

Abstract
We have identified a mechanism, mediated by the ε isozyme of protein kinase C (PKCε) in peripheral neurons, which may have a role in chronic inflammatory pain. Acute inflammation, produced by carrageenan injection in the rat hindpaw, produced mechanical hyperalgesia that resolved by 72 hr. However, for up to 3 weeks after carrageenan, injection of the inflammatory mediators prostaglandin E2 or 5-hydroxytryptamine or of an adenosine A2agonist into the same site induced a markedly prolonged hyperalgesia (>24 hr compared with 5 hr or less in control rats not pretreated with carrageenan). A nonselective inhibitor of several PKC isozymes and a selective PKCε inhibitor antagonized this prolonged hyperalgesic response equally. Acute carrageenan hyperalgesia could be inhibited by PKA or PKG antagonists. However, these antagonists did not inhibit development of the hypersensitivity to inflammatory mediators. Our findings indicate that different second messenger pathways underlie acute and prolonged inflammatory pain.
Keywords: carrageenan, chronic pain, inflammation, prostaglandin E2, protein kinase Cε, second messenger
In studying mechanisms underlying pain, researchers have been successful in elucidating bases of acute inflammatory pain (for review, see Cesare and McNaughton, 1997; Levine and Reichling, 1999). Although chronic inflammatory pain syndromes (e.g., arthritis, gastritis, colitis, dermatitis, and post-traumatic and repetitive strain injuries) result in enormous morbidity and societal cost, they remain poorly understood. Specifically, it is not known whether novel mechanisms different from those of acute inflammatory pain are involved, which is a critical point for the design of rational therapies.
Because chronic inflammatory pain states can follow an episode of acute inflammation (Lockwood, 1989; MacIntyre et al., 1995; Melhorn, 1998), we investigated whether acute inflammation can induce a long-lasting increase in the sensitivity to inflammatory hyperalgesic mediators. Such an increased sensitivity could underlie the development of a chronic pain syndrome and could be used to identify second messenger systems that contribute to chronic inflammatory states. In these experiments, we studied rats previously treated with the inflammatory agent carrageenan, at a dose that produces only short-lived inflammation and hyperalgesia (Guilbaud et al., 1989; Dawson et al., 1991). We found that this treatment resulted in a long-lasting increase in subsequent sensitivity to hyperalgesic inflammatory mediators. We also evaluated the initiation, duration, and mechanisms, including contributing second messengers, underlying this long-lasting hypersensitivity to proinflammatory mediators.
MATERIALS AND METHODS
Animals. Experiments were performed on male Sprague Dawley rats (200–250 gm; Bantin-Kingman, Fremont, CA). Animals were housed in groups of two under a 12 hr light/dark cycle (lights on at 7:00 A.M.). Food and water were available ad libitum. All behavioral testing was done between 10:00 A.M. and 4:00 P.M. Experiments were performed under approval of the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Behavioral testing. The nociceptive flexion reflex (Randall-Selitto paw-withdrawal test) was quantified with a Basile Analgesymeter (Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the dorsum of the rat's hindpaw. Three readings were taken at 5 min intervals, and their mean was considered the baseline threshold. Groups that were compared with to determine effect of drug administration had similar baseline thresholds. Mechanical threshold was redetermined at various time points after drug administration. These time points were determined based on the latency and duration of action of each drug used in the study. The mean of three readings (taken at intervals of 5 min, the last reading corresponding to the time specified [always taken at least at 30 min and 4 hr for prostaglandin E2(PGE2)] after drug treatment) was used to calculate the percentage change from the baseline threshold. To determine the carrageenan dose to be used in the study, the effect of different doses (0.1–2%) were evalvated studied. The time at which each drug had a maximal effect also was considered in timing the measurement of the paw-withdrawal threshold (maximum effect for carrageenan at 4 hr and for the other drugs at 30 min). To study the onset of carrageenan-induced changes in response to hyperalgesic inflammatory mediators, we injected rats with PGE2 at various times (0.5–96 hr) after injection of carrageenan.
Drug administration. The drugs used in this study were as follows: PGE2 (direct-acting hyperalgesic inflammatory mediator), λ carrageenan (inflammatory agent),NG-methyl-l-arginine (l-NMA) (nitric oxide synthase inhibitor), 2-[(2-bis - [carboxymethyl] amino-5-methylphenoxy) methyl]-6-methoxy-8-bis [carboxymethy] aminoquinoline (Quin-2) (calcium chelator), 3,4,5-trimethoxybenzoic acid 8-(diethylamino) octyl ester (TMB-8) (inhibitor of intracellular Ca2+ transport), and 5-hydroxytryptamine (5-HT, serotonin) (all from Sigma, St. Louis, MO); Walsh inhibitor peptide (WIPTIDE) [protein kinase A (PKA) inhibitor 5–22 amide; Peninsula Laboratories, Belmont CA]; and {1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one} (ODQ) (guanylyl cyclase inhibitor), an inhibitor of protein kinase G (PKG) (peptide with sequence, H-Arg-Lys-Arg-Ala-Arg-Lys-Glu-PH) that corresponds to a nonphosphorylatable analog (Ser32 to Ala32) of histone H2B (residues 29–35), bisindolylmalemide 1 HCl (Bis) [protein kinase C (PKC) inhibitor] (all from Calbiochem-Novabiochem, La Jolla, CA); (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (NMDA receptor antagonist) and carboxyethyl phenethylamino-5′-N-ethylcarboxamido adenosine HCl (CGS-21680) (an adenosine A2 agonist) (both from Research Biochemicals, Natick, MA). HDAPIGYD (pseudo ε RACK (ψεR), a PKCε agonist (Dorn et al., 1999) and PKCεV1–2 peptide, H2N-EAVSLKPT-COOH, a PKCε inhibitor (Gray et al., 1997) were synthesized by SynPep (Dublin, CA). The selection of the drug doses used in this study was based on dose–response curves determined during this as well as previous studies (Aley et al., 1995, 1998; Khasar et al., 1999a). The stock solution of PGE2 (1 μg/2.5 μl) was prepared in 10% ethanol, and additional dilutions were made in saline; the final concentration of ethanol was ≤1%. All other drugs were dissolved in saline except ODQ, which was dissolved in DMSO and diluted with saline (final concentration of DMSO was ≤10%). All drugs were administered intradermally in a volume of 5 μl/paw. For test agents with low cell membrane permeability (i.e., WIPTIDE, PKC inhibitor, and ψεR), 2 μl of distilled water was injected first, in the same syringe as the test agent, to produce hypo-osmotic shock and thus transiently permeabilize the cell membrane (Keeney and Linn, 1990;Lepers et al., 1990; Schulz, 1990). When drug combinations were used, they were administered at 5 min intervals with the drug mentioned first, the antagonist, administered first. All of the drugs except carrageenan were administered using a 30 gauge hypodermic needle. Because of its high viscosity, carrageenan was injected using a 27 gauge needle. All drugs were administered either to control rats or on days 5 or 21 after carrageenan. After injection of hyperalgesic inflammatory mediators, readings of nociceptive threshold were always taken at 30 min and 4 hr and sometimes at other time points to determine the time course.
Statistical analysis. Data are presented as mean ± SEM; means were compared by ANOVA. Differences between pairs of means were analyzed by Scheffe's post hoc test and were considered significant at p < 0.05.
RESULTS
Carrageenan induces a long-term prolongation of inflammatory mediator-induced hyperalgesia
We hypothesized that a low dose of an inflammatory agent such as carrageenan would produce a short-term (several days) hyperalgesia from which the animal would fully recover, but might also induce a long-lasting heightened hyperalgesic response to inflammatory mediators. By measuring the carrageenan dose–response relationship (Fig. 1A; see Materials and Methods), we determined that 5 μl of 1% carrageenan (w/v in physiological saline) resulted in swelling, erythema, and reduced paw-withdrawal threshold to mechanical pressure beginning 30–60 min after injection, reaching a maximum between 2–4 hr, and resolving within 72 hr (Fig. 1A,B).
Fig. 1.

A, Dose (0.1–2%)–response curve of carrageenan (Carr; n = 12) induced mechanical hyperalgesia measured at 4 hr in the hindpaw of the normal rat. The Randall-Selitto paw-withdrawal test is an established method to assess heightened nociception in animals in which this subjective experience of pain cannot be directly determined. Measures using this technique have been shown to correlate with pain-like behaviors in animals. B, Time course of hyperalgesia induced by carrageenan 1% (5 μl, n = 24) in normal rats.
Intradermal injection of the inflammatory mediators PGE2, 5-HT, or the A2adenosine receptor agonist CGS-21680, at the same site into which carrageenan had been injected 5 d earlier, resulted in a prolonged mechanical hyperalgesia lasting >24 hr (Fig.2A). This ability of carrageenan to prolong hyperalgesia induced by inflammatory mediators persisted for at least 3 weeks after carrageenan administration (Fig.2B). In comparison, in control rats exposed to vehicle without carrageenan, PGE2, CGS-21680, and 5-HT produced transient hyperalgesia lasting <4 hr (Fig.2C).
Fig. 2.

A, PGE2 (100 ng,n = 24), 5-HT (1 μg, n = 6), and CGS-21680 (100 ng, n = 6)-induced mechanical hyperalgesia at 30 min, 4 hr, and 24 hr after injection in rats treated 5 d previously with carrageenan. B, Mechanical hyperalgesia induced by PGE2 (n = 12), 5-HT (n = 6), and CGS-21680 (n= 6) at 30 min, 4 hr, and 24 hr after injection in rats treated 21 d previously with carrageenan. C, Time course of PGE2-, 5-HT-, and CGS-21680-induced mechanical hyperalgesia in rats 5 d after vehicle used for carrageenan (n = 12 each).
Novel mechanism of prolonged PGE2-induced hyperalgesia
We next examined the second messengers that mediate the ability of carrageenan to prolong hyperalgesia induced by inflammatory mediators. To examine this issue, we evaluated PGE2-induced hyperalgesia and used inhibitors of second messenger pathways important in peripheral nociception. In control animals, previous treatment with the PKA inhibitor (WIPTIDE) or the nitric oxide synthase inhibitor (l-NMA) attenuated PGE2-induced mechanical hyperalgesia, whereas Bis (PKC inhibitor) or PKCεV1–2 (PKCε inhibitor) was without effect (Fig. 3A). PKGI (PKG inhibitor), ODQ (guanylyl cyclase inhibitor), TMB-8 and Quin-2 (calcium antagonists), and MK-801 (NMDA receptor antagonist) were also without effect (data not shown). In rats treated with carrageenan 5 d previously, WIPTIDE or l-NMA inhibited the early phase of PGE2-stimulated hyperalgesia 30 min after injection of PGE2. This was similar to what was observed in control animals not pretreated with carrageenan (Fig.3A). In contrast, neither WIPTIDE norl-NMA inhibited the late phase (4 hr after injection) of PGE2-induced hyperalgesia observed in carrageenan pretreated rats (Fig. 3B). Bis and the PKCε inhibitor did not reduce early PGE2-stimulated hyperalgesia in control or carrageenan pretreated rats (Fig.3A). However, these agents inhibited the late phase of PGE2 hyperalgesia seen in carrageenan pretreated rats (Fig. 3B). The PKCε inhibitor also inhibited the late phases of 5-HT and CGS-21680 hyperalgesia in carrageenan pretreated rats (data not shown). TMB-8 and Quin-2 or MK-801 had no effect on PGE2-induced hyperalgesia in carrageenan pretreated or control rats (data not shown).
Fig. 3.
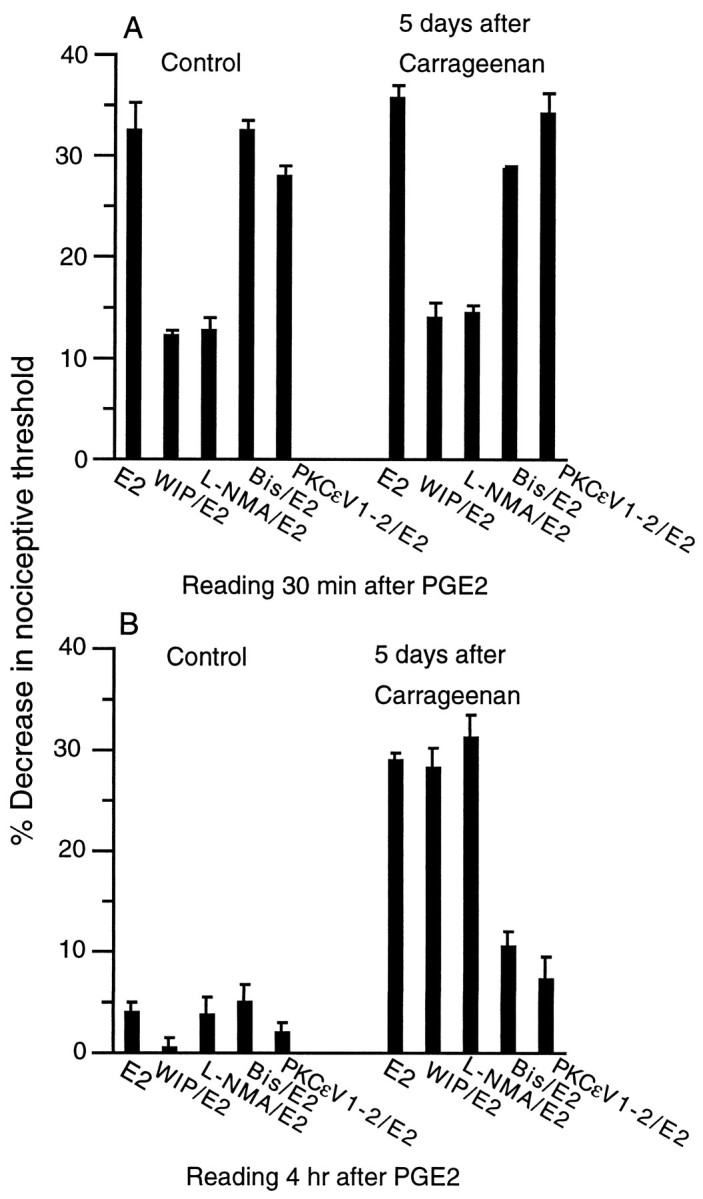
A, Effect of PKA inhibitor WIPTIDE (WIP/E2; 1 μg/100 ng, n = 24), nitric oxide synthase inhibitorNG-methyl-l-arginine (L-NMA/E2; 1 μg/100 ng, n = 12), PKC inhibitor bisindolylmalemide 1 hydrochloride (Bis/E2; 1 μg/100 ng, n = 12), PKCε inhibitor (PKCεV1–2/E2; 1 μg/100 ng,n = 12), administered 5 min before PGE2, on PGE2(E2)-induced mechanical hyperalgesia measured at 30 min after PGE2 injection in control rats and in rats treated 5 d previously with carrageenan. B, Effect of PKA inhibitor WIP/E2 (n = 24), nitric oxide synthase inhibitor L-NMA/E2 (n= 12), PKC inhibitor Bis/E2 (n = 12), PKCε inhibitor PKCεV1–2/E2) (n = 12), administered 5 min before PGE2, on PGE2(E2)-induced mechanical hyperalgesia measured at 4 hr after PGE2 injection in control rats and in rats treated 5 d previously with carrageenan.
We next investigated whether similar second messenger systems mediate acute carrageenan-induced hyperalgesia and the ability of carrageenan to prolong PGE2 hyperalgesia. Carrageenan-induced hyperalgesia was attenuated by an inhibitor of PKA or PKG (Fig.4A). However, when PGE2 was administered after injection of WIPTIDE plus carrageenan or the PKG inhibitor plus carrageenan (Fig.4B,C), a prolonged hyperalgesic response to PGE2 was still observed beginning 48 hr after carrageenan injection (Fig.4B,C).
Fig. 4.

A, Effect of PKA inhibitor WIPTIDE (WIP/Carr; n = 24) and PKG inhibitor (PKGI/Carr; n = 8) on carrageenan-induced mechanical hyperalgesia in the rat hindpaw. Agents were administered 5 min before carrageenan. All readings were taken 4 hr after carrageenan. B, C, Effect of PGE2 injected at different times (30 min to 96 hr) in different groups of rats after injection of carrageenan plus WIPTIDE (B) or PKGI (C). All readings were taken 4 hr after prostaglandin E2 injection.n = 6 each group.
Administration of a PKCε agonist is sufficient to induce the prolonged hyperalgesic response to PGE2
Because the long-term prolongation of PGE2hyperalgesia was inhibited by PKCε inhibitors, we evaluated whether specific activation of PKCε could, like carrageenan, result in a similar long-term prolongation of PGE2hyperalgesia. To perform these studies, we used the PKCε peptide agonist ψεR (Dorn et al., 1999).
Intradermal injection of the PKCε agonist ψεR into the hindpaw of the rat produced a dose-dependent mechanical hyperalgesia (Fig.5A), inhibitable by a nonselective PKC inhibitor (Bis) and a PKCε selective inhibitor (PKCεV1–2), but not by inhibitors of other second messenger pathways implicated in hyperalgesia (WIPTIDE, PKGI, ODQ, l-NMA, Quin-2, TMB-8, or MK801) (Fig.5D). ψεR-induced hyperalgesia lasted for ∼72 hr (Fig.5C). After recovery from ψεR-induced hyperalgesia, on the fifth day after administration of ψεR, the response to PGE2 was markedly prolonged (lasting >24 hr) (Fig. 5B), similar to that observed after recovery from carrageenan hyperalgesia. As after carrageenan, Bis and PKCε inhibitor, but not WIPTIDE, PKGI, ODQ, l-NMA, Quin-2, TMB-8, and MK801, attenuated the prolonged hyperalgesia induced by PGE2 (Fig. 5E). Instead, as found in carrageenan-pretreated rats, WIPTIDE inhibited only the early (30 min after injection) phase of PGE2 hyperalgesia in ψεR-pretreated rats.
Fig. 5.

A, Dose–response curve of ψεR (0.1–10,000 ng, n = 8)-induced mechanical hyperalgesia measured at 30 min in the hindpaw of the rat.B, PGE2-induced hyperalgesia at 30 min, 4 hr, and 24 hr in rats treated 5 d previously with ψεR (1 μg,n = 6). C, Time course of ψεR (1 μg/paw, n = 12)-induced hyperalgesia (1 μg).D, Role of second messengers important in ψεR-induced hyperalgesia. PKA inhibitor WIPTIDE (WIP/ΨεR; both 1 μg, n = 24), the nitric oxide synthase inhibitor NG-methyl-l-arginine (L-NMA/ΨεR; both 1 μg,n = 12), the PKC inhibitor bisindolylmalemide 1 hydrochloride (Bis/ΨεR; both 1 μg, n = 12), the PKCε inhibitor (PKCεV1–2/ΨεR; both 1 μg,n = 12), the PKG inhibitor (PKGI/ΨεR; both 1 μg,n = 8), the guanylyl cyclase inhibitor ODQ (ODQ/ΨεR; both 1 μg,n = 6), the calcium transport antagonist 3,4,5-trimethoxybezoic acid 8-(diethylamino) octyl ester (TMB/ΨεR; both 1 μg,n = 6), the calcium chelator (2-[(2-bis-[carboxymethyl] amino-5-methylphenoxy) methyl]-6-methoxy-8-bis[carboxymethy] aminoquinoline (Quin/ΨεR; both 1 μg, n = 6), or the NMDA receptor antagonist MK-801 (MK801/ΨεR; both 1 μg,n = 8) 5 min before injection of ψεR (1 μg). All readings were taken 30 min after injection of ψεR.E, Role of second messengers in PGE2-induced hyperalgesia in rats pretreated with ψεR. Rats were administered the PKA inhibitor WIPTIDE (E2/WIP; 100 ng/1 μg,n = 6), the nitric oxide synthase inhibitorl-NMA (E2/L-NMA; 100 ng/1 μg,n = 6), the PKC inhibitor Bis (E2/Bis; 100 ng/1 μg, n = 10), the PKCε inhibitor (E2/PKCεV1–2; 100 ng/1 μg, n = 6), the calcium antagonists Quin-2 and TMB-8 (E2/Quin and E2/TMB; both 100 ng/1 μg, both n = 6), or the NMDA receptor antagonist MK-801 (E2/MK801; 100 ng/1 μg,n = 10) 5 min before injection of PGE2(E2) (100 ng) on the fifth day after receiving ψεR (1 μg). All readings were taken 4 hr after injection of PGE2.
DISCUSSION
Although many mechanisms have been demonstrated to contribute to acute inflammatory pain, very little is known of the cellular changes underlying chronic inflammatory pain states. Recently, it has been suggested that sensitization and sprouting may be important mechanisms by which the CNS contributes to chronic inflammatory pain (Woolf and Doubell, 1994; Baranauskas and Nistri, 1998). We now demonstrate a novel peripheral pronociceptive mechanism initiated by acute inflammation, involving a PKCε-dependent prolongation of hyperalgesic responses to inflammatory mediators lasting several weeks after resolution of the initial acute inflammation. This prolonged response to inflammatory mediators constitutes a dramatic unprecedented plastic change in primary afferent nociceptor function, most striking in the marked, sixfold or greater increase in duration of hyperalgesia after a single injection of PGE2 and in the persistence of this change for a 3 week period or more. In addition, this plastic change in the primary afferent nociceptor is not accompanied by a residual baseline hyperalgesia or by histopathological evidence of ongoing inflammation (Guilbaud et al., 1989; Dawson et al., 1991). Because in the dermis, the site of injection of all the test agents we used, PKCε is present exclusively in nerve processes (Khasar et al., 1999a), this plastic change appears to occur in the primary afferent nociceptor.
Carrageenan, which we used to induce the initial inflammation, is a classic agent for the induction of experimental inflammation and inflammatory pain and is considered relevant to clinically important inflammatory pain states (Di Rosa, 1972; Dawson et al., 1991; Gilroy et al., 1999). In addition, the mediators for which we demonstrated the development of a prolonged response [PGE2, 5HT, and purines (CGS-21680)] are known to be present at increased concentration at sites of inflammation (Foon et al., 1976; Driver et al., 1993; Villena et al., 1999) and are known to produce hyperalgesia by a direct action on primary afferent nociceptors (Taiwo and Levine, 1990, 1992; Gold et al., 1996). Therefore, the hyper-responsive state we describe, dependent on PKCε, is very likely active in peripheral nociceptors in chronic inflammatory pain. Such an exaggerated response to inflammatory mediators may explain the inordinate and lasting responses observed in patients with chronic inflammatory pain syndromes after minor stimuli (Lockwood, 1989; MacIntyre et al., 1995; Melhorn, 1998).
In our model, mild acute carrageenan hyperalgesia could be blocked without inhibiting subsequent enhanced responsiveness to PGE2. This demonstrated that enhanced responsiveness to PGE2 was not dependent on the preceding acute hyperalgesia. This finding allowed us to determine that enhanced responsiveness to PGE2 was present as early as 48 hr after injection of carrageenan. These observations suggest that development of a propensity for persistent chronic inflammatory pain may occur after a period of only minimal hyperalgesia, providing an explanation for instances in which chronic inflammatory pain develops without an episode of preceding overt acute inflammation.
Pronociceptive plastic changes in CNS circuitry are well established (Woolf and Doubell, 1994; Mannion et al., 1996; Baranauskas and Nistri, 1998). The search for such changes in the periphery, however, has received little attention, although there are recent reports of altered gene expression in primary afferent neurons stimulated by NGF and electrical activity (Gilchrist et al., 1991; McCarson and Krause, 1994;Nahin and Byers, 1994; Black et al., 1997; Itoh et al., 1997; Tonra and Mendell, 1998; Fjell et al., 1999; Woolf and Costigan, 1999). The time of onset of ∼48 hr for the development of long-term prolongation of hyperalgesic responses to inflammatory mediators is compatible with gene expression followed by transport or newly synthesized protein to peripheral terminals.
Although our data do not exclude actions by other isozymes of PKC, that the epsilon isozyme alone, of PKC, is responsible is supported by the observations that the prolonged response to inflammatory mediators was totally prevented by injection of a specific PKCε inhibitor and that the injection of a PKCε agonist alone resulted in a similar prolonged hyperalgesic response. PKCε is known also to contribute to acute nociception, specifically to acute hyperalgesia produced by epinephrine (Khasar et al., 1999a,b), which may be present at increased levels during inflammation (Cunha et al., 1991; Mikhailov and Rusanova, 1993). That there is a different function of PKCε in prolonged hyperalgesia compared with acute nociception is suggested by the chronic (>24 hr) nature of the resultant hyperalgesia and the apparent novel coupling of PKCε to the PGE2 receptor, a phenomenon not seen in acute hyperalgesia produced by PGE2(Levine and Reichling, 1999).
In summary, we have established, for the first time, a plastic pronociceptive mechanism, most likely in nociceptors, that is dependent on PKCε and may have a role in chronic inflammatory pain states. The dependence on a mechanism not found with acute hyperalgesia and its presence after even mild inflammation has important clinical implications. Our experimental paradigm provides a model for the investigation of other cellular mechanisms that may contribute to chronic inflammatory pain. It should be possible, using this model, to obtain valuable information for the rational development of targeted therapies for both active disease and remission of chronic inflammatory pain states, such as arthritis, bronchitis, asthma, dermatitis, inflammatory bowel disease, and repetitive strain injuries, which contribute greatly to morbidity worldwide. Finally, because the targeted pronociceptive mechanism is peripheral, at the site of pain, it may be possible to administer analgesic therapies locally with minimal or no systemic side effects.
Footnotes
This work was funded by National Institutes of Health Grant NS21647. We acknowledge many helpful discussions with Drs. David Reichling, Philip Heller, and Paul Green.
Correspondence should be addressed to Dr. K. O. Aley, National Institutes of Health Pain Center, University of California, San Francisco, Box 0440, San Francisco, CA 94143-0440. E-mail:aley@itsa.ucsf.edu.
REFERENCES
- 1.Aley KO, Khasar SG, Levine JD. Multiple second messenger systems act sequentially to mediate rolipram-induced prolongation of prostaglandin E2-induced mechanical hyperalgesia in the rat. Neuroscience. 1995;64:769–776. doi: 10.1016/0306-4522(94)00397-n. [DOI] [PubMed] [Google Scholar]
- 2.Aley KO, McCarter G, Levine JD. Nitric oxide signaling in pain and nociceptor sensitization in the rat. J Neurosci. 1998;18:7008–7014. doi: 10.1523/JNEUROSCI.18-17-07008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baranauskas G, Nistri A. Sensitization of pain pathways in the spinal cord: cellular mechanisms. Prog Neurobiol. 1998;54:349–365. doi: 10.1016/s0301-0082(97)00067-1. [DOI] [PubMed] [Google Scholar]
- 4.Black JA, Langworthy K, Hinson AW, Dib-Hajj SD, Waxman SG. NGF has opposing effects on Na+ channel III and SNS gene expression in spinal sensory neurons. NeuroReport. 1997;8:2331–2335. doi: 10.1097/00001756-199707070-00046. [DOI] [PubMed] [Google Scholar]
- 5.Cesare P, McNaughton P. Peripheral pain mechanisms. Curr Opin Neurobiol. 1997;7:493–499. doi: 10.1016/s0959-4388(97)80028-1. [DOI] [PubMed] [Google Scholar]
- 6.Cunha FQ, Lorenzetti BB, Poole S, Ferreira SH. Interleukin-8 as a mediator of sympathetic pain. Br J Pharmacol. 1991;104:765–767. doi: 10.1111/j.1476-5381.1991.tb12502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson J, Sedgwick AD, Edwards JC, Lees P. A comparative study of the cellular, exudative and histological responses to carrageenan, dextran and zymosan in the mouse. Int J Tissue React. 1991;13:171–185. [PubMed] [Google Scholar]
- 8.Di Rosa M. Biological properties of carrageenan. J Pharm Pharmacol. 1972;24:89–102. doi: 10.1111/j.2042-7158.1972.tb08940.x. [DOI] [PubMed] [Google Scholar]
- 9.Dorn GWI, Sourougon MC, Liron T, Chen C-H, Gray MO, Zhou HZ, Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes ε protein kinase C translocation. Proc Natl Acad Sci USA. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. 1993;148:91–97. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- 11.Fjell J, Cummins TR, Davis BM, Albers KM, Fried K, Waxman SG, Black JA. Sodium channel expression in NGF-overexpressing transgenic mice. J Neurosci Res. 1999;57:39–47. doi: 10.1002/(SICI)1097-4547(19990701)57:1<39::AID-JNR5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 12.Foon KA, Wahl SM, Oppenheim JJ, Rosenstreich DL. Serotonin-induced production of a monocyte chemotactic factor by human peripheral blood leukocytes. J Immunol. 1976;117:1545–1552. [PubMed] [Google Scholar]
- 13.Gilchrist CA, Morrison CF, Chapman KE, Harmar AJ. Identification of nerve growth factor-responsive sequences within the 5′ region of the bovine preprotachykinin gene. DNA Cell Biol. 1991;10:743–749. doi: 10.1089/dna.1991.10.743. [DOI] [PubMed] [Google Scholar]
- 14.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 15.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 17.Guilbaud G, Benoist JM, Eschalier A, Kayser V, Gautron M, Attal N. Evidence for central phenomena participating in the changes of responses of ventrobasal thalamic neurons in arthritic rats. Brain Res. 1989;484:383–388. doi: 10.1016/0006-8993(89)90386-7. [DOI] [PubMed] [Google Scholar]
- 18.Itoh K, Ozaki M, Stevens B, Fields RD. Activity-dependent regulation of N-cadherin in DRG neurons: differential regulation of N-cadherin, NCAM, and L1 by distinct patterns of action potentials. J Neurobiol. 1997;33:735–748. doi: 10.1002/(sici)1097-4695(19971120)33:6<735::aid-neu3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 19.Keeney S, Linn S. A critical review of permeabilized cell systems for studying mammalian DNA repair. Mutat Res. 1990;236:239–252. doi: 10.1016/0921-8777(90)90008-s. [DOI] [PubMed] [Google Scholar]
- 20.Khasar SG, Lin Y-H, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C mutant mice. Neuron. 1999a;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999b;81:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- 22.Lepers A, Cacan R, Verbert A. Permeabilized cells as a way of gaining access to intracellular organelles: an approach to glycosylation reactions. Biochimie. 1990;72:1–5. doi: 10.1016/0300-9084(90)90166-e. [DOI] [PubMed] [Google Scholar]
- 23.Levine JD, Reichling DB. Peripheral mechanisms of inflammatory pain. In: Wall PD, Melzack R, Bonica JJ, editors. Textbook of pain. Churchill Livingston; New York: 1999. pp. 59–84. [Google Scholar]
- 24.Lockwood AH. Medical problems of musicians. N Engl J Med. 1989;320:221–227. doi: 10.1056/NEJM198901263200405. [DOI] [PubMed] [Google Scholar]
- 25.MacIntyre DL, Reid WD, McKenzie DC. Delayed muscle soreness. The inflammatory response to muscle injury and its clinical implications. Sports Med. 1995;20:24–40. doi: 10.2165/00007256-199520010-00003. [DOI] [PubMed] [Google Scholar]
- 26.Mannion RJ, Doubell TP, Coggeshall RE, Woolf CJ. Collateral sprouting of uninjured primary afferent A-fibers into the superficial dorsal horn of the adult rat spinal cord after topical capsaicin treatment to the sciatic nerve. J Neurosci. 1996;16:5189–5195. doi: 10.1523/JNEUROSCI.16-16-05189.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarson KE, Krause JE. NK-1 and NK-3 type tachykinin receptor mRNA expression in the rat spinal cord dorsal horn is increased during adjuvant or formalin-induced nociception. J Neurosci. 1994;14:712–720. doi: 10.1523/JNEUROSCI.14-02-00712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melhorn J. Cumulative trauma disorders and repetitive strain injuries. The future. Clin Orthop. 1998;351:107–126. [PubMed] [Google Scholar]
- 29.Mikhailov VV, Rusanova AG. The interrelationship of the catecholamine and protein content of the tissue of the submandibular salivary glands and the mucosa during the secretory cycle in chronic inflammation of the oral soft tissues. Biull Eksp Biol Med. 1993;116:472–474. [PubMed] [Google Scholar]
- 30.Nahin RL, Byers MR. Adjuvant-induced inflammation of rat paw is associated with altered calcitonin gene-related peptide immunoreactivity within cell bodies and peripheral endings of primary afferent neurons. J Comp Neurol. 1994;349:475–485. doi: 10.1002/cne.903490311. [DOI] [PubMed] [Google Scholar]
- 31.Schulz I. Permeabilizing cells: some methods and applications for the study of intracellular processes. Methods Enzymol. 1990;192:280–300. doi: 10.1016/0076-6879(90)92077-q. [DOI] [PubMed] [Google Scholar]
- 32.Taiwo YO, Levine JD. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience. 1990;38:757–762. doi: 10.1016/0306-4522(90)90068-f. [DOI] [PubMed] [Google Scholar]
- 33.Taiwo YO, Levine JD. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience. 1992;48:485–490. doi: 10.1016/0306-4522(92)90508-y. [DOI] [PubMed] [Google Scholar]
- 34.Tonra JR, Mendell LM. Effects of postnatal anti-NGF on the development of CGRP-IR neurons in the dorsal root ganglion. J Comp Neurol. 1998;392:489–498. [PubMed] [Google Scholar]
- 35.Villena C, Vivas JM, Villar AM. Ocular inflammation models by topical application: croton-oil induced uveitis. Curr Eye Res. 1999;18:3–9. doi: 10.1076/ceyr.18.1.3.5395. [DOI] [PubMed] [Google Scholar]
- 36.Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woolf CJ, Doubell TP. The pathophysiology of chronic pain–increased sensitivity to low threshold A beta-fibre inputs. Curr Opin Neurobiol. 1994;4:525–534. doi: 10.1016/0959-4388(94)90053-1. [DOI] [PubMed] [Google Scholar]