43. Plenum; New York: 1985. [Google Scholar]
- 5.Boyson SJ, McGonigle P, Molinoff PB. Quantitative autoradiographic localization of D1 subtypes of dopamine receptors in rat brain. J Neurosci. 1986;6:3177–3188. doi: 10.1523/JNEUROSCI.06-11-03177.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Muller U, Aguet M, Babinet C, Shih JC, DeMaeyer E. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268:1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cases O, Vitalis T, Seif I, DeMaeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of serotonin excess during the critical period. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 8.Cepeda C, Buchwald A, Levine MS. Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervo L, Samanin R. Effects of dopaminergic and glutamatergic receptor antagonists on the acquisition and expression of cocaine conditioning place preference. Brain Res. 1995;673:242–250. doi: 10.1016/0006-8993(94)01420-m. [DOI] [PubMed] [Google Scholar]
- 10.Chen W-JA, Andersen KH, West JR. Cocaine exposure during the brain growth spurt: studies of neonatal survival, somatic growth, and brain development. Neurotoxicol Teratol. 1993;15:267–273. doi: 10.1016/0892-0362(93)90008-c. [DOI] [PubMed] [Google Scholar]
- 11.Chubakov AR, Gromova EA, Konovalov GV, Sakiseva EF, Chumasov EJ. The effects of serotonin on the morpho-functional development of rat cerebral neocortex in tissue culture. Brain Res. 1986;369:285–297. doi: 10.1016/0006-8993(86)90537-8. [DOI] [PubMed] [Google Scholar]
- 12.Clark D, White FJ. Review: D1 dopamine receptor—the search for a function: a critical evaluation of the D1/D2 dopamine receptor classification and its functional implications. Synapse. 1987;1:347–388. doi: 10.1002/syn.890010408. [DOI] [PubMed] [Google Scholar]
- 13.Coyle JT. Major innervation of newborn rat cortex by monoaminergic neurons. Science. 1977;196:444–447. doi: 10.1126/science.850788. [DOI] [PubMed] [Google Scholar]
- 14.Crandall J, Fischer I. Developmental regulation of microtubule-associated protein 2 expression in regions of mouse brain. J Neurochem. 1989;53:1910–1917. doi: 10.1111/j.1471-4159.1989.tb09261.x. [DOI] [PubMed] [Google Scholar]
- 15.Day NL, Cottreau CM, Richardson GA. The epidemiology of alcohol, marijuana and cocaine use among women of childbearing age and pregnant women. Clin Obstet Gynecol. 1993;36:232–245. doi: 10.1097/00003081-199306000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Demyanenko GP, Tsai AY, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Descarries L, Lemay B, Coucet G, Berger B. Regional and laminar density of the dopamine innervation in adult rat cerebral cortex. Neuroscience. 1987;21:807–824. doi: 10.1016/0306-4522(87)90038-8. [DOI] [PubMed] [Google Scholar]
- 18.Dotti CG, Sullivan CA, Banker GA. The establishment of polarity of hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dow-Edwards DL. Comparability of human and animal studies of developmental cocaine exposure. NIDA Res Monogr. 1996;164:146–174. [PubMed] [Google Scholar]
- 20.Du W, Aloyo VJ, Pazdelski PS, Harvey JA. Effects of prenatal cocaine exposure on amphetamine-induced dopamine release in the caudate nucleus of the adult rabbit. Brain Res. 1999;836:194–198. doi: 10.1016/s0006-8993(99)01567-x. [DOI] [PubMed] [Google Scholar]
- 21.El-Bizri H, Guest I, Varma DR. Effects of cocaine on rat embryo development in vivo and in cultures. Pediatr Res. 1991;29:187–190. doi: 10.1203/00006450-199102000-00017. [DOI] [PubMed] [Google Scholar]
- 22.Fremeau RT, Jr, Duncan GE, Fornaretto M-G, Dearry A, Gingrich JA, Breese GR, Caron MG. Localizatin of D1 dopamine receptor mRNA in rat brain supports a role in cognitive, affective, and neuroendocrine aspects of dopaminergic neurotransmission. Proc Natl Acad Sci USA. 1992;88:3772–3776. doi: 10.1073/pnas.88.9.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman E, Yadin E, Wang H-Y. Effect of prenatal cocaine on dopamine receptor-G protein coupling in mesocortical regions of the rabbit brain. Neuroscience. 1996;70:739–747. doi: 10.1016/s0306-4522(96)83011-9. [DOI] [PubMed] [Google Scholar]
- 24.Fuxe K, Hokfelt T, Johansson O, Jonsson G, Lidbrink P, Ljungdahl A. The origin of the dopamine nerve terminals in the limbic and frontal cortex evidence for meso-cortico dopamine neurons. Brain Res. 1974;82:349–355. doi: 10.1016/0006-8993(74)90618-0. [DOI] [PubMed] [Google Scholar]
- 25.Goldman-Rakic PS, Brown RM. Postnatal development of monoamine content and synthesis in the cerebral cortex of rhesus monkeys. Dev Brain Res. 1982;4:339–349. doi: 10.1016/0165-3806(82)90146-8. [DOI] [PubMed] [Google Scholar]
- 26.Halpain S, Girault J-A, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- 27.House MA. Cocaine. Am J Nurs. 1990;90:41–45. [PubMed] [Google Scholar]
- 28.Jones L, Fischer I, Levitt P. Non-uniform alteration of dendritic development in the cerebral cortex following prenatal cocaine exposure. Cereb Cortex. 1996;6:431–445. doi: 10.1093/cercor/6.3.431. [DOI] [PubMed] [Google Scholar]
- 29.Karler R, Calder LD, Thai LH, Bedingfield JB. A dopaminergic-glutamatergic basis for the action of amphetamine and cocaine. Brain Res. 1994;658:8–14. doi: 10.1016/s0006-8993(09)90003-8. [DOI] [PubMed] [Google Scholar]
- 30.Kosofsky BE, Wilkins AS, Gressens P, Evard P. Transplacental cocaine exposure: a mouse model demonstrating neuroanatomic and behavioral abnormalities. J Child Neurol. 1994;9:234–241. doi: 10.1177/088307389400900303. [DOI] [PubMed] [Google Scholar]
- 31.Lankford K, De Mello FG, Klein WL. A transient embryonic dopamine receptor inhibits growth cone motility and neurite outgrowth in a subset of avian retina neurons. Neurosci Lett. 1987;75:169–174. doi: 10.1016/0304-3940(87)90292-8. [DOI] [PubMed] [Google Scholar]
- 32.Lauder JM. Neurotransmitters as morphogens. Prog Brain Res. 1988;73:365–387. doi: 10.1016/S0079-6123(08)60516-6. [DOI] [PubMed] [Google Scholar]
- 33.Levitt P. Prenatal effects of drugs of abuse on brain development. Drug Alcohol Depend. 1998;51:109–125. doi: 10.1016/s0376-8716(98)00070-2. [DOI] [PubMed] [Google Scholar]
- 34.Levitt P, Rakic P, Goldman-Rakic RS. Comparative assessment of monoamine afferents in mammalian cerebral cortex. In: Descarries L, Reader TR, Jasper HH, editors. Monoamine innervation of the cerebral cortex. Alan R. Liss; New York: 1984. pp. 41–50. [Google Scholar]
- 35.Levitt P, Harvey JA, Friedman E, Simansky K, Murphy EH. New evidence for neurotransmitter influences in brain development. Trends Neurosci. 1997;20:269–274. doi: 10.1016/s0166-2236(96)01028-4. [DOI] [PubMed] [Google Scholar]
- 36.Lidow MS. Prenatal cocaine exposure adversely affects development of the primate cerebral cortex. Synapse. 1995;21:332–341. doi: 10.1002/syn.890210408. [DOI] [PubMed] [Google Scholar]
- 37.Lipton SA, Kater SB. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989;12:265–270. doi: 10.1016/0166-2236(89)90026-x. [DOI] [PubMed] [Google Scholar]
- 38.Madras BK, Kaufman MJ. Cocaine accumulates in dopamine-rich regions of primate brain after I.V. administration: comparison with mazindol distribution. Synapse. 1994;18:261–275. doi: 10.1002/syn.890180311. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP. Neurotransmitters in the regulation of neuronal cytoarchitecture. Brain Res Rev. 1988;13:179–212. doi: 10.1016/0165-0173(88)90020-3. [DOI] [PubMed] [Google Scholar]
- 40.Meyer JS, Dupont SA. Prenatal cocaine administration stimulates fetal brain tyrosine hydroxylase activity. Brain Res. 1993;608:129–137. doi: 10.1016/0006-8993(93)90783-j. [DOI] [PubMed] [Google Scholar]
- 41.Molliver ME. Role of monoamines in the development of the neocortex. Neurosci Res Prog Bull. 1982;20:493–507. [PubMed] [Google Scholar]
- 42.Moody CA, Robinson SR, Spear LP, Smotherman WP. Fetal behavior and the dopamine system: activity effects of D1 and D2 receptor manipulations. Pharmacol Biochem Behav. 1993;44:843–850. doi: 10.1016/0091-3057(93)90015-l. [DOI] [PubMed] [Google Scholar]
- 43.Morrison RS, de Vellis J., Jr . Preparation of chemically defined medium for purified astrocytes. In: Barnes D, Sirbasku D, Sato G, editors. Cell culture methods for molecular biology, Vol 4: methods for serum-free culture of neuronal and lymphoid cells. Alan R. Liss; New York: 1984. pp. 15–22. [Google Scholar]
- 44.Murphy EH, Hammer JG, Schumann MD, Groce MY, Wang X-H, Jones L, Romano AG, Harvey JA. The rabbit as a model for studies of cocaine exposure in utero. Lab Anim Sci. 1995;45:163–168. [PubMed] [Google Scholar]
- 45.Murphy EH, Fischer I, Friedman E, Grayson D, Jones L, Levitt P, O'Brien-Jenkins A, Wang H-Y, Wang X-Y. Cocaine administration in pregnant rabbits alters cortical structure and function in their progeny in the absence of maternal seizures. Exp Brain Res. 1997;114:433–441. doi: 10.1007/pl00005652. [DOI] [PubMed] [Google Scholar]
- 46.National Institute on Drug Abuse (NIDA) National pregnancy and health survey: drug use among women delivering live births: 1992 (National Institutes of Health, Publication No. 96–3819). NIH; Rockville, MD: 1996. [Google Scholar]
- 47.Olsen GD. Potential mechanisms of cocaine-induced developmental neurotoxicity: a minireview. Neurotoxicology. 1995;16:159–168. [PubMed] [Google Scholar]
- 48.Pennypacker K, Fischer I, Levitt P. Early in vitro genesis and differentiation of axons and dendrites by hippocampal neurons analyzed quantitatively with neurofilament-H and microtubule-associated protein 2 antibodies. Exp Neurol. 1991;111:25–35. doi: 10.1016/0014-4886(91)90047-g. [DOI] [PubMed] [Google Scholar]
- 49.Pralong E, Jones RSG. Interactions of dopamine with glutamate- and GABA-mediated synaptic transmission in the rat entorhinal cortex in vitro. Eur J Neurosci. 1993;5:760–767. doi: 10.1111/j.1460-9568.1993.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 50.Rashid NA, Cambray-Deakin MA. N-methyl-d-aspartate effects on the growth, morphology and cytoskeleton of individual neurons in vitro. Dev Brain Res. 1992;67:301–308. doi: 10.1016/0165-3806(92)90231-k. [DOI] [PubMed] [Google Scholar]
- 51.Reader TA, Masse P, De Champlaine J. The intracortical distribution of norepinephrine, dopamine and serotonin in the cerebral cortex of the cat. Brain Res. 1979;177:499–513. doi: 10.1016/0006-8993(79)90467-0. [DOI] [PubMed] [Google Scholar]
- 52.Reinoso BS, Undie AS, Levitt P. Dopamine receptors mediate differential morphological effects on cerebral cortical neurons in vitro. J Neurosci Res. 1996;43:439–453. doi: 10.1002/(SICI)1097-4547(19960215)43:4<439::AID-JNR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 53.Rodrigues P dos S, Dowling JE. Dopamine induces neurite retraction in retinal horizontal cells via diacylglycerol and protein kinase C. Proc Natl Acad Sci USA. 1990;87:9693–9697. doi: 10.1073/pnas.87.24.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romano AG, Harvey JA. Prenatal exposure to cocaine disrupts discrimination learning in adult rabbits. Pharmacol Biochem Behav. 1996;53:617–621. doi: 10.1016/0091-3057(95)02061-6. [DOI] [PubMed] [Google Scholar]
- 55.Romano AG, Kachelries WJ, Simansky KJ, Harvey JA. Intrauterine exposure to cocaine produces a modality-specific acceleration of classical conditioning in adult rabbits. Pharmacol Biochem Behav. 1995;52:415–420. doi: 10.1016/0091-3057(95)00129-k. [DOI] [PubMed] [Google Scholar]
- 56.Seeman P, Niznik HB, Guan H-C, Booth G, Ulpian C. Link between D1 and D2 dopamine receptors is reduced in schizophrenia and Huntington diseased brain. Proc Natl Acad Sci USA. 1989;86:10156–10160. doi: 10.1073/pnas.86.24.10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shearman LP, Collins LM, Meyer JS. Characaterization and localization of [125I]RTI-55-labeled cocaine binding sites in fetal and adult rat brain. J Pharmacol Exp Ther. 1996;277:1770–1783. [PubMed] [Google Scholar]
- 58.Simansky KJ, Kachelries WJ. Prenatal exposure to cocaine selectively disrupts motor responding to d-amphetamine in young and mature rabbits. Neuropharmacology. 1996;35:71–78. doi: 10.1016/0028-3908(95)00151-4. [DOI] [PubMed] [Google Scholar]
- 59.Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci USA. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spealman RD, Bergman J, Madras BK, Kamien JB, Melia KF. Role of D1 and D2 dopamine receptors in the behavioral effects of cocaine. Neurochem Int. 1992;20:147S–152S. doi: 10.1016/0197-0186(92)90228-j. [DOI] [PubMed] [Google Scholar]
- 61.Spear LP, Kirstein CL, Frambes NA. Cocaine effects on the developing central nervous system: behavioral, psychoparmacological and neurochemical studies. Ann NY Acad Sci. 1989;562:290–307. doi: 10.1111/j.1749-6632.1989.tb21027.x. [DOI] [PubMed] [Google Scholar]
- 62.Stensaas LJ. The development of hippocampal and dorsolateral pallial regions of the cerebral hemisphere in fetal rabbits. I. Fifteen millimeter stage, spongioblast morphology. J Comp Neurol. 1967a;129:59–70. doi: 10.1002/cne.901310402. [DOI] [PubMed] [Google Scholar]
- 63.Stensaas LJ. The development of hippocampal and dorsolateral pallial regions of the cerebral hemisphere in fetal rabbits. II. Twenty millimeter stage, neuroblast morphology. J Comp Neurol. 1967b;129:71–84. doi: 10.1002/cne.901300204. [DOI] [PubMed] [Google Scholar]
- 64.Stensaas LJ. The development of hippocampal and dorsolateral pallial regions of the cerebral hemisphere in fetal rabbits. IV. Forty-one millimeter stage, intermediate lamina. J Comp Neurol. 1967c;131:409–422. doi: 10.1002/cne.901310402. [DOI] [PubMed] [Google Scholar]
- 65.Streit P. Glutamate and aspartate as transmitter candidates for systems of the cerebral cortex. In Cerebral cortex, Vol. 2 (Jones EG, Peters A, eds.), pp 119–143. Plenum; New York: 1984. [Google Scholar]
- 66.Todd RD. Neural development is regulated by classical neurotransmitters: dopamine D2 receptor stimulation enhances neurite outgrowth. Biol Psychiatry. 1992;31:794–807. doi: 10.1016/0006-3223(92)90311-m. [DOI] [PubMed] [Google Scholar]
- 67.Vincent SL, Khan Y, Benes FM. Cellular colocalization of dopamine D1 and D2 receptors in rat medial prefrontal cortex. Synapse. 1995;19:112–120. doi: 10.1002/syn.890190207. [DOI] [PubMed] [Google Scholar]
- 68.Vorhees CV, Reed TM, Acuff-Smith KD, Schilling MA, Cappon GD, Fisher JE, Pu C. Long-term learning deficits and changes in unlearned behaviors following in utero exposure to multiple daily doses of cocaine during different exposure periods and maternal plasma cocaine concentrations. Neurotoxicol Teratol. 1995;17:253–264. doi: 10.1016/0892-0362(94)00061-h. [DOI] [PubMed] [Google Scholar]
- 69.Wang X-H, Levitt P, Grayson DR, Murphy EH. Intrauterine cocaine exposure of rabbits: persistent elevatiin of GABA-immunoreactive neurons in anterior cingulate cortex but not visual cortex. Brain Res. 1995a;689:32–46. doi: 10.1016/0006-8993(95)00528-x. [DOI] [PubMed] [Google Scholar]
- 70.Wang H-Y, Runyan S, Yadin E, Friedman E. Prenatal exposure to cocaine selectively reduces D1 dopamine receptor-mediated activation of striatal Gs proteins. J Pharmacol Exp Ther. 1995b;273:492–498. [PubMed] [Google Scholar]
- 71.Wang H-Y, Yeung JM, Friedman E. Prenatal cocaine exposure selectively reduces mesocortical dopamine release. J Pharmacol Exp Ther. 1995c;273:1211–1215. [PubMed] [Google Scholar]
- 72.Weinberger DR. From neuropathology to neurodevelopment. Lancet. 1995;346:552–557. doi: 10.1016/s0140-6736(95)91386-6. [DOI] [PubMed] [Google Scholar]
- 73.Wiggins RC. Pharmacokinetics of cocaine in pregnancy and effects on fetal maturation. Clin Pharmacokinet. 1992;22:85–93. doi: 10.2165/00003088-199222020-00001. [DOI] [PubMed] [Google Scholar]
- 74.Wood RD, Molina VA, Wagner JM, Spear LP. Play behavior and stress responsivity in periadolescent offspring exposed prenatally to cocaine. Pharmacol Biochem Behav. 1995;52:367–374. doi: 10.1016/0091-3057(95)00120-l. [DOI] [PubMed] [Google Scholar]