

Abstract
Huntington's disease (HD) is a progressive neurodegenerative illness for which there is no effective therapy. We examined whether creatine, which may exert neuroprotective effects by increasing phosphocreatine levels or by stabilizing the mitochondrial permeability transition, has beneficial effects in a transgenic mouse model of HD (line 6/2). Dietary creatine supplementation significantly improved survival, slowed the development of brain atrophy, and delayed atrophy of striatal neurons and the formation of huntingtin-positive aggregates in R6/2 mice. Body weight and motor performance on the rotarod test were significantly improved in creatine-supplemented R6/2 mice, whereas the onset of diabetes was markedly delayed. Nuclear magnetic resonance spectroscopy showed that creatine supplementation significantly increased brain creatine concentrations and delayed decreases in N-acetylaspartate concentrations. These results support a role of metabolic dysfunction in a transgenic mouse model of HD and suggest a novel therapeutic strategy to slow the pathological process.
Keywords: creatine, mitochondria, Huntington's disease, transgenic mice, diabetes, N-acetylaspartate
Huntington's disease (HD) is an autosomal dominant progressive neurodegenerative disease that starts in midlife and inexorably leads to death. The mean survival after onset is 15–20 years, and at present there is no known effective treatment for HD. The mutation that causes the illness is an expanded CAG/polyglutamine repeat stretch that has been postulated to confer toxic effects by several different mechanisms (The Huntington's Disease Collaborative Research Group, 1993). The protein product of the HD gene, huntingtin, is expressed ubiquitously in both the nervous system and peripheral tissues (Strong et al., 1993; Landwehrmeyer et al., 1995; Sharp et al., 1995; Ferrante et al., 1997).
A breakthrough in HD research was the development of transgenic mouse models. Transgenic mice expressing exon 1 of the human HD gene with an expanded CAG repeat develop a progressive neurological disorder (Mangiarini et al., 1996). These mice (line R 6/2) have CAG repeat lengths of 141–157 (normal, <35), under the control of the human HD promoter. At ∼6 weeks of age the R6/2 mice show loss of brain and body weight, and at 9–11 weeks they develop an irregular gait, abrupt shuddering, stereotypic movements, resting tremors, and epileptic seizures. The mice show an early decrease of several neurotransmitter receptors (Cha et al., 1998). The brains of R6/2 mice appear normal in most respects, however, neuronal intranuclear inclusions that are immunopositive for huntingtin and ubiquitin are detected in the striatum at 4.5 weeks (Davies et al., 1997). Neuropil, cytoplasmic, and neuronal inclusions are also found in human HD (DiFiglia et al., 1997;Gutekunst et al., 1999; Kuemmerle et al., 1999).
A secondary effect of the gene defect may be impaired energy metabolism that may contribute to neuronal death. Consistent with this hypothesis, we and others found that: (1) lactate is elevated in the cerebral cortex and basal ganglia of patients with HD, (2) there is reduced phosphocreatine/inorganic phosphate in resting muscle of HD patients, (3) mitochondrial toxins produce selective damage in the striatum of animals, which closely resembles the pathology of HD, and (4) there are reductions in mitochondrial electron transport enzymes in HD postmortem tissue (Jenkins et al., 1993; Brouillet et al., 1995; Gu et al., 1996;Browne et al., 1997; Koroshetz et al., 1997). Recent studies show increased susceptibility of mitochondria to depolarization in HD lymphoblasts and fibroblasts (Gutekunst et al., 1996; Sawa et al., 1999). Our studies in R6/2 mice show that they develop marked decreases in N-acetylaspartate (NAA) concentrations before neuronal loss (Jenkins et al., 2000) and increased vulnerability to the mitochondrial toxin 3-nitropropionic acid (Bogdanov et al., 1998). This may be a consequence of mitochondrial dysfunction (Bates et al., 1996).
If mitochondrial impairment plays a role in neuronal dysfunction in the R6/2 mice, then buffering intracellular energy levels may ameliorate the neurodegenerative process in these animals. Creatine kinase and its substrates creatine and phosphocreatine constitute an intricate cellular energy buffering and transport system connecting sites of energy production (mitochondria) with sites of energy consumption (Hemmer and Wallimann, 1993). Creatine administration increases brain concentrations of PCr and inhibits activation of the mitochondrial permeability transition (MPT), both of which may exert neuroprotective effects (Hemmer and Wallimann, 1993; O'Gorman et al., 1996). We previously showed that creatine administration is neuroprotective in mitochondrial toxin models of HD (Matthews et al., 1998). In the present study, we examined whether creatine administration exerts beneficial effects on survival as well as the behavioral and neuropathological phenotype in R6/2 mice.
MATERIALS AND METHODS
Transgenic HD mice of the R6/2 strain and littermate controls were obtained from Jackson Laboratories (Bar Harbor, ME). The male R6/2 mice were bred with females from their background strain (B6 CBAFI/J). The offspring were genotyped by PCR assay of DNA obtained from tail tissue. CAG repeat length, using a PCR radioassay method (Wheeler et al., 1999), was examined to ensure that a drift in number of CAG repeats did not play a role in the outcome of the studies (courtesy of Dr. Marcy MacDonald, Massachusetts General Hospital). The repeat length remained stable within a 151–154 range. Transgenic mice were housed in microisolator cages in a modified barrier facility. A 12 hr light/dark cycle was maintained, and animals were given ad libitumaccess to food and water. Groups (n = 25) of transgene negative and positive R6/2 mice from the same “f” generation were placed on either unsupplemented diets or diets supplemented with 1, 2, or 3% creatine at 21 d of age (Avicena Group, Cambridge, MA). Approximately 200 mice were used in the survival studies.
Behavioral testing (rotarod). Mice were given 2 d to become acquainted with the rotarod apparatus (Columbus Instruments, Columbus, OH). Testing commenced on day 23. Mice were placed on a rod that was rotating at 10 rpm. Each mouse was given three trials for a maximum of 180 sec for each trial. The length of time at which the mouse fell off the rotating rod was used as the measure of competency on this task. Mice were tested twice weekly until the R6/2 mice were unable to perform the task.
Body weights. Mice were weighed twice a week at the same time of day.
Survival. Mice were observed twice daily, in the morning and late afternoon. The criterion for killing was the point in time when the mice were unable to initiate movement after being gently prodded for 10 min. Two independent observers confirmed this criterion, and this point was used as the time of death.
NMR spectroscopy. We performed in vivospectroscopy at 4.7 T (GE Omega CSI; GE, Freemont, CA) using a sinusoidal bird cage coil (diameter, 20 mm). Mice were anesthetized using halothane/N2O/O2anesthesia (1.5% halothane; 2:1 O2/N2O). Body temperature was maintained using two water blankets surrounding the body at 38°C. Localized proton spectroscopy was run using a PRESS sequence (Bottomley, 1987), with an echo time (TE) of 136 msec and a repetition time (TR) of 2 sec. Spectral width was 2 kHz with 1024 complex points. Six hundred averages were acquired for each spectrum. The transmitter frequency was placed between the NAA and creatine resonances. Voxels were placed symmetrically around both basal ganglia with an average voxel size of 6 × 3.5 × 3 μm (63 μl). Spectra were analyzed using the NMR1 software program (New Methods Research, Syracuse, NY). Spectra were integrated, and the choline peak was normalized to the signal-to-noise of a water spectrum run from the same voxel without water suppression, but with a TR/TE of 10,000/20 msec and eight averages. The NAA and total creatine values were then taken as a ratio to the choline peak.
Histological evaluation. At 21 d, R6/2 transgenic mice and negative littermate controls were fed 2% creatine-supplemented and unsupplemented diets. Groups of 20 animals were deeply anesthetized and then transcardially perfused with 4% buffered paraformaldehyde at 21, 28, 42, 63, and 90 d. The brains were removed, post-fixed with the perfusant for 2 hr, weighed, cryoprotected in a graded series of 10 and 20% glycerol/2% DMSO solution, subsequently serially frozen sectioned at 50 μm, stored in six well tissue collection clusters, and stained for Nissl substance (cresyl violet). Serially cut tissue sections were immunostained for huntingtin using an antibody (EM48; dilution, 1:1,000) that recognizes the first 256 amino acids of human huntingtin lacking the polyglutamine and polypeptide stretches (courtesy of Dr. Xiao-Jiang Li) (Gutekunst et al., 1999). The antibody reacts with N-terminal fragments of huntingtin expressed by transfection. It is a sensitive marker of huntingtin aggregation. An antibody to ubiquitin (dilution, 1:200; Dako, Carpinteria, CA) was used in tissue sections to confirm the presence of aggregates.
Stereology and quantitation. Serial-cut coronal tissue sections from the rostral segment of the neostriatum to the level of the anterior commissure (interaural 5.34 mm/bregma 1.54 mm to interaural 3.7 mm/bregma −0.10 mm) (Franklin and Paxinos, 1997), including the primary motor cortex, were used for neuronal and huntingtin aggregate analysis. Unbiased stereological counts of huntingtin-positive aggregates (≥1.0 μm) were obtained from the neostriatum and layer 6 of the neocortex in 10 animals from unsupplemented and 2% creatine-supplemented R6/2 mice at 28, 42, 63, and 90 d using Neurolucida Stereo Investigator software (Microbrightfield, Colchester, VT). The total areas of the neostriatum and motor cortex were defined in serial sections in which counting frames were randomly sampled. The dissector counting method was used in which huntingtin-positive aggregates were counted in an unbiased selection of serial sections in a defined volume of the neostriatum and neocortex. Striatal neuron areas were analyzed by microscopic videocapture using a Windows-based image analysis system for area measurement (Optimas; Bioscan Incorporated, Edmonds, WA). The software automatically identifies and measures profiles. All computer-identified cell profiles were manually verified as neurons and exported to Microsoft Excel. Cross-sectional areas were analyzed using Statview.
Glucose tolerance test. After 6–7 hr of fasting, baseline levels of glucose were measured. The mice were lightly anesthetized with isoflurane gas, and tail vein blood was collected. The mice were subsequently given a bolus injection of glucose (1.5 gm/kg, i.p.), and plasma glucose levels were measured 30 and 60 min later with Lifescan One Touch basic glucose monitoring system (Johnson & Johnson) and validated by semiautomatic glucose oxidase enzyme assay (Beckman).
Statistics. Statistical comparisons for survival were made by the Mantel-Cox log-rank test. Statistical comparisons of other parameters were made by ANOVA or repeated measures ANOVA of other parameters followed by the Fisher's least significant difference test.
RESULTS
The effects of oral administration of creatine in the diet on survival in HD transgenic mice are shown in Table1. Oral administration of 1% creatine or 2% creatine in the diet dose-dependently improved survival. Administration of 3% creatine significantly improved survival, but was not as effective as either 1 or 2% creatine. Oral administration of 2% creatine was significantly more efficacious than 3% creatine (p < 0.0001). The mean survival in controls increased from 97.6 ± 0.7 d to 106.6 ± 0.5 d with 1% creatine (p < 0.0001), to 114.6 ± 0.9 d with 2% creatine (p < 0.0001), and 101.9 ± 1.0 d with 3% creatine (p < 0.0002). The percentage increase in survival for 1, 2, and 3% creatine was 9.4, 17.4, and 4.4%, respectively.
Table 1.
Effects of oral administration of creatine on survival in R6/2 mice
| Unsupplemented R6/2 mice | 1% Creatine-supplemented R6/2 mice | 2% Creatine-supplemented R6/2 mice | 3% Creatine-supplemented R6/2 mice | |
|---|---|---|---|---|
| Days | 97.7 ± 0.7 | 106.6 ± 0.5* | 114.6 ± 0.9* | 101.9 ± 1.01-160 |
Oral administration of 1, 2, and 3% creatine in the diet dose-dependently improved survival in R6/2 transgenic mice. Oral administration of 2% creatine was significantly greater than 3% creatine (p < 0.0001).
p < 0.0001;
F1-160: p < 0.0002.
The effects of oral administration of creatine in the diet on rotarod performance between 21 and 90 d are shown in Figure1. There was a dose-dependent effect of creatine supplementation. Oral administration of 2% creatine significantly improved rotarod performance throughout the entire measured (4–13 weeks) life span of the R6/2 mice in contrast to unsupplemented R6/2 mice (2% creatine, 156 ± 20 sec; unsupplemented, 88 ± 39 sec, p < 0.001, data represents combined means and SDs from 5 to 12.5 weeks). Dietary supplementation with 1% creatine resulted in significant motor improvement from 5 to 10 weeks (1% creatine, 161 ± 14 sec; unsupplemented, 114 ± 28 sec, p < 0.001, data represents combined means and SDs from 5 to 10 weeks), with no significance observed after 10 weeks. Oral supplementation using 3% creatine had no significant effect on rotarod performance. The percent increase in rotarod performance for 1, 2, and 3% creatine was 25, 33, and 6.5%, respectively.
Fig. 1.
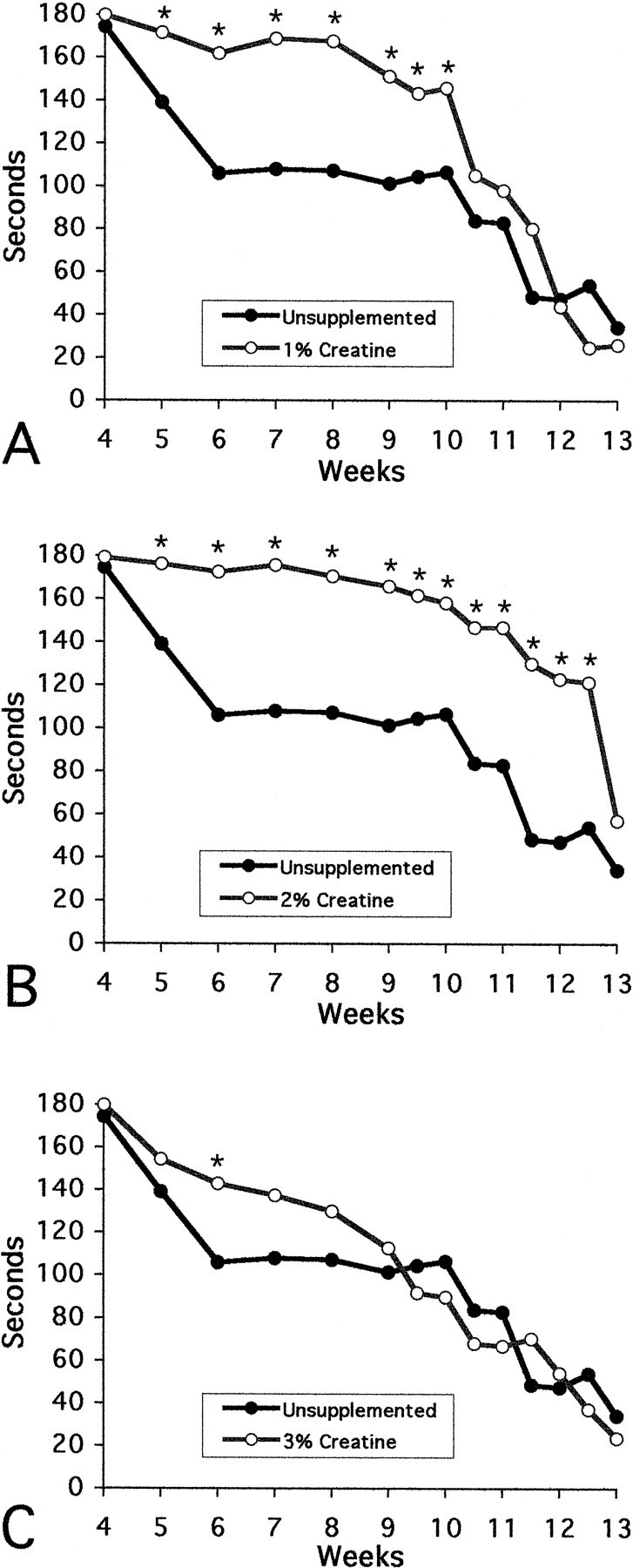
Effects of 1, 2, and 3% creatine on rotarod performance. There was significantly improved performance in R6/2 HD transgenic mice with 2% creatine supplementation throughout the temporal sequence of the experiment (4–13 weeks) (B), from 5–10 weeks in 1% creatine-treated mice (A), with significance only occurring at 6 weeks in the 3% creatine-treated R6/2 mice (C).
The effects of oral administration of creatine on body weight in HD transgenic mice are shown in Figure 2. Whereas all creatine regimens resulted in significant improvement of body weight in comparison to unsupplemented R6/2 mice, 2% dietary creatine supplementation resulted in a significantly greater body weight gain in R6/2 mice (p < 0.01) than either 1 or 3% creatine supplementation (p < 0.02). Significantly greater body weight measurements were present throughout the temporal sequence of measurements (5–13 weeks) in 1 and 2% supplemented R6/2 mice, but were only found from 10–13 weeks in the 3% supplemented group. The total percentage of increase in body weight from 4 to 13 weeks for 1, 2, and 3% creatine-fed R6/2 animals in comparison to unsupplemented R6/2 mice was 6.8, 10.3, and 6.5%, respectively. In comparison to transgene-negative littermate control mice, significant body weight loss in 2% creatine-supplemented mice was delayed until 10 weeks of age. In contrast, significant body weight loss began after 6 weeks in untreated R6/2 mice (data not shown).
Fig. 2.
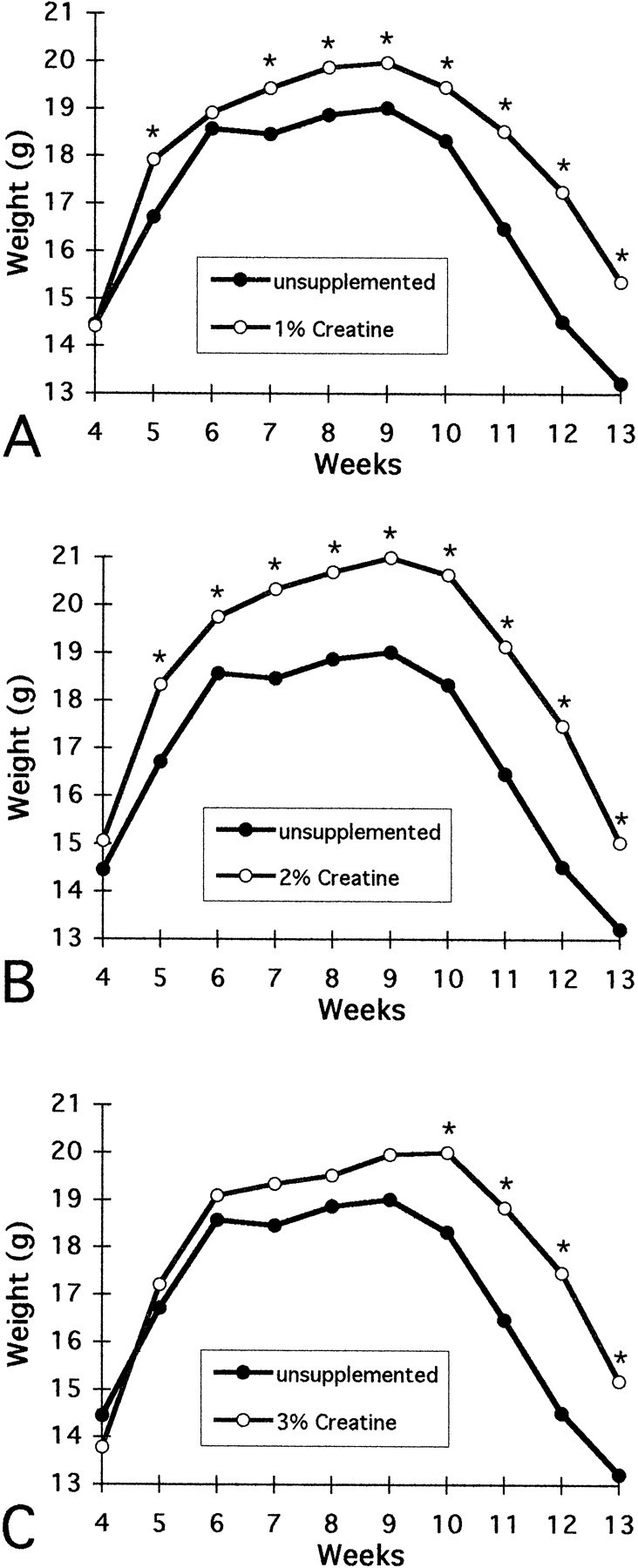
Effects of 1, 2, and 3% creatine on body weight in R6/2 HD transgenic mice. Whereas significantly greater body weight was observed throughout the measured temporal sequence in 1% (except 6 weeks) and 2% creatine-supplemented R6/2 mice, significance was present only from 10 to 13 weeks in the 3% treated mice.
Gross brain weights of unsupplemented R6/2 mice decreased significantly over time until death in comparison to both transgene-negative littermate control mice and 2% creatine-supplemented R6/2 mice at all time points (Table 2). By 90 d, there was a 20.4% reduction in brain weight in contrast to littermate control mice. In comparison, there was no significant decrease in brain weight in the 2% dietary creatine-supplemented R6/2 mice as compared to controls until end stage measurements at 90 d. At that time there was a 6.8% difference in brain weight (Table 2). Coincident with brain weight loss, progressive marked gross atrophy was present in the unsupplemented R6/2 brains, especially within the neostriatum (Fig.3). The striatal atrophy was reminiscent of the neuropathological grading we observed in Huntington's disease (Vonsattel et al., 1985), such that there was bilateral ventricular enlargement with a flattening of the medial aspect of the striatum in the late stages of the illness. Dietary 2% creatine supplementation reduced gross brain atrophy in R6/2 mice in comparison to the untreated R6/2 mice (Fig. 3).
Table 2.
Brain weights/2% creatine-supplemented and unsupplemented R6/2 mice
| Unsupplemented R6/2 mice | 2% Creatine-supplemented R6/2 mice | Littermate controls mice | |
|---|---|---|---|
| 28 d | 402 ± 5* | 414 ± 5 | 412 ± 4 |
| 42 d | 395 ± 6* | 416 ± 4 | 424 ± 6 |
| 63 d | 375 ± 9* | 411 ± 8 | 425 ± 6 |
| 90 d | 348 ± 13* | 407 ± 102-160 | 437 ± 5 |
Paraformaldehyde-fixed brain weights in milligrams from unsupplemented, 2% creatine-supplemented, and littermate transgene negative R6/2 mice.
p < 0.002;
F2-160: p < 0.005.
Fig. 3.
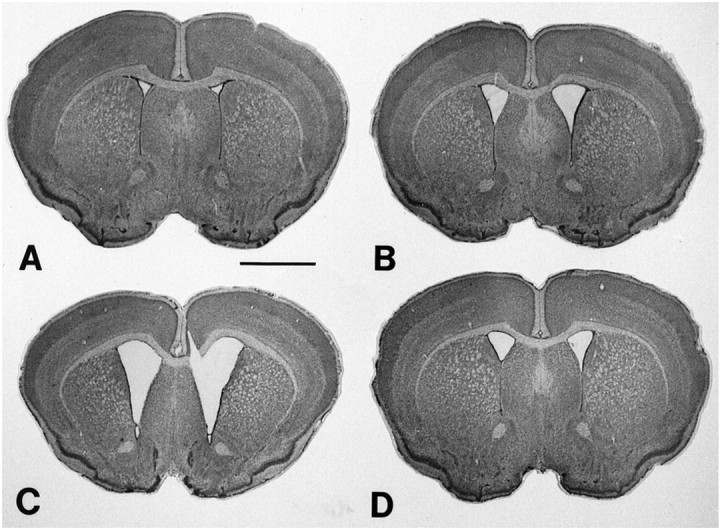
Photomicrographs of coronal sections through the rostral neostriatum at the level of the anterior commissure in R6/2 HD transgenic mice at 42 (A), 63 (B), and 90 (C) d. Note the generalized gross atrophy of the brain over time along with enlargement of the lateral ventricles. In contrast, a 2% creatine-supplemented R6/2 mouse at 90 d (D) shows significantly less atrophy and ventricular enlargement than the unsupplemented mouse (C). Scale bar, 2 mm.
Consistent with the gross brain weight loss and striatal atrophy, there was significant progressive atrophy of striatal neurons from 21 to 90 d in the unsupplemented R6/2 mice with a 37.9% overall decrease in area measurements (striatal neurons R6/2 at 28 d, 88.8 ± 10.7 μm2; striatal neurons R6/2 at 90 d, 55.1 ± 16.8 μm2; p < 0.001) (Figs.4, 5). The cytoprotective effect of 2% creatine significantly delayed striatal neuron atrophy. There were no significant differences in neuronal areas in 2% creatine-supplemented R6/2 mice until endstage measurements at 90 d of age (Fig. 4).
Fig. 4.

Neuronal areas of 2% creatine-supplemented and unsupplemented R6/2 mice in comparison to littermate transgene-negative mice at 28, 42, 63, and 90 d; *p < 0.001.
Fig. 5.

Photomicrographs of Nissl-stained tissue sections from the dorsomedial aspect of the neostriatum (A, C, E, G) and 2% creatine-supplemented (B, D, F, H) R6/2 HD transgene mice at 4 (A, B), 6 (C, D), 9 (E, F), and 13 (G, H) weeks. Note the progressive loss in neuronal size in the unsupplemented R6/2 group, with delayed neuronal atrophy in the 2% creatine-supplemented R6/2 mice. Scale bar, 100 μm.
We examined six mice fed with 2% creatine and eight unsupplemented mice using NMR spectroscopy at 51–57 d of age. As compared with unsupplemented mice, there was a significantly higher NAA/choline ratio in the creatine-fed mice (0.37 ± 0.06 vs 0.48 ± 0.03;p < 0.05). The creatine/choline was significantly increased from 0.67 ± 0.04 to 0.83 ± 0.07 (p < 0.01). In a total of 13 mice fed creatine compared to 14 mice on unsupplemented diets, including mice older than 70 d of age, the NAA/choline ratio was 0.54 ± 0.03 vs 0.45 ± 0.04 (p = 0.09). In the total group the creatine/choline was 0.85 ± 0.04 vs 0.72 ± 0.03 (p < 0.01). When normalized to a water standard, there was a significant 21.3 ± 3.8% increase in brain creatine concentrations. In the creatine-treated mice there was a significant correlation between NAA/choline and Cr/choline (p < 0.01) that was not seen in the unsupplemented mice (Fig. 6).
Fig. 6.

Correlation between creatine and NAA levels in HD transgenic R6/2 mice. Correlation between NAA and CR in unsupplemented mice was not significant.
An analysis of the formation of aggregates in the neostriatum and cortex of R6/2 mice showed an early and progressive accumulation of huntingtin-positive aggregates, as well as an increase in aggregate size, from 21 d of age to the data collection end point at 90 d. Aggregates were much more prominent within the cortex in comparison to the neostriatum. Dietary supplementation with 2% creatine resulted in a significant reduction in striatal aggregate number throughout treatment at each measured time point (Figs.7, 8). At 28, 42, 63, and 90 d, the percentage of decrease in aggregate number as compared with unsupplemented mice was 60, 51, 35, and 39%, respectively. A similar trend toward decreased aggregate number was present within the cortex in 2% creatine-treated R6/2 mice, however these decreases were not significant (Figs. 7,9).
Fig. 7.
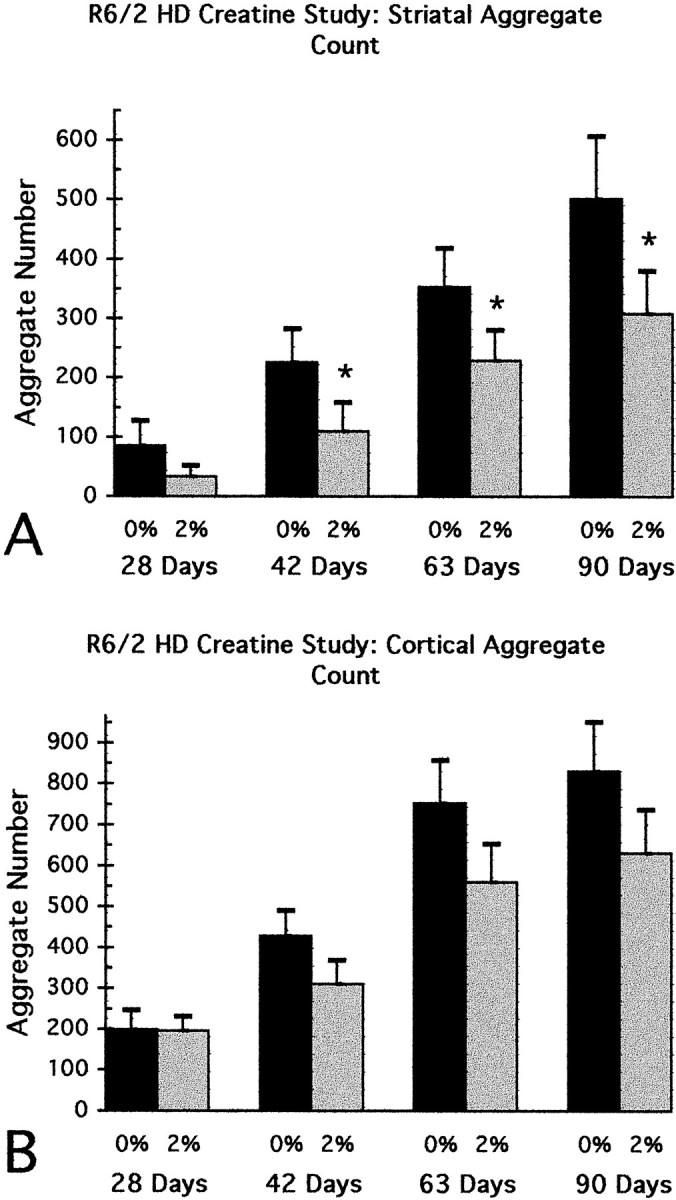
Graphs of the temporal sequence in the number of huntingtin-positive aggregates in the neostriatum (A) and motor neocortex (B) at 4, 6, 9, and 13 weeks. There was a significant delay in the formation of aggregates within the striatum in 2% creatine-supplemented R6/2 mice, in comparison to unsupplemented R6/2 mice. Although a similar trend was observed in the neocortex, significance was not obtained.
Fig. 8.
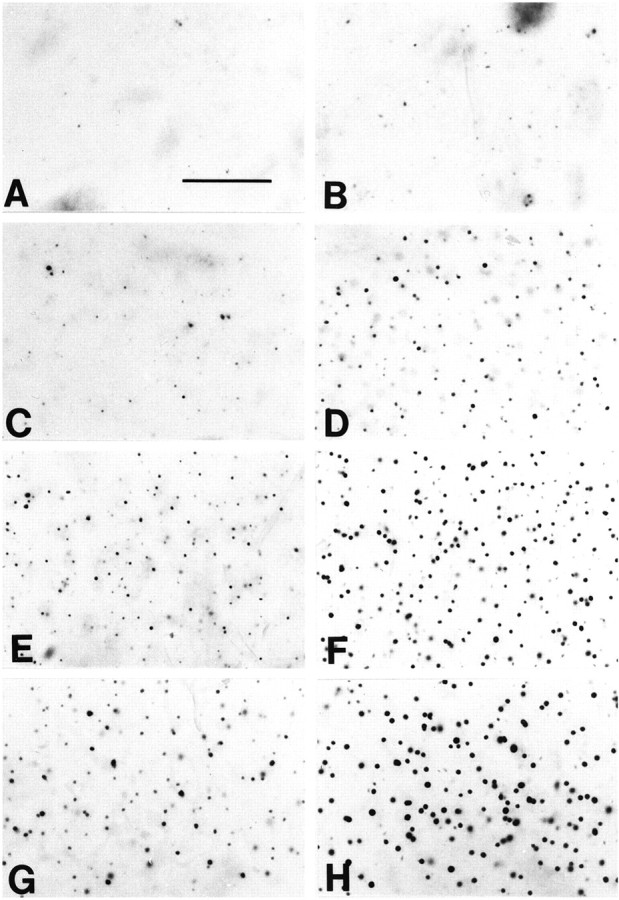
Photomicrographs of huntingtin-immunostained tissue sections from dorsomedial aspect of the neostriatum at the level of the anterior commissure in 2% creatine-supplemented (A, C, E, G) and unsupplemented (B, D, F, H) R6/2 HD transgene mice at 4 (A, B), 6 (C, D), 9 (E, F), and 13 (G, H) weeks. There is a progressive increase in number and size of huntingtin aggregates over time in the unsupplemented R6/2 mice in comparison to the delay in aggregate formation in the 2% creatine-supplemented R6/2 mice. Scale bar, 50 μm.
Fig. 9.

Photomicrographs of huntingtin-immunostained tissue sections from layer 6 of the motor cortex at the level of the anterior commissure in unsupplemented (A, C, E) and 2% creatine-supplemented (B, D, F) R6/2 HD transgene mice at 4 (A, B), 9 (C, D), and 13 (E, F) weeks. There is a progressive increase in number and size of huntingtin aggregates over time in the unsupplemented R6/2 mice in comparison to the delay in aggregate formation in the 2% creatine-supplemented R6/2 mice. Scale bar, 100 μm.
In the unsupplemented R6/2 mice, there was an increase of huntingtin-positive aggregates in the pancreatic Islets of Langerhan over time that was first observed at 42 d and became most prominent at 90 d (Fig. 10). Little or no detectable huntingtin-positive aggregates were observed in the pancreatic stroma. Dietary 2% creatine significantly reduced aggregate number in the pancreas of 90-d-old R6/2 mice (2% creatine, 57 ± 12/field; unsupplemented, 139 ± 15/field;p < 0.001) (Fig. 10). Furthermore, administration of 2% creatine significantly delayed the onset of diabetes as assessed by a glucose tolerance test at 8.5 weeks of age (Fig.11).
Fig. 10.
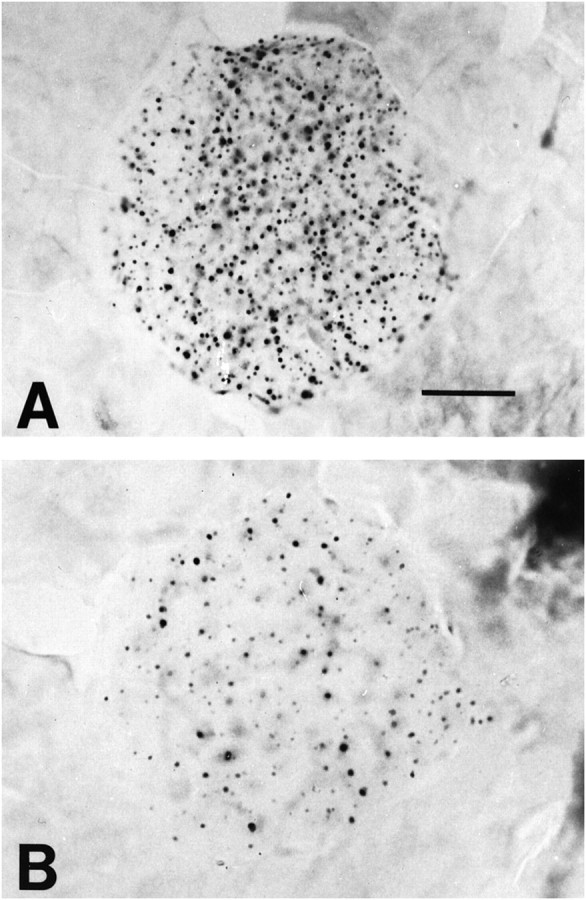
Photomicrographs of islets of Langerhan in the pancreas of 90-d-old unsupplemented (A) and 2% creatine-supplemented (B) R6/2 HD transgenic mice immunostained with EM48 antibody. There is a marked reduction in the huntingtin aggregates within the treated mouse. Scale bar, 50 μm.
Fig. 11.

Effects of 2% creatine supplementation on glucose tolerance in 8.5-week-old R6/2 mice. Creatine administration significantly attenuated abnormal glucose tolerance. *p < 0.05; **p < 0.01.
DISCUSSION
The development of transgenic mouse models of neurodegenerative diseases provides a major advance for studying disease pathogenesis and for developing therapeutics. If therapeutic effects in the transgenic mice are shown to be predictive of beneficial effects in man, then this will allow rapid screening for new therapies. As discussed above, there is substantial evidence that energy dysfunction occurs in HD, and that this may play a role in cell death. Creatine administration, as discussed below, has several potential neuroprotective effects, including buffering of intracellular energy reserves, stabilizing intracellular calcium, and inhibiting activation of the MPT, all of which have been linked to excitotoxic and apoptotic cell death (O'Gorman et al., 1997; Leist and Nicotera, 1998; Lipton and Nicotera, 1998).
The brain isoform of creatine kinase along with the mitochondrial isoform and the substrates creatine and phosphocreatine constitute a system that appears to be critical in regulating energy homeostasis in the brain (Hemmer and Wallimann, 1993). There is evidence for a direct functional coupling of creatine kinase with sodium potassium ATPase, neurotransmitter release, maintenance of membrane potentials, and restoration of ion gradients after depolarization (Dunant et al., 1988;Hemmer and Wallimann, 1993). An important role of creatine kinase in the adult brain is supported by in vivo31P NMR transfer measurements. These show that creatine kinase flux correlates with brain activity as measured by the EEG, as well as with amounts of 2-deoxyglucose uptake in the brain (Sauter and Rudin, 1993; Corbett and Laptook, 1994). We previously showed that administration of creatine increases brain phosphocreatine concentrations and buffers against toxin-induced depletions (Matthews et al., 1998).
Creatine kinase also appears to be coupled directly or indirectly to energetic processes required for calcium homeostasis (Wallimann et al., 1992; Steeghs et al., 1997). Creatine pretreatment delays increases in intracellular calcium produced by 3-nitropropionic acid in cortical and striatal astrocytes in vitro (Deshpande et al., 1997). Another potential neuroprotective mechanism is the ability of phosphocreatine to stimulate synaptic glutamate uptake and thereby reduce extracellular glutamate (Xu et al., 1996).
Lastly, creatine may protect against activation of the MPT, which is associated with both apoptotic and necrotic cell death (Bernardi et al., 1998). The MPT is a Ca2+-dependent increase of the inner membrane permeability to ions and solutes of up to 1500 Da. Mitochondrial creatine kinase is implicated in a functional interaction between the outer membrane voltage-dependent anion channel and the inner membrane adenylate translocator, which are components of the MPT (Brdiczka et al., 1998). Creatine administration can stabilize mitochondrial creatine kinase in an octomeric form, which inhibits activation of the MPT (O'Gorman et al., 1997). Mitochondrial creatine kinase is also a prime target for peroxynitrite-induced damage, which results in dissociation of mitochondrial creatine kinase octomers into dimers (Soboll et al., 1999). Creatine administration inhibits peroxynitrite-induced modification and inactivation (Stachowiak et al., 1998). Creatine administration may also increase ADP concentrations, which inhibits activation of the MPT (Bernardi et al., 1998).
Previous studies showed neuroprotective effects of creatine in vitro and in vivo. Creatine reduces anoxic damage to hippocampal slices in vitro (Carter et al., 1995). We have found that creatine administration exerts neuroprotective effects against animal models of Huntington's disease produced by administration of the mitochondrial toxins malonate and 3-nitropropionic acid (Matthews et al., 1998). Creatine administration also attenuates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopamine depletions and substantia nigra neuronal loss (Matthews et al., 1999). In addition, creatine administration increases survival and improves motor performance in a transgenic mouse model of amyotrophic lateral sclerosis and results in marked neuroprotective effects against the loss of anterior horn motor neurons and substantia nigra dopaminergic neurons (Klivenyi et al., 1999).
In the present study, we therefore investigated whether creatine could exert neuroprotective effects in a transgenic mouse model of HD. We found that creatine dose-dependently improved survival in these mice. The increased survival results are comparable to the report of Ona et al. (1999) in which R6/2 mice were crossed with mice with a dominant-negative inhibitor of caspase 1. In that study the maximal increase in survival was 20 d or 20%, whereas in our study 2% creatine increased survival by 17 d or 17.5%. The effect with 3% creatine, however, was less than that seen with either 1% or 2% creatine, consistent with an inverted dose–response curve. We have made similar observations with malonate and MPTP toxicity (Matthews et al., 1998, 1999). The explanation for the inverted dose–response curve is unclear, and at very high concentrations creatine may be toxic, similar to observations with cyclocreatine (Matthews et al., 1998).
Creatine administration resulted in improved rotarod performance and reduced weight loss in the R6/2 mice. Similar to effects on survival, 2% creatine was most efficacious, with 1% creatine more effective than 3% creatine. Interestingly, the administration of creatine also delayed the onset of diabetes that has been demonstrated in these mice (Hurlbert et al., 1999). Administration of creatine delayed the development of both striatal and pancreatic huntingtin-positive aggregates, consistent with other recent observations that experimental manipulations can slow the development of nuclear intraneuronal inclusions (Ona et al., 1999). Cross-sectional areas of striatal neurons in R6/2 and other transgenic models of HD have been recently reported to be reduced (Reddy et al., 1998; Hodgson et al., 1999;Levine et al., 1999). Consistent with these findings, we found that administration of 2% creatine significantly delayed the development of both neuronal shrinkage, as well as gross atrophy of the brain. Using NMR spectroscopy we showed that 2% creatine significantly increased brain creatine concentrations by 21% and that it significantly attenuated early decreases in N-acetylaspartate concentrations. The increases in creatine peak may largely reflect PCr consistent with our earlier biochemical measurements (Matthews et al., 1998). We recently found a 53% decrease in N-acetylaspartate concentrations in the R6/2 mice starting at 6 weeks of age (Jenkins et al., 2000).N-acetylaspartate is a neuronal marker that decreases in HD (Jenkins et al., 1998), however it may also reflect mitochondrial function (Bates et al., 1996). NMR spectroscopy could therefore be useful in monitoring therapeutic effects of creatine in patients.
These results provide further evidence that creatine is neuroprotective in animal models of neurodegenerative diseases. Creatine administration is well tolerated in man, results in increased PCr levels, and may have benefits in pathological conditions (Balsom et al., 1994; Dawson et al., 1995; Greenhaff, 1997). Long-term administration to patients with gyrate atrophy of the choroid in the retina prevented visual field constriction and resulted in improvement of muscle biopsy findings (Sipila et al., 1981). Several pediatric patients with creatine deficiency accompanied by an extrapyramidal movement disorder showed partial restoration of cerebral creatine concentrations and clinical improvement after oral creatine administration (Stockler et al., 1994,1996). Creatine administration has also resulted in significant improvement of patients with the mitochondrial disorder mitochondrial encephalopathy lactic acidosis and strokes (Tarnopolsky et al., 1997), as well as in patients suffering from neuromuscular disorders (Tarnopolsky and Martin, 1999). The present findings support a role for metabolic dysfunction in a transgenic mouse model of HD and provide further evidence that treatment with creatine might be a novel therapeutic strategy to slow or halt the progression of neurodegeneration in HD.
Footnotes
This work was supported by National Institutes of Health Grants NS38180 (M.F.B.), NS35255 (R.J.F. and S.M.H.), NS37102 and AG13846 (R.J.F.), and AG12992 (M.F.B. and R.J.F.), the Hereditary Disease Foundation, the Huntington's Disease Society of America, and the Veteran's Administration. The secretarial assistance of Sharon Melanson is gratefully acknowledged.
Correspondence should be addressed to Dr. M. Flint Beal, Chairman, Neurology Department, New York Hospital–Cornell Medical Center, 525 East 68th Street, New York, NY 10021. E-mail:fbeal@mail.med.cornell.edu or Dr. Robert J. Ferrante, GRECC Unit, 182B, Bedford VA Medical Center, 200 Springs Road, Bedford, MA 01730. E-mail: rjferr@bu.edu.
REFERENCES
- 1.Balsom PD, Soderlund K, Ekblom B. Creatine in humans with special reference to creatine supplementation. Sports Med. 1994;18:268–280. doi: 10.2165/00007256-199418040-00005. [DOI] [PubMed] [Google Scholar]
- 2.Bates TE, Strangward M, Keelan J, Davey GP, Munro PMG, Clark JB. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. NeuroReport. 1996;7:1397–1400. [PubMed] [Google Scholar]
- 3.Bernardi P, Basso E, Colonna R, Costantini P, Di Lisa F, Eriksson O, Fontaine E, Forte M, Ichas F, Massari S, Nicolli A, Petronilli V, Scorrano L. Perspectives of the mitochondrial permeability transition. Biochim Biophys Acta. 1998;1365:200–206. [Google Scholar]
- 4.Bogdanov MB, Ferrante RJ, Kuemmerle S, Klivenyi P, Beal MF. Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington's disease. J Neurochem. 1998;71:2652–2644. doi: 10.1046/j.1471-4159.1998.71062642.x. [DOI] [PubMed] [Google Scholar]
- 5.Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann NY Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- 6.Brdiczka D, Beutner G, Ruck A, Dolder M, Wallimann T. The molecular structure of mitochondrial contact sites. Their role in regulation of energy metabolism and permeability transition. Biofactors. 1998;8:235–242. doi: 10.1002/biof.5520080311. [DOI] [PubMed] [Google Scholar]
- 7.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MMK, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 9.Carter AJ, Muller RE, Pschorn U, Stransky W. Preincubation with creatine enhances levels of creatine phosphate and prevents anoxic damage in rat hippocampal slices. J Neurochem. 1995;64:2691–2699. doi: 10.1046/j.1471-4159.1995.64062691.x. [DOI] [PubMed] [Google Scholar]
- 10.Cha J-HJ, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corbett RJT, Laptook AR. Age-related changes in swine brain creatine kinase-catalyzed 31P exchange measured in vivo using 31P NMR magnetization transfer. J Cereb Blood Flow Metab. 1994;14:1070–1077. doi: 10.1038/jcbfm.1994.140. [DOI] [PubMed] [Google Scholar]
- 12.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiari L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 13.Dawson TM, Hung K, Dawson VL, Steiner JP, Snyder SH. Neuroprotective effects of gangliosides may involve inhibition of nitric oxide synthase. Ann Neurol. 1995;37:115–118. doi: 10.1002/ana.410370122. [DOI] [PubMed] [Google Scholar]
- 14.Deshpande SB, Fukuda A, Nishino H. 3-Nitropropionic acid increases the intracellular Ca2+ in cultured astrocytes by reverse operation of the Na+-Ca2+ exchanger. Exp Neurol. 1997;145:38–45. doi: 10.1006/exnr.1997.6457. [DOI] [PubMed] [Google Scholar]
- 15.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 16.Dunant Y, Loctin F, Marsal J, Muller D, Parducz A, Rabasseda X. Energy metabolism and quantal acetylcholine release: effects of botulinum toxin, 1-fluoro-2,4-dinitrobenzene, and diamide in the Torpedo electric organ. J Neurochem. 1988;50:431–439. doi: 10.1111/j.1471-4159.1988.tb02930.x. [DOI] [PubMed] [Google Scholar]
- 17.Ferrante RJ, Gutekunst CA, Persichetti F, McNeil SM, Kowall NW, Gusella JF, MacDonald ME, Beal MF, Hersch SM. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci. 1997;17:3052–3063. doi: 10.1523/JNEUROSCI.17-09-03052.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 19.Greenhaff PL. The nutritional biochemistry of creatine. J Nutr Biochem. 1997;8:610–618. [Google Scholar]
- 20.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AHV. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 21.Gutekunst C-A, Peng T-I, Whaley WL, Rock B, Hersch SM, Greenamyre JT. Mitochondrial calcium homeostasis in Huntington's disease fibroblasts. Soc Neurosci Abstr. 1996;22:227. [Google Scholar]
- 22.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hemmer W, Wallimann T. Functional aspects of creatine kinase in brain. Dev Neurosci. 1993;15:249–260. doi: 10.1159/000111342. [DOI] [PubMed] [Google Scholar]
- 24.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 25.Hurlbert MS, Zhou W, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington's disease gene develop diabetes. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins B, Koroshetz W, Beal MF, Rosen B. Evidence for an energy metabolism defect in Huntington's disease using localized proton spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins BG, Rosas HD, Chen YC, Makabe T, Myers R, MacDonald M, Rosen BR, Beal MF, Koroshetz WJ. 1H NMR spectroscopy studies of Huntington's disease: correlations with CAG repeat numbers. Neurology. 1998;50:1357–1365. doi: 10.1212/wnl.50.5.1357. [DOI] [PubMed] [Google Scholar]
- 28.Jenkins BG, Klivenyi P, Kustermann E, Andreassen OA, Ferrante RJ, Rosen BR, Beal MF. Non-linear decrease over time in N-acetylaspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington's disease mice. J Neurochem. 2000;74:2108–2119. doi: 10.1046/j.1471-4159.2000.0742108.x. [DOI] [PubMed] [Google Scholar]
- 29.Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of ALS. Nat Med. 1999;5:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- 30.Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington's disease and possible therapy with coenzyme Q10. Ann Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 31.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntingtin aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 32.Landwehrmeyer GB, McNeil SM, Dure I, LS, Ge P, Aizawa H, Huang Q, Ambrose CM, Duyao MP, Bird ED, Bonilla E, de Young M, Avila-Gonzales AJ, Wexler NS, DiFiglia M, Gusella JF, MacDonald ME, Penney JB, Young AB, Vonsattel J-P. Huntington's disease gene: regional and cellular expression in brain of normal and affected individuals. Ann Neurol. 1995;37:218–230. doi: 10.1002/ana.410370213. [DOI] [PubMed] [Google Scholar]
- 33.Leist M, Nicotera P. Apoptosis, excitotoxicity, and neuropathology. Exp Cell Res. 1998;239:183–201. doi: 10.1006/excr.1997.4026. [DOI] [PubMed] [Google Scholar]
- 34.Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- 35.Lipton SA, Nicotera P. Excitotoxicity, free radicals, necrosis, and apoptosis. Neuroscientist. 1998;4:345–352. [Google Scholar]
- 36.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 37.Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington's disease. J Neurosci. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- 39.O'Gorman E, Beutner G, Wallimann T, Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim Biophys Acta. 1996;1276:161–170. doi: 10.1016/0005-2728(96)00074-6. [DOI] [PubMed] [Google Scholar]
- 40.O'Gorman E, Beutner G, Dolder M, Koretsky AP, Brdiczka D, Wallimann T. The role of creatine kinase inhibition of mitochondrial permeability transition. FEBS Lett. 1997;414:253–257. doi: 10.1016/s0014-5793(97)01045-4. [DOI] [PubMed] [Google Scholar]
- 41.Ona VO, Li M, Vonsattel JPG, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li X-J, Stieg PE, Yuan J, Penney JB, Young AB, Cha J-HJ, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 42.Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 43.Sauter A, Rudin M. Determination of creatine kinase parameters in rat brain by NMR magnetization transfer: correlation with brain function. J Biol Chem. 1993;268:13166–13171. [PubMed] [Google Scholar]
- 44.Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr, Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 45.Sharp NH, Loev SJ, Schilling G, Li S-H, Li X-J, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A, Hedreen J, Sisodia S, Snyder SH, Dawson TM, Ryugo DK, Ross CA. Widespread expression of the Huntington's disease gene (IT-15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- 46.Sipila I, Rapola J, Simell O, Vannas A. Supplementary creatine as a treatment for gyrate atrophy of the choroid and retina. N Engl J Med. 1981;304:867–870. doi: 10.1056/NEJM198104093041503. [DOI] [PubMed] [Google Scholar]
- 47.Soboll S, Brdiczka D, Jahnke D, Schmidt A, Schlattner U, Wendt S, Wyss M, Wallimann T. Octamer-dimer transitions of mitochondrial creatine kinase in heart disease. J Mol Cell Cardiol. 1999;31:857–866. doi: 10.1006/jmcc.1998.0925. [DOI] [PubMed] [Google Scholar]
- 48.Stachowiak O, Dolder M, Wallimann T, Richter C. Mitochondrial creatine kinase is a prime target of peroxynitrite- induced modification and inactivation. J Biol Chem. 1998;273:16694–16699. doi: 10.1074/jbc.273.27.16694. [DOI] [PubMed] [Google Scholar]
- 49.Steeghs K, Benders A, Oerlemans F, de Haan A, Heerschap A, Ruitenbeek W, Jost C, van Deursen J, Perryman B, Pette D, Bruckwilder M, Koudijs J, Jap P, Veerkamp J, Wieringa B. Altered Ca2+ responses in muscles with combined mitochondrial and cytosolic creatine kinase deficiencies. Cell. 1997;89:93–103. doi: 10.1016/s0092-8674(00)80186-5. [DOI] [PubMed] [Google Scholar]
- 50.Stockler S, Holzbach U, Hanefeld F, Marquardt I, Helms G, Requart M, Hanicke W, Frahm J. Creatine deficiency in the brain: a new, treatable inborn error of metabolism. Pediatr Res. 1994;36:409–413. doi: 10.1203/00006450-199409000-00023. [DOI] [PubMed] [Google Scholar]
- 51.Stockler S, Hanefeld F, Frahn J. Creatine replacement therapy in quanidinoacetate methyltransferase deficiency, a novel inborn error of metabolism. Lancet. 1996;348:789–790. doi: 10.1016/s0140-6736(96)04116-5. [DOI] [PubMed] [Google Scholar]
- 52.Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, Kaatz KW, Collins FS, Albin RL. Widespread expression of the human and rat Huntington's disease gene in brain and nonneural tissues. Nat Genet. 1993;5:259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- 53.Tarnopolsky M, Martin J. Creatine monohydrate increases strength in patients with neuromuscular disease. Neurology. 1999;52:854–857. doi: 10.1212/wnl.52.4.854. [DOI] [PubMed] [Google Scholar]
- 54.Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20:1502–1509. doi: 10.1002/(sici)1097-4598(199712)20:12<1502::aid-mus4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 55.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 56.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 57.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281:21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V, Weaver M, Gusella JF, Joyner AL, MacDonald ME. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- 59.Xu CJ, Klunk WE, Kanfer JN, Xiong Q, Miller G, Pettegrew JW. Phosphocreatine-dependent glutamate uptake by synaptic vesicles. J Biol Chem. 1996;271:13435–13440. doi: 10.1074/jbc.271.23.13435. [DOI] [PubMed] [Google Scholar]