

Abstract
We investigated the expression, activation, and distribution of c-Jun N-terminal kinases (JNKs), p38 mitogen-activated protein kinases (p38s) and extracellular signal-regulated kinases (ERKs) using Western blotting and immunohistochemistry in gerbil hippocampus after transient forebrain ischemia to clarify the role of these kinases in delayed neuronal death (DND) in the CA1 subfield. Immunoblot analysis demonstrated that activities of JNK, p38, and ERK in whole hippocampus were increased after 5 min of global ischemia. We used an immunohistochemical study to elucidate the temporal and spatial expression of these kinases after transient global ischemia. The immunohistochemical study showed that active JNK and p38 immunoreactivities were enhanced at 15 min of reperfusion and then gradually reduced and disappeared in the hippocampal CA1 region. On the other hand, in CA3 neurons, active JNK and p38 immunoreactivities were enhanced at 15 min of reperfusion and peaked at 6 hr of reperfusion and then gradually reduced but was continuously detected 72 hr after ischemia. Active ERK immunoreactivity was observed transiently in CA3 fibers and dentate gyrus. Pretreatment with SB203580, a p38 inhibitor, but not with PD98059, an ERK kinase 1/2 inhibitor, reduced ischemic cell death in the CA1 region after transient global ischemia by inhibiting the activity of p38. These findings indicate that the p38 pathway may play an important role in DND during brain ischemia in gerbil. Components of the pathway are important target molecules for clarifying the mechanism of neuronal death.
Keywords: mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (p38), extracellular signal-regulated kinase (ERK), transient global ischemia, delayed neuronal death (DND), hippocampus, gerbil
The members of the mitogen-activated protein kinase (MAPK), which are characterized as proline-directed serine-threonine-protein kinases, in particular, c-Jun N-terminal kinases (JNKs), p38 mitogen-activated protein kinases (p38s), and extracellular signal-regulated kinases (ERKs), play important roles in transducing stress-related signals in eukaryotic cells (Kyriakis and Avruch, 1996). On activation by phosphorylation on both Thr and Tyr residues, these kinases phosphorylate intracellular enzymes and transcription factors. The JNK and p38 are activated by stress signals such as inflammatory cytokines, heat shock, ultraviolet light, and ischemia (Kyriakis and Avruch, 1996; Davis, 1999). Because JNK and p38 are generally activated by the same stress signals, they have been collectively referred to as the stress kinases (Kyriakis and Avruch, 1996). Phosphorylation of p38 results in MAPK-activated protein 2 and activating transcription factor 2 (ATF-2) activation, whereas phosphorylation of JNK results in c-Jun induction (Robinson and Cobb, 1997). On the other hand, the ERK is activated in response to growth factors (Boulton et al., 1991; Nishida and Gotoh, 1993), oxidative stress (Aikawa et al., 1997), increases in intracellular calcium levels, or glutamate receptor stimulation (Bading et al., 1993; Fiore et al., 1993; Rosen et al., 1994; Kurino et al., 1995; Sgambato et al., 1998). Phosphorylation of ERK links to immediate early gene induction and hyperphosphorylation of Elk-1 and cAMP/calcium-responsive element-binding protein (Sgambato et al., 1998).
Recent studies demonstrate that the members of MAPK, in particular JNK and p38, are activated in heart, kidney, and brain after ischemia and ischemia–reperfusion (Hu and Wieloch, 1994; Mizukami et al., 1997; Yin et al., 1997; Herdegen et al., 1998; Walton et al., 1998) and suggest that the signaling pathway of these kinases may be important and responsible for tissue injury after ischemia–reperfusion.
A brief period of global ischemia causes delayed neuronal death (DND) in the CA1 region of hippocampus in gerbil (Kirino, 1982; Nitatori et al., 1995), but the mechanism of DND has not been fully elucidated.
Although there are some in vivo reports about MAPK cascade and brain ischemia (Hu and Wieloch, 1994; Herdegen et al., 1998; Walton et al., 1998), the expression and distribution of phosphorylated forms of JNK, p38, and ERK in the hippocampus after ischemic insult have never been systematically reported in gerbil. The effectiveness of inhibitors of p38 and ERK on DND in the CA1 region of the gerbil also has not been demonstrated.
In the present study, we investigated the activations of JNK, p38, and ERK during transient forebrain ischemia in gerbil to clarify the roles of these kinases in DND in the hippocampal CA1 region. We demonstrated for the first time the activation and distribution of MAPKs after transient forebrain ischemia in gerbil hippocampus. Moreover, inhibitors of p38 and ERK were used to confirm the involvement of these kinases on DND in the CA1 region.
MATERIALS AND METHODS
Induction of global ischemia. Adult male Mongolian gerbils (Shimizu Laboratory Supplies Co. Ltd., Kyoto, Japan) weighing 60–80 gm were used. They were kept in a temperature-controlled (23 ± 1°C) and light/dark cycle-controlled animal room (lights on at 8 A.M., off at 8 P.M.).
Animals were deprived of food overnight before the induction of ischemia to exclude the influence of hyperglycemia on ischemic brain damage (Pulsinelli et al., 1982; Ginsberg et al., 1987). All experiments were performed under spontaneous respiration. Anesthesia was induced with 4% halothane in a gas mixture of 30% oxygen and 70% nitrous oxide administered via face mask and maintained at 2% halothane in the same gas mixture. Bilateral common carotid arteries were dissected and occluded with microaneurysmal clips for 5 min, and then halothane was reduced to 1%. In the sham-operated group, the same procedure was performed without carotid occlusion.
Pretreatment with SB203580 and PD98059. To examine the effect of SB203580 (Calbiochem, La Jolla, CA), a p38 inhibitor (Lee et al., 1994; Cuenda et al., 1995), on brain ischemia, SB203580 (0, 1, 10, and 100 μm) was administered into the right lateral ventricle stereotaxically 30 min before the ischemic insult under general anesthesia (described earlier). The heads of gerbils were secured in a stereotaxic frame, and a 27 gauge needle was inserted into the right lateral ventricle. Coordinates were 2 mm lateral and 2 mm ventral from the dural surface using bregma as a landmark (Thiessen and Goar, 1970). Twenty-five microliters of each drug solution were injected over 10 min. SB203580 was dissolved in 1% dimethyl sulfoxide (DMSO; Nacalai Tesque, Kyoto, Japan).
To examine the effect of PD98059 (Calbiochem), an ERK kinase 1/2 (MEK1/2) inhibitor (Pang et al., 1995; Waters et al., 1995), on brain ischemia, PD98059 (0, 3, 30, and 300 μm) was administered in the same way. PD98059 was also dissolved in 1% DMSO.
Tissue and sample preparation. For Western blot analysis of the total amounts of JNK, p38, and ERK in hippocampus, the gerbils were killed 0, 15, and 30 min and 2, 6, 24, and 72 hr after ischemia. For immunoblotting of the active forms of JNK, p38, and ERK, the gerbils were killed 0 and 30 min after ischemia and 30 min after sham operation (each n = 3). The whole hippocampus was rapidly excised and frozen in liquid nitrogen.
For immunohistochemical study, 0, 15, and 30 min and 2, 6, 18, 24, 48, and 72 hr (each n = 3) after reperfusion, the animals were killed and perfused transcardially with a buffer (0.1m PBS, pH 7.4, and 0.2 mm sodium orthovanadate) followed by 4% buffered paraformaldehyde. To examine the effects of SB203580 (0, 1, 10, and 100 μm) on phosphorylation of p38, ATF-2, and JNK and PD98059 (0, 3, 30, and 300 μm) on phosphorylation of ERK after ischemia, gerbils (n = 3 for each dose) were perfused as described earlier at 0 and 15 min after ischemia. The brains were rapidly removed and cryoprotected in 25% sucrose in 0.1 m PBS overnight at 4°C.
Antibodies. Anti-JNK1 (FL) antibody, anti-p38 (c-20) antibody, anti-ERK2 (c-14) antibody, anti-phosphorylated ATF-2 (p-ATF-2) (F-1) antibody (Santa Cruz Biotechnologies, Santa Cruz, CA) and antibodies for the active forms of (anti-ACTIVE) JNK, p38, and MAPK (ERK; Promega, Madison, WI) were used in the present study. Anti-ACTIVE JNK polyclonal antibody recognizes the dually phosphorylated JNK2 enzyme but also reacts with the dually phosphorylated JNK1 enzyme. Anti-ACTIVE ERK polyclonal antibody recognizes the dually phosphorylated ERK2 enzyme.
Absorption test. The specificity of the antibodies against JNK1, p38, and ERK2 (Santa Cruz) were verified using whole-brain homogenate of gerbils and rats as control (male SD rats weighing 280–320 gm; Shimizu Laboratory Supplies). The absorption test was performed using the antibodies against p38 and ERK2 incubated 30 min at 37°C with 0, 1, 10, 100, and 1000 times molar concentration of p38 and ERK2, respectively. The procedure of electrophoresis and immunoblotting was then performed in the same way as described in the next section. With regard to JNK1, an absorption test was not able to be performed because a control peptide of JNK1 was not available (Santa Cruz).
The antibodies recognized JNK1, p38, and ERK2 and each band corresponding to the JNK1, p38, and ERK2 molecular weight in the gerbil brain homogenate, respectively. The antibodies against JNK1 and ERK2 also recognized the bands corresponding to JNK2 and ERK1, respectively. The absorption test showed that no bands corresponding to p38 and ERK2 were demonstrated when the antibodies were preincubated with 1 and 1000 times molar concentrations of the antigens, respectively (data not shown).
The antibodies against active forms of JNK, p38, and ERK (Promega) were raised against a dually phosphorylated peptide sequence representing the catalytic core of the active JNK, p38, and ERK enzymes, respectively. The phosphorylated amino acid residues correspond to Thr-183 and Tyr-185 of these enzymes in mammalians. Because the rodents including gerbils and rats belong to the mammalians, these antibodies should recognize the homogenate of the gerbil brain (Walton et al., 1998).
Western immunoblot analysis for JNK1, p38, and ERK2. For gel electrophoresis and Western blotting of JNK1, p38, and ERK2, the hippocampi were homogenized with 0.5 ml of ice-cold buffer containing 20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 5 mm MgCl2, 1 mm DTT, 20 μg/ml aprotinin, and 1 mm phenylmethylsulfonyl fluoride (PMSF). Aliquots of total tissue homogenate were frozen and kept at −80°C. The protein content was determined using the method ofBradford (1976). SDS-PAGE (11%) was performed as described byLaemmli (1970). Equal amounts of protein (20 μg in all assays) were loaded in each lane with loading buffer containing 0.125 mTris-HCl, pH 6.8, 20% glycerol, 4% SDS, 10% mercaptoethanol, and 0.002% bromphenol blue. Samples were heated at 95°C for 5 min before gel loading. Protein analyzed on gel electrophoresis was transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon; Millipore, Bedford, MA) using an electrophoretic transfer system (Semi-Dry Transfer System; Biocraft) at 12 V for 1 hr. The membranes were then washed with Tris-buffered saline solution, pH 7.4, and 0.05% Tween 20 (TTBS) and then blocked in TTBS containing 5% skim milk for 60 min. Membranes were then incubated overnight at 4°C with the primary anti-rabbit polyclonal antibody against JNK1 (Santa Cruz) diluted 1:1000 in TTBS containing 3% bovine serum albumin (Nacalai Tesque). After washing, the membranes were incubated with secondary antibody (anti-rabbit IgG-alkaline phosphatase conjugate; Santa Cruz) diluted 1:10,000 for 1 hr at room temperature and washed again, and the blots were finally developed with the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate (170 μg/ml) along with nitro blue tetrazorium (330 μg/ml; Nacalai Tesque) in sodium glycinate buffer, pH 9.5, in the presence of 5 mmMgCl2 (Leary et al., 1983). Other membranes were incubated with the antibodies against p38 (Santa Cruz) diluted 1:1000 and ERK2 (Santa Cruz) diluted 1:20,000.
Western immunoblot analysis for ACTIVE JNK, p38, and ERK.For gel electrophoresis and Western blotting of active forms of JNK, p38, and ERK, the hippocampi were homogenized with 0.5 ml of ice-cold buffer containing 20 mm Tris-HCl, pH 7.5, 1 mmEDTA, 5 mm MgCl2, 1 mmDTT, 20 μg/ml aprotinin, 1 mm PMSF, and 2 mmsodium orthovanadate. After SDS-PAGE, blotting to PVDF membranes, and blocking overnight at 4°C with 5% skim milk, the membranes were incubated 2 hr at room temperature with the primary anti-rabbit polyclonal active forms of JNK, p38, and ERK (catalog #8031). Polyclonal anti-ACTIVE p38 antibody was used at a dilution of 1:1000; JNK and ERK were used at a dilution of 1:2000. After washing, the membranes were incubated for 1 hr at room temperature with secondary antibody (Donkey anti-rabbit IgG-alkaline phosphatase conjugate; Promega) diluted 1:10,000 for active forms of JNK and p38 and 1:5000 for ERK. The other procedure was performed as described above.
Measurement of the densities of the immunoblot bands. For quantitative analysis of the densities of the immunoblot bands (JNK1, p38, ERK2, p-JNK2, p-p38, and p-ERK2), the densities were measured by NIH Image analyzer 1.61 (Dr. Wayne Rasband, National Institutes of Health, Bethesda, MD). The data of the densities (JNK1, p38, and ERK2) were statistically analyzed by ANOVA followed by post hocBonferroni test between groups using Stat View II (Abacus Concepts, Berkeley, CA). Data are presented as mean ± SD, and whenp < 0.05, differences were considered significant.
Immunohistochemistry. Frozen coronal sections (40 μm in thickness) of the brains were prepared using a microtome. For immunohistochemistry, antibodies against active forms of JNK, p38, and ERK (catalog #6671; Promega) were used. The sections were processed by the free-floating method. After washing three times in 0.1m PBS, quenching endogenous peroxidase in 2% H2O2 in 60% methanol, and blocking with 5% goat serum, the sections were incubated overnight at 4°C with polyclonal antibodies against active forms of JNK, p38, and ERK. The sections were washed three times in 0.1 m PBS and incubated in biotinylated anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA) at a 1:200 dilution for 2 hr and in avidin–biotin complex (ABC kit; Vector) for 60 min. Peroxidase was demonstrated with a DAB substrate kit (Vector). Negative control sections received identical treatment except for the primary antibody. For evaluation of morphological change, adjacent sections were stained with cresyl violet. Each section was mounted on glass slides, air-dried, dehydrated in ascending ethanol series, immersed in xylene, and coverslipped with M·X (Matsunami Glass Industries, Ltd., Osaka, Japan).
Measurement of survival neurons in CA1 and CA3. To examine the effect of intraventricular administration of SB203580 30 min before the ischemia on the development of DND in CA1 and neuronal death in CA3, the animals were anesthetized with diethyl ether and perfused transcardially with a buffer (0.1 m PBS, pH 7.4, and 0.2 mm sodium orthovanadate) followed by 4% buffered paraformaldehyde 7 d after the operation. Twenty-one gerbils were used (n = 6, 5, 5, and 5 for vehicle and doses of 1, 10, and 100 μm, respectively). As for PD98059, 15 gerbils were used (n = 5, 5, and 5 for doses of 3, 30, and 300 μm, respectively). Frozen coronal sections (20 μm in thickness) of the brains were prepared (described earlier) and stained with cresyl violet.
The severity of neuronal damage in CA1 and CA3 regions was evaluated by the number of surviving neurons. The mean number of surviving neurons of the pyramidal cell layer per 450 μm length was calculated in the CA1 region for each group. In the CA3 region, the number of surviving neurons per 0.2 mm2 indicated by the rectangular fields in Figure 11 was calculated (see Fig.11C). Cell counting was performed using the light microscope equipped with a 10× objective by independent observers in a blind manner (Tokime et al., 1996). The data were statistically analyzed as described earlier. The values were expressed as mean ± SD. The results were considered significant if p < 0.05.
Fig. 11.

Effects of SB203580 and PD98059 on neuronal death in CA1 and CA3 regions. Numbers of survival neurons in CA1 (A) and CA3 (B) regions of the hippocampus after SB203580 and PD98059 treatments at various doses are shown. SB203580 showed neuroprotective effects in a dose-dependent manner in the CA1 subfield (A). Neuroprotective effects with 10 and 100 μm SB203580 were significant compared with vehicle (1% DMSO) (*p < 0.0001). PD98059 showed no neuroprotective effects in the CA1 region (A). In the CA3 region, both drugs showed no significant effects on neuronal survival (B). Cell count in the CA3 subfield was done in the squaresindicated in the diagram(C).
Physiology. In randomly selected animals treated with 100 μm SB203580, 30 μm PD98059, or 1% DMSO (n = 4 in each group), arterial blood pressure was monitored by using an RMP-6004M transducer amplifier (Nihon Kohden, Tokyo, Japan) through a femoral artery catheterized with a PE-8 polyethylene tube. Arterial blood samples (50 μl) were collected through the tube and analyzed by using a blood gas–pH analyzer (Corning 248; Ciba-Corning Diagnostics, Tokyo, Japan). During the operation, and until animals became awake, a rectal temperature was maintained at 37–38°C with a thermoregulator (Animal Blanket Controller ATB-1100; Nihon Kohden) and a heating pad and lamp and did not change significantly during the operative procedure and postoperative period (∼2 hr).
RESULTS
Total amounts of MAPKs were not changed after transient global ischemia
To determine whether the total amounts of JNK1, p38, and ERK2 were altered by ischemia, whole hippocampal extracts were subjected to Western analysis using the anti-JNK1, p38, and ERK2 antibodies. No significant changes in the amounts of JNK1 (Fig.1A,B), p38 (Fig.1A,C), and ERK2 (Fig. 1A,D) at each time point were observed after global ischemia compared with ischemic control animals (0 min; each n = 3).
Fig. 1.

Total amounts of JNK, p38, and ERK were not changed after 5 min of transient forebrain ischemia. A, Western blotting of hippocampal homogenates of postischemic gerbils at 0 min (ischemic control, lane 1), 15 min (lane 2), 30 min (lane 3), 2 hr (lane 4), 6 hr (lane 5), 24 hr (lane 6), and 72 hr (lane 7) after 5 min of transient forebrain ischemia (top panel, JNK1;middle panel, p38; bottom panel, ERK2).B–D, Quantitative representation of immunoblots (mean ± SD) showing no significant change of total amounts of JNK1 (B), p38 (C), and ERK2 (D) in the hippocampal extracts.
These results indicate that the induction of immunoreactivities of active forms of JNK, p38, and ERK might not be attributable to the increase of total protein amounts.
MAPKs are activated after transient global ischemia
To determine whether the phosphorylated forms of JNK, p38, and ERK were altered by ischemia, whole hippocampal extracts were subjected to Western analysis using the antibodies against active forms of JNK, p38, and ERK. No activations of JNK, p38, and ERK were detected in sham-operated animals (Fig. 2,A, lane 1, B). Significant increases in the activities of JNK and p38 were observed 30 min after global ischemia (Fig. 2, A, lane 3, B) compared with the ischemic control animal (0 min; Fig. 2, A,lane 2, B). The activity of ERK was increased immediately after global ischemia (Fig. 2, A, lane 2, B, triangle) and slightly decreased 30 min of reperfusion (Fig. 2, A, lane 3, B, triangle). The data presented are representative of at least three separate experiments.
Fig. 2.

Increased phosphorylation of JNK, p38, and ERK after 5 min of transient forebrain ischemia. A, Western immunoblots of hippocampal homogenates of sham-operated gerbils (lane 1) and postischemic gerbils at 0 min (lane 2) and 30 min (lane 3) after 5 min of transient forebrain ischemia (top panel, active JNK; middle panel, active p38; bottom panel, active ERK;p-, phosphorylated). B, Quantitative representation of immunoblots showing activation of JNK1 and p38 30 min after ischemia and ERK2 immediately after ischemia in the hippocampal extracts. The data presented are representative of at least three separate experiments.
These results indicate that transient global ischemia induced the activations of JNK, p38, and ERK.
DND occurs after transient global ischemia
Cresyl violet staining demonstrated that only a few neurons showed ischemic changes, such as pyknosis and cell shrinkage, in the hippocampal CA1 region after 24 hr of reperfusion (Fig.3C). At 72 hr of reperfusion, almost all neurons showed ischemic changes in the CA1 region (Fig.3E). After 7 d of reperfusion, almost all neurons died in the CA1 subfield (Fig. 3G).
Fig. 3.
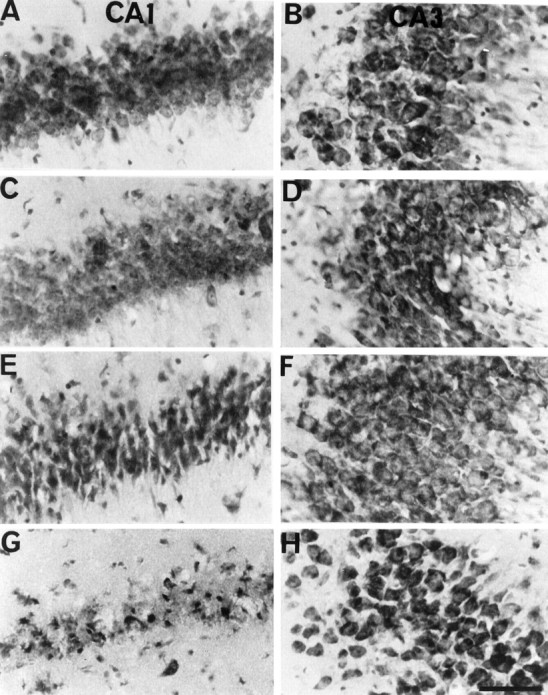
Nissl staining after transient global ischemia. Cresyl violet staining in CA1 (A, C, E, G) and CA3 (B, D, F, H) of the hippocampus is shown. Almost all neurons were preserved in the hippocampal CA1 region 24 hr after reperfusion (C). At 72 hr of reperfusion, almost all neurons showed ischemic changes in the CA1 region (E). Almost all neurons died in the CA1 region 7 d after the ischemia (G). A, B, 5 min of ischemia; C, D, 5 min of ischemia followed by reperfusion for 24 hr; E, F, 5 min of ischemia followed by reperfusion for 72 hr; G, H, 5 min of ischemia followed by reperfusion for 7 d. Scale bar, 100 μm.
In the CA3 region, no remarkable ischemic change was observed after transient global ischemia (Fig. 3B,D,F,H).
Immunolocalization for active JNK and p38 after transient global ischemia
Few neurons in the hippocampus showed immunoreactivity for active JNK and p38 in sham-operated animals (results not shown) and immediately after the ischemia (Figs. 4,5A,B). After 15 min of reperfusion, active JNK, and p38 immunoreactivities were enhanced in neurons in CA1 (Figs. 4, 5C) and CA3 (Figs. 4,5D). After 6 hr of reperfusion, active JNK and p38 immunoreactivities reduced in CA1 (Figs. 4, 5E), and the immunoreactivities almost disappeared after 18 hr of reperfusion (Figs.4, 5G). In the CA3 region, the immunoreactivities were intensely enhanced after 6 hr of reperfusion (Figs. 4,5F), and then the immunoreactivities reduced (Figs.4, 5H) but were continuously observed up to 72 hr of reperfusion (results not shown).
Fig. 4.
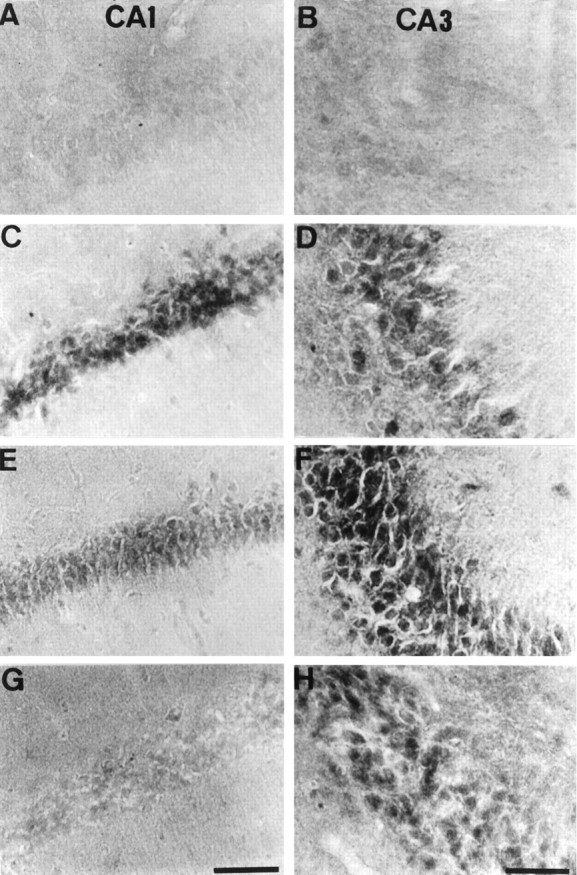
Phosphorylated JNK in CA1 and CA3 after ischemia. Immunohistochemical staining for the active form of JNK in CA1 (A, C, E, G) and CA3 (B, D, F, H) of the hippocampus is shown. The photographs represent 0 min (A, B), 15 min (C, D), 6 hr (E, F), and 18 hr (G, H) after 5 min of transient forebrain ischemia. Scale bar, 100 μm.
Fig. 5.

Phosphorylated p38 in CA1 and CA3 after ischemia. Immunohistochemical staining for the active form of p38 in CA1 (A, C, E, G) and CA3 (B, D, F, H) of the hippocampus is shown. The photographs represent 0 min (A, B), 15 min (C, D), 6 hr (E, F), and 18 hr (G, H) after 5 min of transient forebrain ischemia. Scale bar, 100 μm.
Immunolocalization for active ERK after transient global ischemia
Few neurons in the hippocampus showed immunoreactivity for active ERK in sham-operated animals (Fig.6A). Immediately after transient global ischemia, active ERK immunoreactivity was detected in CA3 fibers (Fig. 6B). After 15 min, active ERK immunoreactivity was enhanced in dentate gyrus (Fig. 6C,D) but not in the CA3 region (Fig. 6C). After 30 min, the immunoreactivity reduced in dentate gyrus (Fig. 6E). After 48 hr, no immunoreactivity for active ERK was detected in all hippocampal regions including dentate gyrus (results not shown). In subiculum, there were a few neurons that showed only faint ERK-positive stainings at 30 min after the ischemia (Fig. 6G). In the CA1 region, immunoreactivity for active ERK was never detected after global ischemia (Fig. 6F).
Fig. 6.
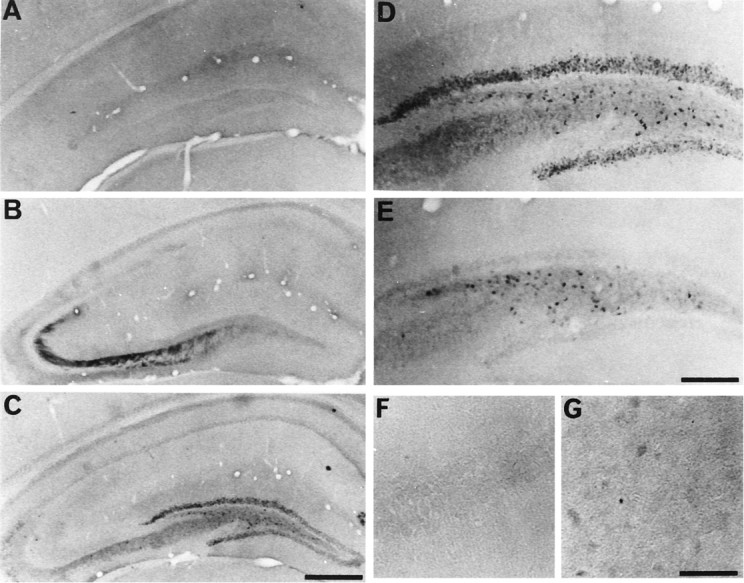
Phosphorylated ERK in hippocampus after ischemia. Immunohistochemical staining for the active form of ERK in whole hippocampus (A–C), dentate gyrus (D, E), CA1 (F), and subiculum (G) of the hippocampus is shown. The photographs represent sham-operated gerbil (A) and 0 min (B), 15 min (C, D), and 30 min (E-G) after 5 min of transient forebrain ischemia. Scale bars: A–C, 1 mm; D, E, 100 μm;F, G, 400 μm.
Although the activity of ERK was moderately detected 30 min after the transient forebrain ischemia in immunoblot analysis (Fig.2C), immunoreactivity for active ERK reduced in dentate gyrus at the same time point (Fig. 6E). The discrepancy might be attributable to the differences of sensitivities of the antibodies (catalog #6671 and #8031) or to the inclusion of whole hippocampal tissues in samples for Western blot analysis.
Effect of SB203580 on activation of p38, ATF-2, and JNK
Few neurons showed immunoreactivity for active p-ATF-2 in sham-operated animals (Fig.7A). Consistent with the activation of the p38 signaling cascade, an increase in phosphorylated ATF-2 levels was observed 15 min after global ischemia in the CA1 region (Fig. 7B). Intraventricular administration of SB203580 (0, 1, 10, and 100 μm) 30 min before the ischemic insult could not reduce the immunoreactivity for active p38 15 min after ischemia (results not shown) but reduced the immunoreactivity for p-ATF-2, a substrate of p38 (Fig. 7C), because SB203580 is not able to inhibit the phosphorylation of p38 but inhibits the activity of the phosphorylated form of p38 (Larsen et al., 1997). SB203580 also moderately reduced the immunoreactivity for active JNK (Fig. 8B), but its reduction might be attributable to the side effect of high doses of SB203580 (Whitmarsh et al., 1997), because a low dose of SB203580 (10 μm) could also reduce the immunoreactivity for p-ATF-2 but not reduce the one for active JNK (results not shown). Concerning the MEK–ERK pathway, SB203580 could not reduce the immunoreactivity for active ERK immediately and 15 min after the ischemia (results not shown).
Fig. 7.

Representative photomicrographs of immunohistochemical study for phospho-ATF-2 in CA1 of the hippocampus of sham-operated animals (A) and at 15 min after ischemic insult in animals that received intraventricular injection of vehicle (B) and 100 μm SB203580 (C) 30 min before ischemic insult. Intraventricular injection of SB203580 reduced the immunoreactivity 15 min after global ischemia (B). Scale bar, 100 μm.
Fig. 8.
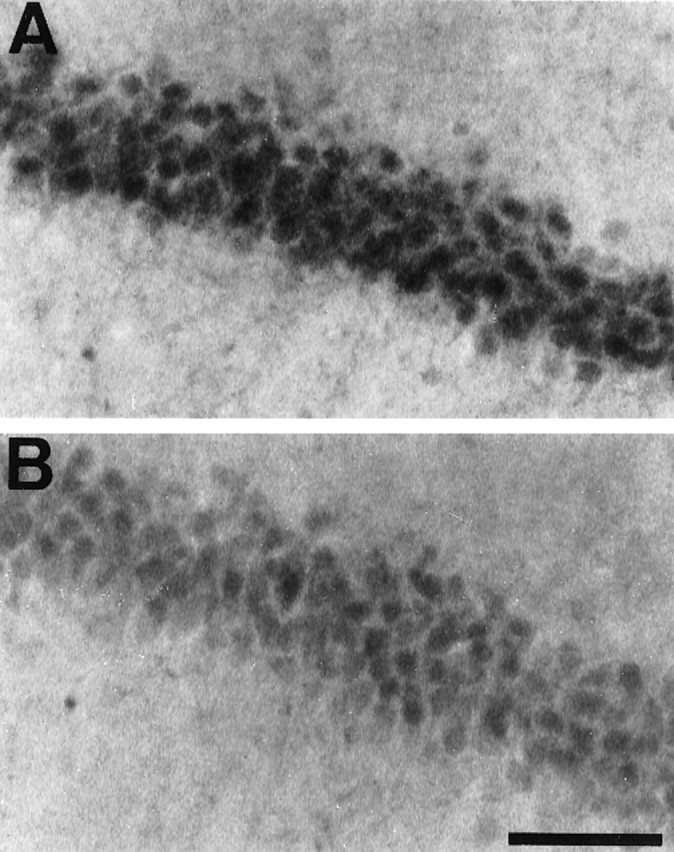
Representative photomicrographs of immunohistochemical study for active JNK in CA1 of the hippocampus 15 min after ischemic insult in animals that received intraventricular injection of vehicle (A) and 100 μmSB203580 (B) 30 min before ischemic insult. Intraventricular injection of SB203580 moderately reduced the immunoreactivity 15 min after global ischemia (B). Scale bar, 100 μm.
Effect of PD98059 on activation of ERK
Intraventricular administration of PD98059 (0, 3, 30, and 300 μm) 30 min before the ischemic insult reduced the immunoreactivity for active ERK immediately after ischemia in a dose-dependent manner; the maximum reduction of the immunoreactivity for active ERK was obtained with a dose of 30 μm (Fig.9B). Concerning p38- or JNK-dependent cascades, PD98059 could not reduce the activation of p38, ATF-2, and JNK in the immunohistochemical study (results not shown).
Fig. 9.
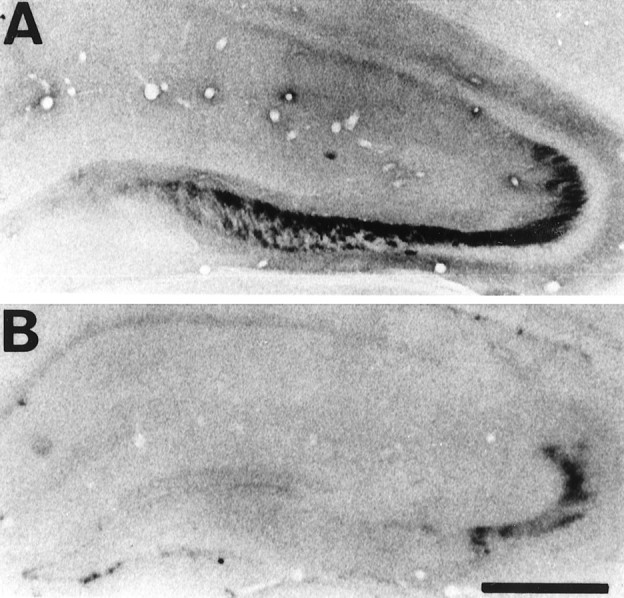
Representative photomicrographs of immunohistochemical study for active ERK in whole hippocampus immediately after ischemic insult in animals that received intraventricular injection of vehicle (A) and 30 μm PD98059 (B) 30 min before ischemic insult. In animals that received vehicle, immunoreactivity of the active form of ERK was the same as in animals that received only ischemic insult (A). Intraventricular injection of PD98059 markedly reduced the immunoreactivity (B). Scale bar, 1 mm.
SB203580, a p38 inhibitor, is neuroprotective, resulting in a decrease in DND
In the sham-operated group, no significant neuronal damage was detected. The mean number of surviving neurons in the CA1 region was 126.3 ± 5.5 (n = 6). Intraventricular administration of SB203580 (0, 1, 10, and 100 μm) 30 min before the ischemic insult significantly reduced DND in the hippocampal CA1 region 7 d after the ischemia in a dose-dependent manner, and the maximum effect was obtained at a dose of 100 μm (Figs.10G, 11A). We tested ≥300 μm doses of SB203580, but the protective effect was not increased significantly in our preliminary data. In the CA3 region, no significant effects in survival were observed (Fig. 11B). Rectal temperature was not significantly different between groups during the operative procedure and the postoperative period (data not shown).
Fig. 10.
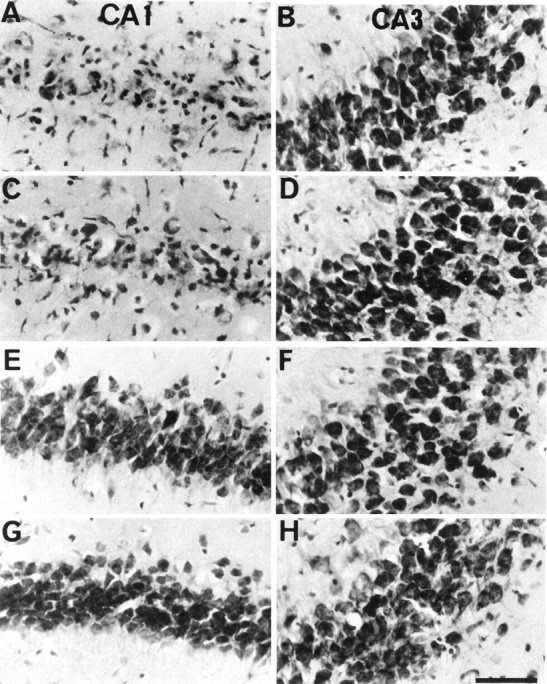
Photomicrographs of Nissl staining of the hippocampus after SB203580 treatment. Cresyl violet staining in CA1 (A, C, E, G) and CA3 (B, D, F, H) of the hippocampus after transient forebrain ischemia is shown. Almost all neurons died in the hippocampal CA1 region pretreated with 1% DMSO (A) and 1 μm SB203580 (C). When pretreated with 10 μm (E) and 100 μm (G) SB203580, more than half of the neurons survived in the CA1 region after forebrain ischemia. A, B, Pretreated with 1% DMSO; C, D, pretreated with 1 μm SB203580; E, F, pretreated with 10 μm SB203580; G, H, pretreated with 100 μm SB203580. Scale bar, 100 μm.
PD98059, a MEK1/2 inhibitor, is not neuroprotective on neuronal death in CA1 and CA3
Intraventricular administration of PD98059 (0, 3, 30, and 300 μm) 30 min before the ischemic insult could not significantly reduce DND in the hippocampal CA1 region 7 d after the ischemia (Fig. 11A). In the CA3 region, no significant effects in survival were observed (Fig.11B). Rectal temperature was also not significantly different between groups during the operative procedure and the postoperative period (data not shown).
SB203580 and PD98059 do not alter respiratory and cardiovascular parameters and body temperatures
SB203580 and PD98059 did not affect respiratory and cardiovascular parameters or produce hypothermia. Mean arterial blood pressure, arterial gas analysis (PCO2, PO2, and pH), blood glucose, and body temperature were not significantly affected by the presence or absence of 100 μm SB203580 or 30 μm PD98059 5 min before the ischemia (Table 1).
Table 1.
Physiological parameters before transient global ischemia under halothane anesthesia
| Treatment | Temp. (°C) | MABP (mmHg) | PCO2 (mmHg) | PO2 (mmHg) | pH | glucose (mg/dl) |
|---|---|---|---|---|---|---|
| DMSO (1%) (n = 4) | 37.5 ± 0.3 | 87.5 ± 4.4 | 37.8 ± 9.5 | 145.5 ± 23.1 | 7.43 ± 0.05 | 106.5 ± 21.2 |
| SB203580 (100 μm) (n = 4) | 37.6 ± 0.2 | 83.3 ± 2.2 | 33.8 ± 7.8 | 155.9 ± 7.2 | 7.39 ± 0.02 | 101.5 ± 21.6 |
| PD98059 (30 μm) (n = 4) | 37.5 ± 0.2 | 87.5 ± 2.4 | 36.5 ± 4.1 | 143.7 ± 22.9 | 7.43 ± 0.03 | 97.0 ± 4.8 |
The rectal temperature (Temp.), mean arterial blood pressure (MABP), arterial blood gas (PCO2, PO2, and pH), and glucose were measured 5 min before transient global ischemia. Data are presented as mean ± SD.
DISCUSSION
Transient forebrain ischemia causes DND in the CA1 region of hippocampus in gerbil (Kirino, 1982). DND occurs within 1–2 d after the initiation of reperfusion following ischemia (Kirino, 1982;Pulsinelli et al., 1982). Although recent reports showed that DND was related to the glutamate receptor activation (Mitani et al., 1998), tyrosine phosphorylation (Kindy, 1993), Bcl-xl–Bax interaction (Antonawich et al., 1998), and caspase-3 activation (Himi et al., 1998) and that it might be apoptotic (Kirino, 1982; Nitatori et al., 1995), the mechanism of DND has not been fully elucidated. The present work focused on the role of MAPKs in ischemic damage of hippocampus after transient forebrain ischemia.
In response to extracellular stimulation such as growth factors (Boulton et al., 1991; Nishida and Gotoh, 1993), oxidative stress (Aikawa et al., 1997), and glutamate receptor stimulation (Fiore et al., 1993; Kurino et al., 1995), the ERK is activated. Previous reports showed that a constitutively active and nuclear form of ERK2 was sufficient for neurite outgrowth and cell transformation (Fukuda et al., 1995; Robinson et al., 1998). Flood et al. (1998) showed that the distribution of MEK1 and ERK2 proteins in rat brain only partially overlapped with that of MEK4 (activator of JNK1) and JNK1. They suggest that the growth–differentiation and death–stress pathways affected by these kinases may not necessarily act to counterbalance each other in response to extracellular stimuli (Flood et al., 1998).
Unlike ERK, JNK and p38 are not associated with mitogenesis or differentiation (Cano and Mahadevan, 1995; Cobb and Goldsmith, 1995;Seger and Krebs, 1995), and cellular signaling by proinflammatory cytokines and extracellular stress activates JNK and p38 (Kyriakis and Avruch, 1996; Davis, 1999). Activation of stress kinases has been suggested to be involved in non-neuronal cell apoptosis (Kyriakis and Avruch, 1996; Davis, 1999). However, Huang et al. (1997) demonstrated that p38 did not participate in Fas/MAP kinase kinase 6b-mediated apoptosis of Jurkat T cells. Moreover, a recent report by Toyoshima et al. (1997) has shown that the Fas-induced activation of the JNK and p38 signaling pathways is not required for cytoplasmic apoptotic events or the cell death itself. These reports suggest that p38 may not necessarily be involved apoptotic cell death in Jurkat T cells and KB cells. On the other hand, it has been reported that phosphorylation of tyrosine 182 of p38 correlates with the protective action of preconditioning in the heart (Weinbrenner et al., 1997; Nagarkatti and Sha'afi, 1998). Assefa et al. (1999) demonstrated that activation of JNK and p38 protected HeLa cells from apoptosis after photodynamic therapy with hypericin. The latter two reports showed that activation of JNK and p38 might afford some cytoprotective effect in non-neuronal cells.
The role of JNK and p38 in neuronal death is also still controversial.Xia et al. (1995) demonstrated that activation of JNK and p38 and concurrent inhibition of ERK are critical for induction of apoptosis in PC12 cells. The absence of neuronal apoptosis after kainic acid-induced seizures has been reported in the JNK-3 knock-out mouse (Yang et al., 1997). JNK has been reported to be activated during NGF-deprived cell death in PC12 cells (Kummer et al., 1997). Kawasaki et al. (1997)showed the activation of p38 in glutamate-induced apoptosis in cerebellar granule cells. These observations indicate that activation of JNK and p38 may initiate neuronal apoptosis. On the contrary,Morooka and Nishida (1998) have reported that the activation of p38 was required to induce neurite growth in PC12 cells. Giasson et al. (1999)have recently reported that protease inhibition caused neurite outgrowth as a result of activation of JNK in PC12 cells. The role of JNK and p38 in neuronal death still remains to be elucidated.
Several in vivo studies have been recently reported regarding the participation of the MAPK cascade in brain ischemia (Hu and Wieloch, 1994; Ferrer et al., 1997; Herdegen et al., 1998; Walton et al., 1998; Ozawa et al., 1999), but there is some confusion about the difference between the activation and the increase in total amounts of the protein. Hu and Wieloch (1994), Herdegen et al. (1998), Walton et al. (1998), and Ozawa et al. (1999) demonstrated that phosphorylated forms of ERK, JNK, p38, and MAPKs elevated in hippocampus after brain ischemia, respectively. In the meantime, Ferrer et al. (1997) has shown that the total amounts of the protein including both phosphorylated and nonphosphorylated forms of JNK1 increased after ischemia. The activation of MAPKs is induced by phosphorylation on both Thr and Tyr residues but not by the increase of total amounts of the protein. These two phenomena should be considered separately.
In the present study, we demonstrated that the activation of JNK and p38 was induced 15 min after global ischemia in CA1 and CA3 and gradually reduced in CA1 but was continuously observed in CA3 at 72 hr of reperfusion. The decrease in immunoreactivities of JNK and p38 preceded histological changes in CA1. The activation of ERK was mainly transient in CA3 fibers and dentate gyrus. We speculate that transient activation of JNK and p38 in CA1 may be harmful effects on survival of CA1 neurons from ischemic cell death. The activation of ERK might not be significantly related to the determination of the fate of neurons to live or die, because the immunoreactivity was detected mainly temporarily in CA3 and dentate gyrus and was never detected in the CA1 region.
To evaluate the significance of the activation of p38 in transient forebrain ischemia, we tested the effect of SB203580, a p38 inhibitor, on DND in the CA1 region of hippocampus. Intraventricular administration of SB203580 (10 or 100 μm) 30 min before ischemic insult significantly reduced DND in the CA1 region. Kawasaki et al. (1997) demonstrated that SB203580 tended to inhibit apoptosis more effectively when glutamate induced apoptosis efficiently in cerebellar granule cells. DND in the CA1 region has been suggested to be apoptotic cell death (Kirino, 1982; Nitatori et al., 1995). Taken together, SB203580 might inhibit apoptotic neuronal death in CA1 after transient forebrain ischemia through the inhibition of the p38-dependent pathway.
To evaluate the significance of the activation of ERK in transient forebrain ischemia, we tested the effect of PD98059, a MEK1/2 inhibitor, on DND in the CA1 region. Intraventricular administration of PD98059 30 min before ischemic insult could not ameliorate DND in the CA1 region. We suggested that this enzyme may not participate in gerbil hippocampal neuronal survival after brain ischemia, because the inhibition of ERK by PD98059 in the CA3 region did not affect the neuronal survival. Alessandrini et al. (1999) have reported that the MEK–ERK pathway was activated during focal cerebral ischemia and may play a neurotoxic role in inducing ischemic damage. On the other hand,Gonzalez-Zulueta et al. (2000) recently reported that activation of the Ras/ERK cascade was a critical mechanism for the development of ischemic tolerance in cortical neurons. The roles of ERK in neurons are still controversial. In the present study, we did not observe any protective or harmful effects on hippocampal neuronal survival after transient global ischemia in both CA1 and CA3 subfields. The differences of the roles of ERK between the present study and the reports listed above might be attributable to the differences of the experimental ischemic models such as the durations of ischemia and reperfusion and their species. We speculate that activation of ERK may not affect the hippocampal neuronal death or survival in gerbil brain.
In the CA3 region, no significant neuronal death was observed in the presence or absence of SB203580 or PD98059 after ischemia. We suggest that there might be a selective vulnerability of the CA1 region to active p38 that might not be present in the CA3 region. We also speculate that different upstream or downstream cascades of MAP kinases might be activated in CA1 and CA3 after ischemia in gerbil brain.
Up to now, a specific inhibitor of JNK has not been available (Maroney et al., 1998). It remains to be elucidated that the inhibition of transient activation of JNK may protect against DND in CA1 pyramidal cells after transient forebrain ischemic insult.
The present study demonstrated for the first time that JNK, p38, and ERK were activated in gerbil hippocampus after transient forebrain ischemia in vivo, that the activation of JNK and p38 in CA1 neurons after ischemia was seen earlier than previously reported (Walton et al., 1998) both by immunoblot analysis and by immunohistochemistry, and that the inhibition of the activity of p38 protected against DND in CA1 pyramidal cells. Because high doses of SB203580 inhibited the activation of JNK moderately, the possibility that activation of JNK might contribute to DND in CA1 may not be able to be excluded completely.
In conclusion, JNK, p38, and ERK were activated after transient forebrain ischemia in gerbil hippocampus in vivo. We suggest that p38 may play an important role in hippocampal neuronal survival and death during brain ischemia. Components of the pathway are important target molecules in elucidating the mechanism of ischemic neuronal death.
Footnotes
Correspondence should be addressed to Dr. Kazuhiko Nozaki, Department of Neurosurgery, Graduate School of Medicine, Kyoto University, 54 Kawahara-cho, Shogoin, Sakyo-ku, Kyoto 606-8507, Japan. E-mail:noz@kuhp.kyoto-u.ac.jp.
REFERENCES
- 1.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–1821. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonawich FJ, Krajewski S, Reed JC, Davis JN. Bcl-xl Bax interaction after transient global ischemia. J Cereb Blood Flow Metab. 1998;18:882–886. doi: 10.1097/00004647-199808000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Assefa Z, Vantieghem A, Declercq W, Vandenabeele P, Vandenheede JR, Merlevede W, de Witte P, Agostinis P. The activation of c-Jun N-terminal kinase and p38 mitogen-activated protein kinase signaling pathways protects HeLa cells from apoptosis following photodynamic therapy with hypericin. J Biol Chem. 1999;274:8788–8796. doi: 10.1074/jbc.274.13.8788. [DOI] [PubMed] [Google Scholar]
- 5.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 6.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- 7.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 8.Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- 9.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 10.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 11.Davis RJ. Signal transduction by the c-Jun N-terminal kinase. Biochem Soc Symp. 1999;64:1–12. doi: 10.1515/9781400865048.1. [DOI] [PubMed] [Google Scholar]
- 12.Ferrer I, Ballabriga J, Pozas E. Transient forebrain ischemia in the adult gerbil is associated with a complex c-Jun response. NeuroReport. 1997;8:2483–2487. doi: 10.1097/00001756-199707280-00014. [DOI] [PubMed] [Google Scholar]
- 13.Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM. Activation of mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- 14.Flood DG, Finn JP, Walton KM, Dionne CA, Contreras PC, Miller MS, Bhat RV. Immunolocalization of the mitogen-activated protein kinases p42MAPK and JNK1, and their regulatory kinases MEK1 and MEK4, in adult rat central nervous system. J Comp Neurol. 1998;398:373–392. doi: 10.1002/(sici)1096-9861(19980831)398:3<373::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda M, Gotoh Y, Tachibana T, Dell K, Hattori S, Yoneda Y, Nishida E. Induction of neurite outgrowth by MAP kinase in PC12 cells. Oncogene. 1995;11:239–244. [PubMed] [Google Scholar]
- 16.Giasson BI, Bruening W, Durham HD, Murshynski WE. Activation of stress-activated kinases correlates with neurite outgrowth induced by protease inhibition in PC12 cells. J Neurochem. 1999;72:1081–1087. doi: 10.1046/j.1471-4159.1999.0721081.x. [DOI] [PubMed] [Google Scholar]
- 17.Ginsberg MD, Prado R, Dietrich WD, Busto R, Watson BD. Hyperglycemia reduces the extent of cerebral infarction in rats. Stroke. 1987;18:570–574. doi: 10.1161/01.str.18.3.570. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci USA. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herdegen T, Claret F-X, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Himi T, Ishizaki Y, Murota S. A caspase inhibitor blocks ischaemia-induced delayed neuronal death in the gerbil. Eur J Neurosci. 1998;10:777–781. doi: 10.1046/j.1460-9568.1998.00073.x. [DOI] [PubMed] [Google Scholar]
- 21.Hu BR, Wieloch T. Tyrosine phosphorylation and activation of mitogen-activated protein kinase in the rat brain following transient cerebral ischemia. J Neurochem. 1994;62:1357–1367. doi: 10.1046/j.1471-4159.1994.62041357.x. [DOI] [PubMed] [Google Scholar]
- 22.Huang S, Jiang Y, Li Z, Nishida E, Mathias P, Lin S, Ulevitch RJ, Nemerow GR, Han J. Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity. 1997;6:739–749. doi: 10.1016/s1074-7613(00)80449-5. [DOI] [PubMed] [Google Scholar]
- 23.Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- 24.Kindy MS. Inhibition of tyrosine phosphorylation prevents delayed neuronal death following cerebral ischemia. J Cereb Blood Flow Metab. 1993;13:372–377. doi: 10.1038/jcbfm.1993.50. [DOI] [PubMed] [Google Scholar]
- 25.Kirino T. Delayed neuronal death on the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 26.Kummer JL, Rao PK, Heidenreich KA. Apoptosis induced by withdrawal of trophic factors is mediated by p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:20490–20494. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- 27.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 28.Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- 29.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Larsen JK, Yamboliev IA, Weber LA, Gerthoffer WT. Phosphorylation of the 27 kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am J Physiol. 1997;273:L930–L940. doi: 10.1152/ajplung.1997.273.5.L930. [DOI] [PubMed] [Google Scholar]
- 31.Leary JJ, Brigati DJ, Ward DC. Rapid and sensitive colorimetric method for visualizing biotin-labeled DNA probes hybridized to DNA or RNA immobilized on nitrocellulose: bio-blots. Proc Natl Acad Sci USA. 1983;80:4045–4049. doi: 10.1073/pnas.80.13.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 33.Maroney AC, Glicksman MA, Basma AN, Walton KM, Ernest K, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitani A, Namba S, Ikemune K, Yanase H, Arai T, Kataoka K. Postischemic enhancement of N-methyl-d aspartic acid (NMDA) and non-NMDA receptor-mediated responses in hippocampal CA1 pyramidal neurons. J Cereb Blood Flow Metab. 1998;18:1088–1098. doi: 10.1097/00004647-199810000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Mizukami Y, Yoshioka K, Morimoto S, Yoshida K. A novel mechanism of JNK1 activation. J Biol Chem. 1997;272:16657–16662. doi: 10.1074/jbc.272.26.16657. [DOI] [PubMed] [Google Scholar]
- 36.Morooka T, Nishida E. Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J Biol Chem. 1998;273:24285–24288. doi: 10.1074/jbc.273.38.24285. [DOI] [PubMed] [Google Scholar]
- 37.Nagarkatti DS, Sha'afi RI. Role of p38 MAP kinase in myocardial stress. J Mol Cell Cardiol. 1998;30:1651–1664. doi: 10.1006/jmcc.1998.0733. [DOI] [PubMed] [Google Scholar]
- 38.Nishida E, Gotoh Y. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci. 1993;18:128–131. doi: 10.1016/0968-0004(93)90019-j. [DOI] [PubMed] [Google Scholar]
- 39.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozawa H, Shioda S, Dohi K, Matsumoto H, Mizushima H, Zhou CJ, Funahashi H, Nakai Y, Nakajo S, Matsumoto K. Delayed neuronal cell death in the rat hippocampus is mediated by the mitogen-activated protein kinase signal transduction pathway. Neurosci Lett. 1999;262:57–60. doi: 10.1016/s0304-3940(99)00034-8. [DOI] [PubMed] [Google Scholar]
- 41.Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 42.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 43.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 44.Robinson MJ, Stippec SA, Goldsmith E, White M, Cobb MH. A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol. 1998;8:1141–1150. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- 45.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 46.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 47.Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiessen DD, Goar S. Stereotaxic atlas of the hypothalamus of the mongolian gerbil. J Comp Neurol. 1970;140:123–128. doi: 10.1002/cne.901400108. [DOI] [PubMed] [Google Scholar]
- 49.Tokime T, Nozaki K, Kikuchi H. Neuroprotective effect of FK506, an immunosuppressant, on transient global ischemia in gerbil. Neurosci Lett. 1996;206:81–84. doi: 10.1016/s0304-3940(96)12438-1. [DOI] [PubMed] [Google Scholar]
- 50.Toyoshima F, Moriguchi T, Nishida E. Fas induces cytoplasmic apoptotic responses and activation of the MKK7-JNK/SAPK and MKK6–p38 pathways independent of CPP32-like proteases. J Cell Biol. 1997;139:1005–1015. doi: 10.1083/jcb.139.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walton KM, DiRocco R, Bartlett BA, Koury E, Marcy VR, Jarvis B, Schaefer EM, Bhat RV. Activation of p38MAPK in microglia after ischemia. J Neurochem. 1998;70:1764–1767. doi: 10.1046/j.1471-4159.1998.70041764.x. [DOI] [PubMed] [Google Scholar]
- 52.Waters SB, Holt KH, Ross SE, Syu LJ, Guan KL, Saltiel AR, Koretzky GA, Pessin JE. Desensitization of Ras activation by a feedback disassociation of the SOS-Grb2 complex. J Biol Chem. 1995;270:20883–20886. doi: 10.1074/jbc.270.36.20883. [DOI] [PubMed] [Google Scholar]
- 53.Weinbrenner C, Liu G-S, Cohen MV, Downey JM. Phosphorylation of tyrosine 182 of p38 mitogen-activated protein kinase correlates with the protection of preconditioning in the rabbit heart. J Mol Cell Cardiol. 1997;29:2383–2391. doi: 10.1006/jmcc.1997.0473. [DOI] [PubMed] [Google Scholar]
- 54.Whitmarsh AJ, Yang S-H, Su MS-S, Sharrocks AD, Davis RJ. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 56.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–867. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 57.Yin T, Sandhu G, Wolfgang CD, Burrier A, Webb RL, Rigel DF, Hai T, Whelan J. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J Biol Chem. 1997;272:19943–19950. doi: 10.1074/jbc.272.32.19943. [DOI] [PubMed] [Google Scholar]