

Abstract
Long-term potentiation (LTP) has several different phases, and there is general agreement that the late phase of LTP requires the activation of adenylyl cyclase (AC) and cAMP-dependent protein kinase (PKA). In contrast, several studies indicate that the early LTP is not affected by interfering with the cAMP pathway. We have further tested the role of the cAMP pathway in early LTP using several types of inhibitors. Bath application of the PKA inhibitor H89 suppressed the early LTP induced by a single tetanus. Similarly, the LTP induced by a pairing protocol was decreased by postsynaptic intracellular perfusion of the peptide PKA inhibitor PKI(6–22) amide. The decrease of LTP produced by these inhibitors was evident immediately after induction. These results indicate that PKA is important in early LTP, that its locus of action is postsynaptic, and that it does not act merely by enhancing the depolarization required for LTP induction. The failure of some other inhibitors of the cAMP pathway to affect the early phase of LTP might be attributable to the saturation of some step in the cAMP pathway during a tetanus. In agreement with this hypothesis we found that application of the AC inhibitor SQ 22536 by itself did not affect the early phase of LTP, but did produce a reduction if the cAMP pathway was already attenuated by the PKA inhibitor H89. Our analysis of the results of genetic modifications of the cAMP pathway, especially the work on AC knock-outs, indicates that the genetic data are generally consistent with the pharmacological results showing the importance of this pathway in early LTP.
Keywords: adenylyl cyclase, CA1, cAMP-dependent protein kinase, early LTP, H89, PKI(6–22) amide, SQ 22536
Long-term potentiation (LTP) and long-term depression (LTD) are activity-dependent modifications of synaptic strength that are thought to underlie some forms of memory. Both LTP and LTD induction requires the elevation of intracellular Ca2+. This elevation, in turn, activates a biochemical cascade that modifies synaptic strength. The intermediate steps of this cascade are not well established. There is general agreement that Ca2+-dependent protein kinases and phosphatases are involved. Other second messenger pathways have also been implicated, but their role is less clear (for review, see Malenka and Nicoll, 1999).
In this paper, we focus on the role of the cAMP pathway in LTP at the Schaffer collateral synapses on CA1 hippocampal pyramidal cells. It is known that LTP induction produces a rise in cAMP (Chetkovich and Sweatt, 1993), activation of PKA (Blitzer et al., 1995; Roberson and Sweatt, 1996), and phosphorylation of some PKA substrates (Blitzer et al., 1998), but the role of this pathway in LTP remains unclear despite extensive genetic and pharmacological work. The issue is complex because LTP is itself not a unitary phenomenon, but rather appears to involve late, intermediate, and early phases (Frey et al., 1993; Huang and Kandel, 1994; Winder et al., 1998) relevant to the mechanism of memory consolidation (Bourtchouladze et al., 1998). Late LTP begins ∼3 hr after induction, and there is general agreement that it depends on cAMP and protein synthesis (Frey et al., 1993; Huang and Kandel, 1994; Winder et al., 1998), probably related to the formation of new synapses (Bolshakov et al., 1997; Ma et al., 1999). It has been reported that there is also an intermediate phase of LTP that begins approximately an hour after induction and is dependent on cAMP, but not protein synthesis (Winder et al., 1998).
The issue of whether early LTP depends on cAMP is less clear. The standard measure of early LTP is the potentiation observed 30–40 min after induction, a time at which short-term plasticity processes induced by a tetanus have decayed. It is believed that early LTP does not depend on AC or PKA (Frey et al., 1993; Huang and Kandel, 1994), and there are several pharmacological and genetic studies that show that interfering with the cAMP pathway does not affect early LTP (Qi et al., 1996; Abel et al., 1997; Otmakhova and Lisman, 1998; Winder et al., 1998). However, this conclusion cannot be considered definitive because many of the genetic modifications did not lead to a substantial change in measured enzyme activity, probably because of presence of multiple enzyme isoforms and because the degree to which pharmacological inhibitors actually inhibited the enzyme activity in living cells could not be directly determined. We have reexamined the role of the cAMP pathway using a broader range of inhibitors, combinations of inhibitors, and more direct methods for introducing inhibitors into the postsynaptic cell. The general conclusion we have reached from our data and from reevaluation of the published genetic evidence is that the cAMP pathway does have an important role in early LTP.
MATERIALS AND METHODS
Transverse hippocampal slices (350- to 400-μm-thick) were prepared from 17- to 25-d-old Long–Evans rats. After preparation, slices were preincubated in an inverse interface chamber for 2–6 hr before an experiment. The inverse interface conditions were created by placing a slice in a drop of the artificial CSF (ACSF) on a Millipore filter (pore diameter, 8 μm). The filters were held in the humidified atmosphere of 95% O2 and 5% CO2. In this condition the slice-gas interface was at the bottom side of the slice rather then at the top side in the standard interface chamber. In the experiment using field potential recordings, slices were placed on a nylon net and perfused on both sides with ACSF using a pump with flow rate 1.5–2.25 ml/min. ACSF contained (in mm): NaCl 120, NaHCO426, NaH2PO4 1, KCl 2.5, MgSO4 1.3, CaCl2 2.5, andd-glucose 10. Before entry into the recording chamber, ACSF was saturated with a gas mixture of 95% oxygen and 5% carbon dioxide and heated to the temperature 29–30oC. When whole-cell patch-clamp experiments were performed, slices were held submerged on the glass bottom of the recording chamber and were continuously superfused (2 ml/min). Whole-cell recording was performed at room temperature (∼22–25oC). ACSF was slightly modified: 4 mm CaCl2, 4 mm MgSO4. In addition, 50 μm picrotoxin was used to block GABAA inhibition.
Field potential experiments. A glass recording electrode filled with ACSF (resistance, 0.2–0.3 MΩ) was placed in stratum radiatum of the CA1 region. To stimulate independent inputs, two monopolar stimulating electrodes (glass pipettes filled with ACSF; resistance, 0.25–0.35 MΩ) were positioned on both sides of the recording electrode ∼200–250 μm apart from each other. Data were acquired by personal computer with Digidata-1200 interface (Axon Instruments, Foster City, CA) by a custom-made AXOBASIC program. The strength of stimulation was adjusted so that field EPSP (fEPSP) amplitude was 50% of the population spike threshold. The same stimulus strength was used for both test and tetanic stimulation. The tetanus used for the induction of early LTP consisted of 40 stimuli and lasted 0.6 sec (10 bursts of four stimuli at 100 Hz with 30 msec intervals between bursts). In some experiments three of such tetani 10 min apart were given. Test stimulation alternated between two stimulating electrodes throughout the experiment at constant interval (10 sec). LTP was induced after at least 15 min of a stable baseline and observed for 30–40 min after the induction. SQ 22536 (Research Biochemicals, Natick, MA) was dissolved in ACSF and applied in the perfusion media during the experiment. H89 (Calbiochem, La Jolla, CA) was dissolved in DMSO (10 mm), diluted in regular ACSF to 20 μm, and used during preincubation of slices for 1–2 hr before an experiment.
Whole-cell patch-clamp. Whole-cell recording was performed using an Axopatch-1D amplifier (Axon Instruments) with low-pass filter set at 1 kHz. The patch pipettes had a resistance of 2.5–3.5 MΩ when filled with pipette solution. The pipette solution contained (in mm): Cs-methanesulfonate, 120; CsCl, 20; HEPES, 10; MgATP, 4; Na3GTP, 0.3; EGTA, 0.2; and phosphocreatine, 10; pH, 7.3; osmolarity, 300 mOsm. Patching was performed under visual control using infrared oblique illumination and a CCD video camera. Recordings were made from cell bodies in the CA1 pyramidal layer, 20–80 μm beneath the slice surface. Whole-cell currents were measured in voltage-clamp mode at a holding potential of −65 mV. Series and input resistances were monitored every 6 sec by measuring the peak and steady-state currents in response to 2–4 mV, 38 msec hyperpolarizing steps. Recorded signals were digitized at 5–10 kHz and then stored and analyzed using custom software written in Axobasic (Axon Instruments). Two stimulating electrodes (glass pipettes filled with ACSF, 300 kΩ) were placed in stratum radiatum to activate two independent inputs of Schaffer collaterals. The electrodes were positioned in the dendritic region at ∼70 and 150 μm from the cell body and ∼50 μm lateral from the dendritic tree of the recorded cell. Each input was stimulated every 6 sec with a 2–50 μA, 150 μsec square pulse delivered through current isolation units (Isolator-11; Axon Instruments). The two inputs were stimulated alternatively. LTP was induced using a pairing procedure in which a cell was depolarized to 0 mV, and 200 stimuli were delivered to one of the inputs at 1.4 Hz.
Intracellular perfusion. Perfusion was performed as described elsewhere (Otmakhov et al., 1997). This method allows reliable intracellular introduction of substances without altering the electrophysiological characteristics of the recorded cell. Stock solutions of AC3 control peptide and PKI(6–22) amide (PKI), (20 mm both; QCB, Hopkinton, MA) were prepared in deionized water and stored in aliquots at −70°C. AC3 control peptide has a reversed sequence relative to AC3 inhibitory peptide of Ca2+-calmodulin-dependent protein kinase 2 (CaMKII) (Otmakhov et al., 1997). On the day of an experiment a fresh aliquot was diluted in standard internal solution concentrated by 10%, and the osmolarity was adjusted to 300 mOsm. All peptides were applied in 2 mm concentrations. At similar concentration (1 mm) peptide inhibitors were shown to block the PKA-dependent inhibition of afterhyperpolarization (AHP) (Pedarzani and Storm, 1993, 1996). The AHP is though to be generated closer to the cell body (Sah and Bekkers, 1996) than the synaptic processes we have studied. We could not allow a long time for diffusion of peptide in dendrites because of the possibility of “washout” of LTP. Direct measurements of diffusion of dye in the dendrites show that the equilibration takes ∼40 min (Otmakhov et al., 1997). The dye used, however, had a much lower molecular weight as compared to peptides we use in these experiments. The intracellular concentration of peptides can also be decreased by a proteolysis (Otmakhov et al., 1997). Thus, the actual concentration of peptide at synapses at the time of LTP induction (only 12 min of perfusion) would be much lower than that in the pipette solution.
Statistical analysis. For statistical analysis of fEPSP experiments responses were first collected and averaged in 5 min blocks: 15 min of baseline and 40 min after the tetanus. Field EPSP slope (millivolts per millisecond) and fiber volley amplitude (millivolts) were calculated, and data for each experiment were normalized relative to baseline. The amplitude of a synaptic response in whole-cell voltage clamp experiments was calculated by subtracting the average value of the data points in a 15 msec window before the stimulus from the average value of the data points in a 3 msec window at the peak of the synaptic response. In figures, the amplitude measurements from individual experiments were first averaged over 1 min periods, then normalized to the baseline period before pairing, and finally averaged across experiments. Two way ANOVA for repeated measurements (df = 1 for drug and df = 5 for time after the tetanus factors) and a two-tailed t test for means as apost hoc criterion in the Excel program package with α < 0.05 were used as criteria for significance. When results from different slices (or cells) were compared, we performed a one-way ANOVA or multiple two-tailed unpaired t tests at the same time intervals. For graphic presentations, data were collected and averaged in 1 min blocks. Figures show means ± SEs.
RESULTS
We first performed experiments using a PKA inhibitor, H89 (Chijiwa et al., 1990). Slices were pretreated (1–2 hr) with the drug, as previously described (Thomas et al., 1996; Otmakhova and Lisman, 1998). Each day 3 slices from the same animal were used. One slice was used as a control, one was presoaked for 2 hr in 20 μm H89 (dissolved in 0.2% DMSO), and one was presoaked for the same time in the vehicle (0.2% DMSO in ACSF). Slices were then transferred to the experimental chamber and perfused with ACSF. To induce LTP, slices were stimulated with a single tetanus. As shown in Figure1, at all times after induction, LTP was smaller in H89 than in vehicle-treated or control slices (p < 0.05; n = 7). At the 30 min point, LTP was decreased by 52% relative to DMSO (p < 0.02).
Fig. 1.

The PKA inhibitor H89 suppressed the early LTP induced by a single tetanus. In slices pretreated with H89 (20 μm), early LTP was strongly reduced as compared to both control and vehicle groups (p < 0.05;n = 7 for each group). There was no difference between control and vehicle (0.2% DMSO)-treated slices.Insets represent averaged (n = 6) fEPSP from individual experiments before and 30 min after the tetanus. Field EPSP slopes were normalized relative to the baseline. The figure shows means and SE. The arrow indicates the time of the tetanus.
The action of H89 is unlikely to be a secondary effect of changes in excitability because, by several measures, excitability was unaffected. The standard stimulus was adjusted in all three groups to evoke a fEPSP slope half that required to elicit a population spike. This slope, the amplitude of the fEPSP, and the amplitude of the fiber volley before the tetanus were all statistically indistinguishable (p > 0.5; p > 0.5; andp > 0.3 in a one-way ANOVA) between control, DMSO, and H89 groups. Field EPSP maximal slopes were 0.58 ± 0.03, 0.59 ± 0.03, and 0.54 ± 0.04 mV/msec; fEPSP amplitudes were 0.87 ± 0.08, 0.78 ± 0.12, and 0.94 ± 0.07 mV; fiber volleys were 0.34 ± 0.04, 0.34 ± 0.03, and 0.28 ± 0.025 mV, respectively. We also did not see any differences in field potential dynamics during the tetanus. The degree of fEPSP suppression (last relative to first fEPSP in the train) during tetanus were similar in all three groups (p > 0.6). Population spike was rarely observed during the tetanus, and the incidence of population spikes did not differ between control, DMSO, and H89 groups (p > 0.5 in a one-way ANOVA).
To study the involvement of PKA in early LTP by a second, independent method, we applied the PKA inhibitor PKI into the postsynaptic cell through the patch pipette. This inhibitor is a membrane-impermeable peptide, so any effect of postsynaptic application can be specifically attributed to postsynaptic targets. LTP was induced by a pairing procedure that combined synaptic stimulation (1.4 Hz, 200 stimuli) with simultaneous depolarization of the postsynaptic cell to 0 mV. We induced LTP by pairing in one of two inputs 6 min after the initiation of whole-cell recording (baseline) (Fig.2A). Two minutes after this pairing, PKI (2 mm) was perfused into the cell so that its effect on subsequent LTP induction (12 min later) in a second input could be determined. In the presence of PKI (Fig.2A), LTP was significantly smaller than without PKI (by 30% on average: ANOVA, F = 190.5;p < 0.001; n = 8). In control experiments we tested the effects of a different peptide that does not have a PKA-specific action, the AC3 peptide. Under the same experimental conditions, the AC3 peptide did not affect pairing-induced LTP (Fig. 2B; ANOVA, F = 0.85;p > 0.35; n = 6). In both experiments holding current, series, and input resistances were stable at the −65 mV membrane potential at which the EPSC were recorded. Figure2C shows that the LTP induced in the presence of PKI was smaller (by 32% at 30 min after pairing; p < 0.01;t test) than in the presence of control peptide.
Fig. 2.
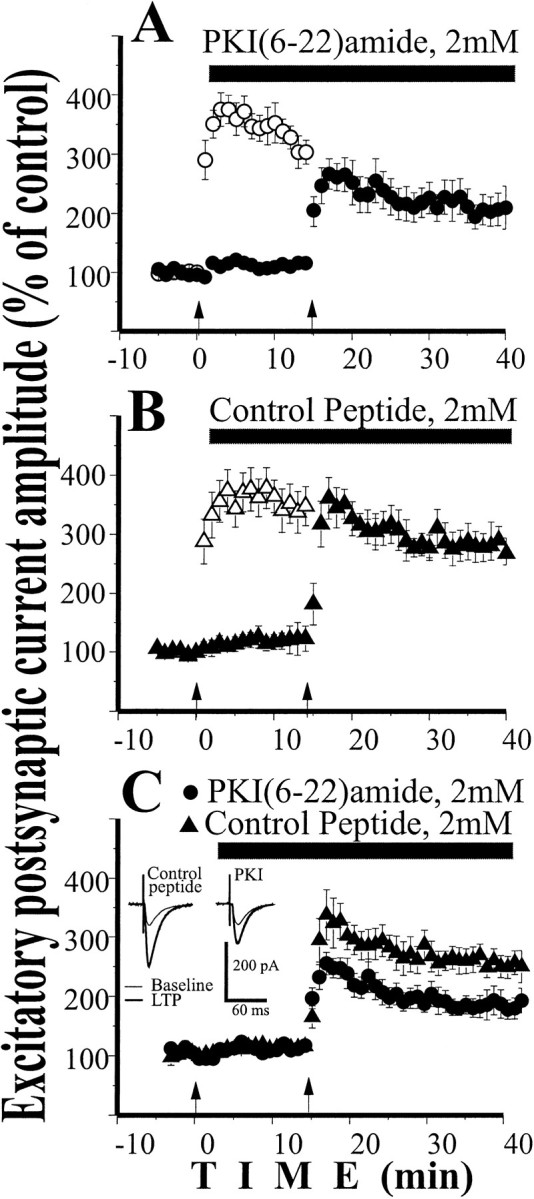
Intracellular perfusion of the PKA inhibitor PKI(6–22) amide (2 mm) decreased the early LTP induced by pairing. A, In the first input, pairing procedure induced large LTP. Two minutes after pairing, PKI was perfused inside the cell. A pairing procedure applied to the second input in the presence of PKI produced significantly smaller LTP (n = 8; p < 0.001).B, In similar experiments, AC3 control peptide (2 mm) was perfused into the cell after the first pairing. A pairing procedure applied to the second input (in the presence of AC3) produced LTP of the same size as a first pairing (n= 6; p > 0.35). C, Replot of LTP induced by second pairing procedure shows significant inhibition of LTP by PKI as compared to control peptide (p < 0.01). Insets represent averaged EPSC (n = 40) from individual experiments 5 min before and 30 min after the pairing.
We next sought to understand why we and others failed to detect changes in LTP produced by other inhibitors of the cAMP/PKA pathway. In initial experiments, we checked whether we could repeat our previous finding (Otmakhova and Lisman, 1998) that bath application of the AC inhibitor, SQ 22536, did not affect early LTP. A concentration of 100 μm was used because previous work showed that this concentration inhibited the AHP that follows a train of action potentials (Madison and Nicoll, 1986) and facilitated depotentiation (Otmakhova and Lisman, 1998). Application of SQ 22536 produced a small, reversible increase in the baseline responses (6.8 ± 1.2%;p < 0.01; two-tailed paired t test). In each slice LTP was induced by a single tetanus in one input under control conditions, in the second input after 10–20 min of application of SQ 22536. As shown in Figure3A, SQ 22536 did not significantly affect early LTP (ANOVA; F = 1.104;p > 0.3; n = 7), consistent with our previous results (Otmakhova and Lisman, 1998).
Fig. 3.

Inhibition of AC significantly decreased early LTP when combined with the inhibition of PKA. A, The AC inhibitor SQ 22536 (100 μm) alone did not affect early LTP induced by a single tetanus (n = 7;p > 0.3). B, In slices pretreated with the PKA inhibitor H89 (2 hr, 20 μm), application of the AC inhibitor SQ 22536 (100 μm) significantly decreased the early LTP induced by a single tetanus (n = 7; p < 0.001).Insets represent averaged (n = 6) fEPSP from individual experiments taken at 5 min before and 30 min after the tetanus. All calculations and markings are as in Figure 1. Note that LTP after the application of H89 in B is larger than the H89 condition in Figure 1. This difference should not be considered significant because the experiments were done at different times, and variability in LTP magnitude is common.
One possible explanation of the lack of effect of SQ 22536 may be that the inhibition of AC is only partial and that the remaining elevation of cAMP during a tetanus is still sufficient to fully perform the downstream functions required for LTP induction (i.e., the downstream pathway is normally saturated). If this is the case, an effect of SQ 22536 should become apparent if the pathway is already attenuated by some other inhibitor, thus removing the pathway from saturation. We therefore studied the effect of SQ 22536 under conditions where the slice was pre-exposed to 20 μm H89 (2 hr). In one of the inputs, LTP was induced by a single tetanus without additional drug application (Fig. 3B, H89); in the other input LTP was induced by a single tetanus after the application of 100 μm SQ 22536 (Fig. 3B, H89 + SQ 22536). The results show that under these conditions, SQ 22536 produced a substantial additional decrease of early LTP (ANOVA,F = 12.038; p < 0.001;n = 7). This decrease was statistically significant (p < 0.05 in post hoc pairedt test) in all but the first 5 min time interval after the tetanus. At 30 min, SQ 22536 decreased LTP by 40% relative to H89 alone (p < 0.01). Thus when the AC/PKA pathway is already partially inhibited, further inhibition by SQ 22536 produces a large reduction of early LTP.
Because of data indicating that agents that inhibit PKA decrease the LTP induced by three tetani more strongly than that induced by a single tetanus (Huang and Kandel, 1994), it was of interest to determine whether this was also the case for an AC inhibitor. In these experiments, LTP was produced by three tetani given 10 min apart. SQ 22536 (100 μm) was applied from 20 min before the first tetanus until the end of the third tetanus. Control and drug conditions were measured sequentially in two pathways in the same slice. As in other studies (Huang and Kandel, 1994; Winder et al., 1998), under control conditions, three tetani produced LTP that was almost twice as large as that produced by a single tetanus (p < 0.05; n = 7 in each group). As shown in Figure4, SQ 22536 alone significantly inhibited three tetani LTP (27% on average; ANOVA, F = 19.4;p < 0.001; n = 7).
Fig. 4.

The AC inhibitor SQ 22536 inhibited early LTP induced by three tetani. The LTP induced by three tetani was significantly larger than the LTP induced by a single tetanus (p < 0.05). AC inhibitor SQ 22536 (100 μm) inhibited the early LTP induced by three tetani (p < 0.001; n = 7).Insets represent averaged (n = 6) fEPSP from individual experiments taken at 5 min before and 30 min after the tetanus. All calculations and markings are as in Figure 1. Field EPSP between tetani are not shown.
DISCUSSION
The results presented here demonstrate that three different pharmacological agents (H89, PKI, and SQ 22536) that inhibit the AC/PKA pathway produce a substantial decrease of the early phase of LTP. The inhibitors we have used have different targets and were applied in different ways, allowing us to make several points about the involvement of this pathway. We found that bath application of H89 produced a ∼50% inhibition of the early LTP induced by a single tetanus. This inhibition was apparent immediately after induction and was maintained for as long as we monitored the slice. We also applied the membrane-impermeable PKA inhibitor PKI directly to the postsynaptic neuron. This produced a ∼30% decrease in LTP. These experiments thus indicate that a locus of PKA action on early LTP is postsynaptic (see also Blitzer et al., 1995). Because a pairing procedure was used in these latter experiments, the results indicate that the AC/PKA pathway is directly involved in the biochemical machinery of synaptic modification rather than in generating the depolarization required for LTP induction. As has been noted (Sanes and Lichtman, 1999), a general weakness of experiments that use LTP induction by tetanic stimulation is that the number of factors that can influence induction is very large, including the action of interneurons, frequency-dependent presynaptic properties, and postsynaptic conductances. In contrast, when LTP is induced by pairing, these complexities do not come into play because inhibition is blocked by picrotoxin, depolarization is imposed by clamp, and only low-frequency stimulation is used.
It is not yet possible to say whether LTP can be only partially inhibited by interfering with the cAMP pathway or whether it can be totally inhibited. It was shown before (Blitzer et al., 1995) that the early LTP induced by three tetani can be almost completely blocked by the PKA inhibitor Rp-cAMPS. Similarly in a separate study (Otmakhov et al., 1999), we have found that postsynaptic application of Rp-cAMPS (4 mm) completely blocked the LTP induced by pairing. However, our results (Otmakhov et al., 1999) suggest that Rp-cAMPS might act on some other target in addition to PKA. Thus, based on the current evidence, all that can be said is that inhibition of PKA produces at least a 30–50% reduction of LTP.
Our results provide new insight into why some manipulations of the cAMP pathway might not affect early LTP. We have replicated our previous results (Otmakhova and Lisman, 1998) that early LTP was not affected by the adenylyl cyclase inhibitor SQ 22536. However, we now find that when SQ 22536 is applied under conditions when the AC/PKA pathway has been already attenuated by H89, SQ 22536 produce a large additional decrease of LTP. This suggests that the failure of SQ 22536 to be effective by itself might be attributable to a saturation effect; the elevation of cAMP during a tetanus may be so large that it saturates downstream processes. In this case, partial inhibition of AC may have little effect. However, if the downstream processes are brought out of saturation by PKA inhibitor, the effect of AC inhibitor can be revealed. The lack of effectiveness of PKA inhibitor KT 5720 on 1-tetanus LTP (Huang and Kandel, 1994) might have a similar explanation. The possibility of such saturation may seem to contradict the fact that LTP can be enhanced by agents that increase cAMP (Otmakhova and Lisman, 1996, 1998; Barad et al., 1998), however these agents are not strictly comparable. Specifically, agents that inhibit the pathway will primarily decrease the amplitude of the activation, whereas agents that increase the cAMP concentration also prolong its period of action (some agents such as forskolin produce a tonic elevation of cAMP; other agents, such as the phosphodiesterase inhibitor rolipram presumably prolong the tetanically induced elevation).
Several different enzymes that affect the cAMP/PKA pathway have been altered genetically, and it is important to assess whether the conclusion derived from these studies are in agreement or disagreement with the conclusion we have derived from pharmacological studies. The strongest evidence for involvement of the cAMP pathway in early LTP has come from work on adenylyl cyclase knock-outs. Initial work with a knockout of the Ca2+-activated AC1 (Wu et al., 1995), showed that late LTP was unaffected while early LTP was slightly decreased. However, biochemical experiments showed only a modest (46%) decrease in Ca2+-dependent cyclase activity in the hippocampus. The reason for relatively small biochemical effect was recently demonstrated in experiments in which the two major Ca2+-dependent adenylyl cyclases (AC1 and AC8) were knocked out (Wong et al., 1999). In this case, Ca2+-dependent cyclase activity in the hippocampus was completely abolished. The focus of this study was on late LTP, so the authors did not explicitly discuss their findings on early LTP. However, their results (Wong et al., 1999, their Fig. 3D) clearly show that early LTP was dramatically decreased (by ∼ 35% at 30 min). These results thus strongly support the involvement of AC in early LTP.
Several genetic perturbations of PKA have also been made, but no knock-out has yet been found that strongly affects the total PKA activity. The knock-out of regulatory subunit R1 beta(Brandon et al., 1995) did not produce measurable changes in total PKA activity and did not show any effect on early or late LTP. This might be because it is the R2 type of regulatory subunit, anchored by AKAP-79 to the PSD (Carr et al., 1992), which is the form important for synaptic function (Rosenmund et al., 1994). In other experiments involving knock-out of C beta1 catalytic subunits (Qi et al., 1996) there was no measurable change in total PKA activity, but there was modest (∼20%) decrease of the early LTP induced by three tetani (Qi et al., 1996, their Fig. 2B). In yet another study, total PKA activity was reduced 20–30% by expression of the R(AB) inhibitory form of regulatory subunit (Abel et al., 1997). This produced a substantial reduction (30–50%) of the early LTP induced by three tetani (Abel et al., 1997, their Fig. 4C). We conclude that the genetic work with PKA provides some support for a role of PKA in early LTP and is certainly not inconsistent with such a role.
An important question that remains to be investigated is why agents that interfere with the cAMP pathway have a greater effect on the LTP induced by three tetani than that induced by a single tetanus (Huang and Kandel, 1994; Abel et al., 1997). This pattern of effects has been recently reported for early LTP (Blitzer et al., 1995) using Rp-cAMPS, and we have replicated this finding with SQ 22536. Our results thus in no way contradict the idea that PKA may participate in multiple processes, some of which might be only involved in late LTP. Indeed it would seem quite likely that an initial period of activity would produce changes in the cell that could affect subsequent activity-dependent plasticity. This phenomenon has been termed metaplasticity (Abraham and Bear, 1996).
It will be of interest to determine what reactions are involved in the regulation of early LTP by PKA. One possibility is suggested by recent work showing that PKA is closely linked to the NMDA channel by the synaptic scaffolding protein yotiao (Westphal et al., 1999). These results raise the possibility that inhibition of PKA reduces the NMDA conductance and thereby decreases LTP. Alternatively, disinhibition of protein phosphatase 1 by PKA inhibitors may decrease the autophosphorylation of CaMKII which is required for the induction of LTP (Lisman, 1994; Coussens and Teyler, 1996; Blitzer et al., 1998). Further experiments will be required to distinguish between these and other possible models of PKA involvement in early LTP.
Footnotes
This work was supported by National Institutes of Health Grants 2R01 NS27337/11 and 1R01 NS35083/03 and Alzheimer Association Grants RG3-96-015 to J.L., and National Institutes of Health Grant 5F32 MH11720-02, National Alliance for Research on Schizophrenia and Depression Young Investigator Award, and the Scottish Rite Schizophrenia Research Program, NMJ grants to N.O. Howard Hughes Undergraduate Initiative 71199-513104 00 supported work of L.M. We also appreciate the support from W. M. Keck Foundation.
Correspondence should be addressed to John E. Lisman, Biology Department, Center for Complex Systems, Brandeis University, 415 South Street, Waltham, MA 02454. E-mail: Lisman@binah.cc.brandeis.edu.
REFERENCES
- 1.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 3.Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci USA. 1998;95:15020–15025. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 5.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 6.Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP- dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 7.Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem. 1998;5:365–374. [PMC free article] [PubMed] [Google Scholar]
- 8.Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1995;92:8851–8855. doi: 10.1073/pnas.92.19.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992;267:16816–16823. [PubMed] [Google Scholar]
- 10.Chetkovich DM, Sweatt JD. NMDA receptor activation increases cyclic Amp in area Ca1 of the hippocampus via calcium/calmodulin stimulation of adenylyl cyclase. J Neurochem. 1993;61:1933–1942. doi: 10.1111/j.1471-4159.1993.tb09836.x. [DOI] [PubMed] [Google Scholar]
- 11.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5- isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 12.Coussens CM, Teyler TJ. Protein kinase and phosphatase activity regulate the form of synaptic plasticity expressed. Synapse. 1996;24:97–103. doi: 10.1002/(SICI)1098-2396(199610)24:2<97::AID-SYN1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 13.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y-Y, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- 15.Lisman J. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 16.Ma L, Zablow L, Kandel ER, Siegelbaum SA. Cyclic AMP induces functional presynaptic boutons in hippocampal CA3- CA1 neuronal cultures [see comments]. Nat Neurosci. 1999;2:24–30. doi: 10.1038/4525. [DOI] [PubMed] [Google Scholar]
- 17.Madison DV, Nicoll RA. Cyclic adenosine 3′,5′-monophosphate mediates beta-receptor actions of noradrenaline in rat hippocampal pyramidal cells. J Physiol (Lond) 1986;372:245–59. doi: 10.1113/jphysiol.1986.sp016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malenka RC, Nicoll RA. Long-term potentiation-A decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 19.Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long- term potentiation. J Neurosci. 1997;17:5357–5365. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otmakhov N, Chen H-X, Lisman J. Postynaptically applied cAMP analogs block induction of pairing-induced LTP, possibly by a PKA independent mechanism. Soc Neurosci Abstr. 1999;25:989. [Google Scholar]
- 21.Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otmakhova NA, Lisman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- 24.Pedarzani P, Storm JF. Evidence that Ca/calmodulin-dependent protein kinase mediates the modulation of the Ca2+-dependent K+ current, IAHP, by acetylcholine, but not by glutamate, in hippocampal neurons. Pflügers Arch. 1996;431:723–728. [PubMed] [Google Scholar]
- 25.Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 27.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 28.Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long- term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanes JR, Lichtman JW. Can molecules explain long-term potentiation? Nat Neurosci. 1999;2:597–604. doi: 10.1038/10154. [DOI] [PubMed] [Google Scholar]
- 30.Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 31.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 32.Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- 33.Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- 34.Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci USA. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]