

Abstract
G-Protein receptor kinase 1 (GRK1) (“rhodopsin kinase”) is necessary for the inactivation of photoactivated rhodopsin, the light receptor of the G-protein transduction cascade of rod photoreceptors. GRK1 has also been reported to be present in retinal cones in which its function is unknown. To examine the role of GRK1 in retinal cone signaling pathways, we measured in mice having null mutations of GRK1 (GRK1 −/−) cone-driven electroretinographic (ERG) responses, including an a-wave component identified as the field potential generated by suppression of the circulating current of the cone photoreceptors. Dark-adapted GRK1 −/− animals generated cone-driven ERGs having saturating amplitudes and sensitivities in both visible and UV spectral regions similar to those of wild-type (WT) mice. However, after exposure to a bright conditioning flash, the cone-driven ERGs of GRK1 −/− animals recovered 30–50 times more slowly than those of WT mice and similarly slower than the cone-driven ERGs of mice homozygously null for arrestin (Arrestin −/−), whose cone (but not rod) response recoveries were found to be as rapid as those of WT. Our observations argue that GRK1 is essential for normal deactivation of murine cone phototransduction and provide the first functional evidence for a major role of a specific GRK in the inactivation of vertebrate cone phototransduction.
Keywords: G-protein coupled receptor, G-protein coupled receptor kinase, phototransduction, cone photoreceptors, photoresponse recovery kinetics, mouse genetics, electroretinography
The initial amplifier in visual transduction is a photoactivated state (R*) of a photopigment G-protein-coupled receptor (GPCR); in vertebrate rod photoreceptors, the GPCR is rhodopsin. Biochemical evidence has long supported the conclusion that inactivation of R* in vertebrate rod photoreceptors involves phosphorylation by the G-protein-coupled receptor kinase GRK1 (“rhodopsin kinase”) of one or more serine residues on the rhodopsin C terminus (Wilden et al., 1986; Kuhn and Wilden, 1987; Ohguro et al., 1996).
Proof that GRK1 binding to and/or phosphorylation of R* is required for normal inactivation of R* in rods in vivo has come from recordings of photocurrent responses of single rods of mice expressing mutant rhodopsin lacking the C terminus (Chen et al., 1995) and more recently of responses of rods from mice having null mutations in GRK1 (CK Chen et al., 1999).
The expression of GRK1 in human rod and cone photoreceptors has been documented (Zhao et al., 1998). Lack of functionally active GRK1 in humans has been shown to cause congenital night blindness (Oguchi disease) characterized by profoundly slowed rod dark adaptation (Yamamoto et al., 1997). A human patient with a homozygous null mutation in GRK1 has been shown to have a reliable but “slight slowing of cone deactivation kinetics” (Cideciyan et al., 1998) and, in evaluating the latter evidence, the hypothesis has been proposed that human cones might rely mainly on pigment regeneration for deactivation of the photoactivated pigment (Cideciyan et al., 1998).
To further examine the possible role of GRK1 in mammalian cone function, we have investigated the cone-driven responses of mice with the GRK1 gene inactivated (CK Chen et al., 1999). Wild-type (WT) mice and mice with the arrestin gene inactivated (Arrestin −/−) (Xu et al., 1997) were also investigated. Arrestin −/− mice serve as controls not only for the genetic inactivation of a retina-specific (and rod-specific) protein involved in R* inactivation but also for the adaptational state produced by the very strong and prolonged activation of the rods in the GRK1 −/− mice (relative to WT) during the experiments.
MATERIALS AND METHODS
All experimental procedures were done in compliance with National Institutes of Health guidelines, as approved by the respective Institutional Animal Care and Use Committees of the University of Pennsylvania and the California Institute of Technology. GRK1 −/− mice and Arrestin −/− were derived at the California Institute of Technology on C57BL/6-129/SvJ background as described previously (Xu et al., 1997; CK Chen et al., 1999). C57BL/6 (Charles River Laboratories, Wilmington, MA) mice were used to derive WT controls. All animals used for electroretinographic (ERG) recordings were born and maintained under controlled ambient illumination on a 12 hr light/dark cycle with the illumination level at 2.5 photopic lux as described previously (Pugh et al., 1998; Lyubarsky et al., 1999). ERG recordings were made when animals were between 8 and 12 weeks of age. Mice used for histological analysis were raised in complete darkness (CKChen et al., 1999).
In situ hybridization. Eyes of killed mice were enucleated, and eyecups were prepared by removal of the cornea and lens. The eyecups were fixed overnight at 4°C with 4% paraformaldehyde in PBS, pH 7.4, and then impregnated with 30% sucrose in PBS at 4°C for 8 hr. They were then embedded and frozen in OCT (Sakura Finetek, Torrance, CA). Frozen sections 15-μm-thick were cut and mounted on precleaned superfrost slides (Fisher Scientific, Houston, TX). The first 660 nucleotides of the murine GRK1 coding sequences was cloned into pGEM-T vector (Promega, Madison, WI) and served as a template for in vitro transcription. An SP6/T7 transcription kit and DIG RNA Labeling Mix (Boehringer Mannheim, Indianapolis, IN) were used to produce digoxigenin-labeled sense and antisense riboprobes of GRK1 according to the manufacturer's instructions. Sections were washed three times with PBS, 5 min each, before prehybridization in Hyb buffer [50% formamide, 5× SSC (0.9 m NaCl and 90 mm sodium citrate, pH 7.0), 1 mg/ml yeast RNA, 100 gm/ml heparin, 1× Denhardt's solution (0.1% w/v each Ficoll, polyvinylpyrrolidone, and bovine serum albumin), 0.1% Tween 20, 0.1% CHAPS, and 5 mm EDTA] at 65°C for 4–8 hours. Probes were used at a concentration of 100 ng/ml in Hyb buffer. Hybridization was performed at 65°C for 36–48 hr. After hybridization, the sections were washed in 5× SSC at 60°C for 5 min and then washed three times in 0.2× SSC at 60°C for a total of 60 min. After cooling to room temperature, the sections were washed with MAB buffer (0.1 m maleic acid, pH 7.5, and 0.15m NaCl) for 5 min and then blocked by 1% BLOCK (Boehringer Mannheim) in MAB buffer at room temperature for 2 hr. To detect the hybridized probes, alkaline phosphatase-conjugated anti-digoxigenin antibody (used at 1:5000 dilution; Boehringer Mannheim) was added to the sections and incubated at room temperature for 2 hr. The sections were then washed twice with MAB buffer, 30 min each, and washed once for 5 min with AP buffer (0.1 m Tris-HCl, pH 9.5, 50 mm MgCl2, and 100 mm NaCl). Sixty-seven microliters of nitroblue-tetrazolium-chloride (50 mg/ml; Promega) and 35 μl of 5-bromo-4-chloro-indolyl phosphate (50 mg/ml; Promega) were added to 10 ml of AP buffer, and the resulting solution was used to develop the sections immediately after the washes. Typical developing time for GRK1 was 12–24 hr. To stop the development, the sections were washed in TE buffer (10 mmTris-HCl, pH 7.5, and 10 mm EDTA) and sealed in 50% glycerol with coverslips.
Immunocytochemistry. Eyecups of mice raised in complete darkness were fixed with 4% paraformaldehyde at 4°C for 1 hr, impregnated with 30% sucrose in PBS, embedded, and frozen in OCT. Sections were cut 15-μm-thick and mounted on precleaned superfrost slides. The sections were washed once with PBS for 5 min before blocking by 1% BLOCK in MAB buffer for 1 hr at room temperature. GRK1-specific polyclonal antibody 8585 (generously provided by Dr. Robert Lefkowitz, Duke University, Durham, NC) was used at 1:100 dilution and biotin-labeled peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA) was used at 1:200 dilution in 1% BLOCK in MAB buffer. After blocking, the sections were incubated with 8585, PNA, or both at room temperature for 1 hr and then washed with PBS three times for a total of 15 min. Sections stained with 8585 were then incubated in dark at room temperature for 1 hr with fluorescein-conjugated goat antibody against rabbit IgG (1:100 dilution; Vector Laboratories). Sections stained with biotin-labeled PNA were incubated in dark at room temperature for 1 hr with Texas Red-conjugated streptavidin (used at 1:100 dilution; Life Technologies, Gaithersburg, MD). Sections stained with both 8585 and biotin-labeled PNA were incubated in darkness at room temperature with both the fluorescein-conjugated goat antibody against rabbit IgG and Texas Red-conjugated streptavidin. After incubation, sections were then washed three times with PBS for a total of 30 min and sealed in VECTASHIELD (Vector Laboratories) with coverslips. Micrographs were taken with an Axiovert 35 microscope (Zeiss, Oberkochen, Germany) equipped with incident-light fluorescence.
Electroretinography. The experimental apparatus, methods of light stimulation and quantification, electroretinogram (ERG) recording, and cone signal isolation have been described in detail previously (Lyubarsky and Pugh, 1996; Lyubarsky et al., 1999). In brief, mice that had been dark adapted overnight were anesthetized under dim red light with an intraperitoneal injection of a solution containing (in μg/gm body weight): 25 ketamine (KetaJect; Phoenix Pharmaceutical, Mountain View, CA), 10 xylazine (XylaJect; Phoenix Pharmaceutical), and 1000 urethane (Sigma, St. Louis, MO). The anesthetized animal was immobilized in a holder so that the right eye was pointing upward; the pupil of the right eye was then dilated with 1% tropicamide solution (Mydriacil; Alconox, New York, NY). Next, a drop of methylcellulose solution (Goniosol; Iolab Pharmaceutical, Indianapolis, IN) was placed on the eye for protection and electrical contact, and a recording platinum wire electrode was put into electrical contact with the cornea. A tungsten needle reference electrode was next inserted subcutaneously on the forehead. The holder with the animal was then placed inside a light-proof aluminum Faraday cage whose interior was lined with aluminum foil to maximize UV reflectivity (Lyubarsky et al., 1999). Light stimuli were delivered through several ports in the walls of the cage. The intensity of stimulation and its spectral composition were controlled with neutral density and bandpass interference filters. For the latter, the wavelength cited corresponds to the median of the filter spectral transmission function (Lyubarsky et al., 1999). Baffles against the ports were used to avoid direct illumination of the tested eye. At the position occupied by the mouse eye during experiments, directional variations of light intensity did not exceed ±15% of the average intensity.
ERGs were amplified, bandpass filtered at 0.1–1000 Hz, sampled at 5 kHz, averaged, and stored on a personal computer using a Digidata 1200 acquisition board and Axotape2 software (Axon Instruments, Foster City, CA). For some analyses, oscillatory potentials (Gorgels and Norren, 1992; Peachey et al., 1993;Lyubarsky et al., 1999) were removed by digital filtering with a gaussian filter having a bandwidth of 11 Hz (3 dB). We refer to the peak amplitude of the response filtered in this manner as the cone b-wave magnitude (bmax). Before experiments, mice were dark-adapted overnight and all the preparations were performed under dim red light. Before electrical recordings commenced animals were kept in the Faraday cage in complete darkness for at least 15 min. The overall duration of a recording session was 80–100 min.
Stimulus quantification: conversion of light stimuli to numbers of photoisomerizations and relative amounts of visual pigment isomerized in rods and cones. Previous work has established that, in addition to rhodopsin in rods, the mouse retina expresses two different cone visual pigments with λmax values near 360 and 510 nm in two types of cones having distinctive regional distributions over the retina (Jacobs et al., 1991; Szel et al., 1992; Calderone and Jacobs, 1995; Lyubarsky et al., 1999). We identify the two types of cones as the “M cones” and the “UV cones” after the dominant species of pigment in each.
For a monochromatic light stimulus isomerizing a small fraction (≪1) of pigment in a specific class of photoreceptor, the number (Φ) of photoisomerizations per photoreceptor produced by a light flash in a ganzfeld is proportional to the quantal flux density Q(λ) (photons μm−2, at the cornea):
| Equation 1 |
where Apupil and Aretina are the areas of the mouse pupil and retina, respectively, ηSC is a scale factor (<1) introduced to account for the Stiles–Crawford effect of the first kind (Stiles and Crawford, 1933; Snyder and Pask, 1973), aC(λ) is the collecting area at the retina for the specific photoreceptor class, and τ(λ) is the transmissivity of the prereceptor eye media. A standard expression for aC(λ) has been developed by Baylor et al. (1979, their Eq. 14) and was adopted previously with appropriate parameter values for the mouse (Lyubarsky and Pugh, 1996; Lyubarsky et al., 1999, their Eq. 5). In our previous presentation of Equation 1, the factor ηSCfor the Stiles–Crawford effect was not made explicit but was discussed as modifying the effective pupil area (Lyubarsky et al., 1999).
One can collapse the eye- and wavelength-dependent terms of Equation 1into two parameters: one for the efficacy of light at the wavelength of maximum absorbance (λmax) and a second accounting for the spectral sensitivity of the receptor, i.e., the sensitivity at the variable wavelength λ. Thus,
| Equation 2 |
where
| Equation 3 |
is the apparent collecting area of the specific photoreceptor type at the cornea in a ganzfeld for light of wavelength λ, and
| Equation 4 |
is the normalized spectral sensitivity at the cornea of the photoreceptor under consideration. A potential problem with these formulas is the neglect of the Stiles–Crawford effect of the second kind, i.e., the wavelength dependence of ηSC. This dependence is not known for mouse, but in experiments using very broadband flashes it can be expected that such dependence will have only a second-order effect on our calculations, relative to the SC-I effect for wavelengths near the cone λmax.
Because we used broadband (“white”) flash stimulation, the calculation of the number of photoisomerizations per flash must sum the effects of different wavelengths. Thus, Equation 2 must be modified as follows:
| Equation 5 |
where q(λ) is spectral density of quantal flux (photons μm−2 nm−1 at the cornea), measured as described by Lyubarsky et al. (1999) at the position of the mouse cornea in the recording chamber. We used Equation 5 with the measured values of the spectral flux density and with spectral sensitivities and parameter values as described in the succeeding paragraph to estimate Φ for all flashes used in the experiments reported here.
We will assume the following values and conventions for parameters in Equations 1-5, as explained previously (Lyubarsky and Pugh, 1996): collecting areas at the visual pigment λmax values at the retina aC(λmax), 1.3 μm2 for rods and 2.4 μm2 for cones; Apupil/ Aretina= 0.22 (fully dilated pupil) (Pennesi et al., 1998); ηSC = 1 for rods and 0.33 for cones; λmax of 498 nm for rods, and for cones, 355 and 508 nm, and normalized cone spectral sensitivities, S̄(λ), as described previously (Lyubarsky et al., 1999, their Fig. 6B). The prereceptor spectral transmissivity τ(λ) of the eye media for the mouse has not been measured. As an improved approximation for the transmissivity, we have used data on the spectral transmittance of the rat lens in the spectral range of 330–700 nm reported by Gorgels and Norren (1992) assuming that (1) the lens, which occupies most of the preretinal optical path, is the major prereceptor light absorber and (2) absorption by the lens in the mouse eye is half of its value in rat, because the size of the mouse eye is about half that of the rat (Remtulla and Hallett, 1985). With these assumptions, τ(λmax) is 0.78 at the peak of the UV pigment and 0.96 at the peak of the M pigment, and aC,cornea(λmax) are 0.14 and 0.17 μm2 for the UV and M cones, respectively.
The fraction of visual pigment isomerized, fisomerized, in each receptor class by a specified flash was computed from the estimated number of photoisomerizations Φ as follows:
| Equation 6 |
where Ntotal is the total number of pigment molecules per photoreceptor, C the is molar concentration of visual pigment in the outer segment (∼3.5 mm; see Harosi, 1975), NA is Avogadro's number (6.02 × 1023) and VOS is the envelope volume of the outer segment (liters). The volume used for mouse cone outer segments was based on the anatomical investigation ofCarter-Dawson and LaVail (1979): rod outer segments were assumed to be 25 μm long with uniform diameter of 1.8 μm; cone outer segments were assumed to have length of 13 μm and diameter of 1.5 μm at their base, tapering to 1.0 μm at their tips. Combining these factors, one obtains for rods VOS = 64 μm3, Ntotal = 1.3 × 108 and fisomerized = 7.7 × 10−9 Φ, and for cones, VOS = 16 μm3, Ntotal = 3.5 × 107and fisomerized = 2.9 × 10−8 Φ.
Recent histological evidence has been presented that both mouse cone visual pigments are coexpressed in many of the cones, in a manner that may vary with retinal location (Gloesman and Ahnelt, 1998), and electrophysiological data have provided support for the hypothesis that the M cones coexpress a small amount of UV pigment (Lyubarsky et al., 1999). Although coexpression of pigments presents a serious complication for stimulus quantification in experiments using monochromatic stimulation with wavelengths far from the pigment λmax, it is unlikely to greatly alter the estimation of the number of photoisomerizations per cone produced by the flashes used in the experiments reported here and will be neglected. The justifications for such neglect are that results to date indicate that the level of coexpression is relatively low, ∼3% for the UV pigment in the M cones (Lyubarsky et al., 1999) and that we used either narrowband stimulation near the λmax values of the cone pigments or a broadband white flash. The relative number of isomerizations from the coexpressed pigment are estimated to be negligible for either of these flash stimuli. Moreover, any error in estimation will be such that more pigment will be isomerized per cone per flash than we compute, and this will not differentially affect our comparison between the recoveries of the cone-driven ERGs of WT and mutant animals.
RESULTS
GRK1 is present in rods and cones of WT mice but absent in GRK1 −/−
Previous investigations have shown that GRK1 is present in both rod and cone photoreceptors of human, bovine, and chicken retinas (Palczewski et al., 1993; Zhao et al., 1998). Consistent with these previous results, we found GRK1 mRNA to be present in the photoreceptor inner segment layer in the retina of WT mice, demonstrated by the strong binding of an antisense probe (Fig. 1,middle); as expected, GRK1 mRNA was not detectable in GRK1 −/− retina (Fig. 1, right) nor was GRK1 message detectable in any other layer of WT retina (middle). Also, consistent with observations made in retinas of other species, we found GRK1 to be expressed in murine cones (Fig. 2). The localization of GRK1 in murine cones led us to examine the hypothesis that GRK1 plays a role in cone-driven signaling similar to that which it plays in rod signaling.
Fig. 1.

Localization of GRK1 mRNA expression in the retina. In situ hybridization was performed as described in Materials and Methods. Each panel is from a different retinal section of a mouse with genotype as labeled above the panel. The GRK1 antisense probe, which binds to the complementary message, strongly labels the layer corresponding to the photoreceptor inner segments of the WT mouse retina (GRK1 +/+, middle); antisense labeling is missing in the GRK1 −/− retina (right). The GRK1 sense probe serves as a control for nonspecific labeling (left). Scale bar, 30 μm. OS, Outer segment; IS, inner segment;ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer.
Fig. 2.

Localization GRK1 in rod and cone photoreceptor outer segments. Immunocytochemistry was performed as described in Materials and Methods on retinal sections from WT mice; 1–3show images made from a single section of WT retina and 4–6from a single section of GRK1 −/− retina. GRK1-specific antibody 8585 was raised in rabbit against the first 50 amino acids of bovine GRK1; its binding was visualized with fluorescein-conjugated goat anti-rabbit antibody (1, 3, 4, 6). Biotin-conjugated PNA, which binds to cone membranes, was used to localize cone photoreceptors; its binding was visualized by conjugation with Texas Red-conjugated streptavidin (2, 3, 5, 6). The outer segment layer of the retina of WT mice is strongly labeled with 8585 antibody (1); WT cones seen to be labeled with PNA in 2are also clearly labeled with 8585 (arrows in1–3). Both rod and cone outer segments of GRK1 −/− mouse are negative for GRK1 (4). Scale bar, 30 μm. Layer abbreviations are given in Figure 1.
Rods of fully dark-adapted GRK1 −/− mice generate nearly normal circulating currents in situ but are very slow to recover from strong light stimuli
Figure 3A shows ERGs elicited from a WT, a GRK1 −/−, and an Arrestin −/− mouse with a flash estimated to isomerize ∼1% of the rhodopsin. Tracesa–c were obtained when the animals were fully dark-adapted. The initial corneal-negative component of these traces is the a-wave; the immediately following positive-going potential is a mixture of the b-wave and oscillatory potentials. As illustrated schematically in Figure 3B, the a-wave is the transient field potential generated by suppression of photoreceptor circulating currents: the amplitude of the a-wave in response to such intense flashes is directly proportional to the photoreceptor circulating current present immediately before the flash, and in WT mice, more than 95% of this amplitude is from suppression of rod circulating current (Lyubarsky and Pugh, 1996; Pugh et al., 1998; Lyubarsky et al., 1999). The amplitude of the initial a-wave of the GRK1 −/− animal (215 μV) is 63% that of WT (342 μV) and 77% of that of the Arrestin −/− mouse (280 μV), indicating somewhat diminished rod circulating current for both mutant strains under our experimental conditions. Nonetheless, these a-wave responses show that the rods of dark-adapted GRK1 −/− and Arrestin −/− animals generate circulating currents of near normal magnitude and that the activation phase of the rod phototransduction cascade in them is unaffected by the null mutation (Xu et al., 1997; CK Chen et al., 1999). The diminished a-wave magnitudes of the GRK1 −/− and Arrestin −/− mice in Figure 3may have arisen from shortened outer segments (J Chen et al., 1999) or from incomplete recovery of the rod circulating current after the exposure to red light during the anesthetization and placement of the corneal electrode. (The necessarily limited period of anesthesia, coupled with the experimental design of the present experiments, aimed at testing cone function, precluded examination of the nature of the differences between animals in the amplitudes of the rod a-wave to the initial saturating flash delivered in this investigation.)
Fig. 3.

ERGs (transretinal field potentials) from WT, GRK1 −/−, and Arrestin −/− mice, and cellular basis of the a- and b-waves of the ERG. A, A white flash isomerizing ∼1% of the rhodopsin in the retina was delivered in a ganzfeld to each dark-adapted animal, generating responses a–c; the response to the same flash was then recorded again after 2 min in darkness for the WT (d), after 15 min for the GRK1 −/− (e), and after 20 min for the Arrestin −/− (f) mouse. The initial corneal-negative component clearly seen ina–d is the a-wave, and the corneal-positive deflections that follow (and truncate) the a-wave are a mixture of rod- and cone-driven b-waves and the so-called oscillatory potentials (Gorgels and Norren, 1992). B, In the dark, the circulating currents of the rods and cones (arrows atbottom left) flow in the extracellular spaces of the outer nuclear layer (ONL), inner segment (IS), and outer segment (OS) layers toward the receptor outer segment tips, creating a vitreal- (and thus corneal-) positive transretinal field potential (Hagins et al., 1970) represented by the + and − symbols near the worddark. An intense ganzfeld flash of light initially completely suppresses the receptor circulating currents; the consequent collapse of their field potential generates the vitreal-negative-going a-wave, recordable in diminished magnitude but unaltered kinetics at the cornea (Hagins et al., 1970; Hood and Birch, 1995; Cideciyan and Jacobson, 1996; Smith and Lamb, 1997; Pugh et al., 1998). The suppression of their circulating currents hyperpolarizes the photoreceptors, diminishing their glutamate release at their synapses, leading to the opening of nonspecific cation channels in the dendrites of ON bipolar cells [two of which are shown spanning the inner nuclear layer (INL)]. Thus, a strong light exposure causes ON bipolar cells to generate circulating currents that flow in the inner plexiform layer (IPL) and inner nuclear layer toward cationic sinks in the outer plexiform layer (OPL); the consequent, vitreal-positive field potentials (symbolized by the + and −symbols near the word light) are now understood to underlie the b-waves (for review, see Pugh et al., 1998). The oscillatory potentials have been hypothesized to originate in a feedback circuit that involves certain amacrine cells (Wachmeister, 1998). The retinal schematic is modified from Dowling and Boycott (1966); the proper layer thicknesses of the mouse retina are seen in Figure 1.
In contrast to the modest diminution of its saturated, dark-adapted amplitude, the recovery time course of the a-wave amplitude of GRK1 −/− and Arrestin −/− mice was greatly retarded relative to that in WT. In the experiments of Figure 3, after exposure to the initial flash, the mice were left to dark adapt and then restimulated with the same flash (traces d–f). Whereas the ERG from the WT mouse shows a completely recovered a-wave (and thus rod circulating current) after 2 min, those of the GRK1 −/− and Arrestin −/− mice exhibit only very small a-waves (∼10 μV) and a substantially reduced-amplitude b-wave after 15 and 20 min of dark adaptation, respectively. The absence of all but a small fraction of the initial a-wave in the GRK1 −/− and Arrestin −/− mutants shows that the recovery phase of the rod transduction cascade has been greatly slowed, consistent with the hypotheses that GRK1 and Arrestin are essential for normal inactivation of photoactivated rhodopsin (Wilden et al., 1986; Kuhn and Wilden, 1987; Palczewski et al., 1993; Chen et al., 1995; Ohguro et al., 1996; Xu et al., 1997; Yamamoto et al., 1997; Zhao et al., 1998;Cideciyan et al., 1998; CK Chen et al., 1999).
Previous investigations of murine ERG responses obtained when the a-wave is strongly suppressed by intense flashes or steady “rod-saturating” backgrounds has led to the conclusion that the responses obtained under such conditions are driven by signals originating in cones (Peachey et al., 1993; Pugh et al., 1998; Lyubarsky et al., 1999). Based on these previous observations, we attribute responses of the GRK1 −/− and Arrestin −/− mice in Figure 3A (e, f) to the activity of cone-driven retinal neurons and proceed to characterize these responses.
Both UV and M cone-driven retinal responses are functional in GRK1 −/− and Arrestin −/− mice
WT mice have both midwave (M)-sensitive (λmax≈ 510 nm) and UV-sensitive cones (λmax ≈ 355 nm) (Jacobs et al., 1991; Calderone and Jacobs, 1995; Lyubarsky et al., 1999), and so we inquired whether the sensitivity of the cone-driven responses of GRK1 −/− and Arrestin −/− animals are comparable with those of WT mice. Figure 4A presents families of ERGs elicited from a GRK1 −/− mouse by monochromatic 361 and 513 nm flashes of increasing intensity. The saturating magnitudes of the cone-driven response components were similar for WT, GRK −/−, and Arrestin −/− mice (Table 1), indicating that cones and cone-driven secondary neurons (Fig.2B) are present in normal numbers and are functional. Intensity–response relationships derived from the data of Figure4A are shown in 4B(filled circles), together with data from three additional GRK1 −/− and four Arrestin −/− mice; from such data, the ratio of peak spectral sensitivities,S̄UV/S̄M, of the two cone types at the cornea can be derived (Lyubarsky et al., 1999). The sensitivity ratios so derived were also similar among the different animals (Fig. 4C; Table 1). Generally, the absolute sensitivities exhibited greater variability than their ratios; possible reasons for this include inter-animal variability in transparency of eye tissues and pupil size.
Fig. 4.

A, Response families of cone-driven b-waves for 361 and 513 nm flashes for a GRK1 −/− mouse; each trace is the average of three to five individual responses. The 361 nm flash intensities were (from lowest to highest intensity) 740, 1400, 4300, 7200, and 13,500 photons μm−2 at the cornea (estimated to produce from 104 to 1900 photoisomerizations in the UV cones), and the 513 nm intensities were 2100, 4200, 8800, 21,500, and 90,000 photons μm−2 at the cornea (estimated to produce 360 to 15,000 photoisomerizations in the M cones). Thetopmost trace in the 361 nm column is the response to a “white” saturating flash isomerizing ∼1.2% of the “green” and 0.09% of the UV cone pigment. B, Amplitude versus intensity data for GRK1 −/− mice (filled symbols, n = 4) and Arrestin −/− mice (open symbols, n = 4) obtained with flashes of 361 nm (symbols including dots) and 513 nm (not dotted), under cone-isolation conditions, as in A. Eachsymbol represents the normalized peak amplitude of a cone-driven b-wave response (points derived from the responses illustrated in A are shown as filled orfilled + dotted circles); peak amplitudes were measured after filtering responses at 11 Hz to remove oscillations (see Materials and Methods). The peak amplitudes were normalized by dividing them by the saturating amplitude, obtained in response to the white flash (W on abscissa). The flash intensities for each animal's data were scaled by a single, common factor; this factor was the intensity at 513 nm (I513) estimated by linear interpolation to produce a response of 20% saturated amplitude (dashed line). Two saturation functions (unbroken lines), having the form rpeak/rmax = 1 − exp (−S̄λĪλ) have been plotted through the data, where Iλ is the scaled flash intensity and S̄λ is a wavelength-dependent sensitivity factor; the black curve was arranged to intercept the dashed line at the abscissa value 1.0; thegray curve is shifted left by the average relative sensitivity of cone-driven responses of WT mice to these two wavelengths, i.e., by the factorS̄UV/S̄M= 5.2 (Table 1). C, Spectral sensitivity of cone-isolated b-wave responses of GRK1 −/− (filled circles) and Arrestin −/− (open circles) compared with WT. For each GRK1 −/− and Arrestin −/− animal, the lateral shift (in logarithmic units) between the two saturation functions best fitting the 513 and 361 nm data in B was measured; the points plotted at ∼361 nm are the mean ± SD of these shifts (the open symbols have been shifted laterally for clarity). The theoretical spectra give the spectral sensitivities of the UV and M cone-driven b-wave responses of WT mice and are replotted without alteration from Figure 6 of Lyubarsky et al. (1999).
Table 1.
Parameters of cone-driven electroretinograms of WT, GRK1 −/−, and Arrestin −/− mice
| Parameter | amax | bmax | S̄M | S̄UV | S̄UV/S̄M |
|---|---|---|---|---|---|
| Unit | μV | μV | 10−5 (photon μm2)−1 | 10−5(photon μm2)−1 | |
| WT (C57BL/6) | 12.6 ± 5.1 (21 ± 10)* | 100 ± 36 | 3.0 ± 2.0 | 17 ± 12 | 5.2 ± 1.8 |
| n | 4 (9)* | 11 | 7 | 6 | 6 |
| GRK1 −/− | 10.0 ± 5.1 | 97 ± 40 | 5.8 ± 1.9 | 20 ± 4.4 | 3.8 ± 1.6 |
| n | 11 | 13 | 5 | 5 | 5 |
| Arrestin −/− | 9.1 ± 5.6 | 129 ± 13 | 5.4 ± 3.1 | 14.5 ± 9 | 3.7 ± 1.3 |
| n | 14 | 6 | 6 | 6 | 6 |
The top row identifies a measured or computed ERG parameter. amax is the saturating amplitude of the a-wave component of the ERG (Fig. 6), and bmax the saturating amplitudes of the gaussian-filtered b-wave, obtained under cone-isolation conditions (Fig. 4A).S̄M and S̄UVare the relative sensitivities of the cone-driven b-wave in the midwave and UV spectral regions, derived as follows. The peak amplitudes of gaussian-filtered cone b-wave responses were obtained in the linear region of the amplitude versus intensity function and were normalized by the amplitude of the saturating response (Fig. 4B);S̄M was calculated as the average of all data obtained with 492, 501, and 513 nm flashes, andS̄UV is the mean of all data obtained with 346, 356, and 361 nm flashes.S̄UV/S̄M is the ratio, determined separately for each animal. The errors given are SDs. *The observations in parentheses are taken from Lyubarsky et al. (1999) and were obtained in the presence of a steady background estimated to produce 6000 photoisomerizations rod−1sec−1 and shown to suppress at least 95% of the rod circulating current. To obviate a possible residual contribution from the rod a-wave that may have contaminated this previous estimate of the cone amax in some mice, the measurements of the cone a-waves of WT mice reported in this table (n = 4) were all obtained in a double-flash paradigm, which measures the cone response a few seconds after the rod a-wave is suppressed with a very intense flash (Lyubarsky et al., 1999) and which takes advantage of the very rapid recovery of the cone a-wave (Figs. 6,7).
A distinctive feature of cone-driven ERGs in GRK1 −/− mice was the exaggeration and tight synchronization of the oscillatory potentials. In some records after a single saturating flash, we observed hundreds of oscillations, even after averaging; in WT mice, the oscillations in the responses to comparable flashes typically damp out after no more than a half of a dozen cycles (Peachey et al., 1993;Lyubarsky et al., 1999). The oscillatory potentials are generally associated (but not exclusively) with the light-adapted, cone-driven retina (Peachey et al., 1993) and are thought to be generated by a type of inner retinal neurons, such as amacrines with dendritic fields radially extended in the retina or by a multicellular feedback loop (Wachmeister, 1998). We suggest that the most likely basis of the exaggerated phenotype in the GRK1 −/− mice is the strongly and persistently adapted state of the retina during the experiments rather than a “loss of function” of GRK1 in an inner retinal cell, because no GRK1 message or protein has been identified outside the photoreceptor layer (Figs. 1, 2). An interesting alternative hypothesis is that some “miswiring” of the retina may have occurred during development (Banin et al., 1999).
Cone-driven responses of GRK1 −/− animals have profoundly slowed recovery from strong stimulation
Because GRK1 is involved in the inactivation of R* in rods but is also expressed in cones, we investigated the possibility that cone-driven responses recover from strong stimulation more slowly in GRK1 −/− mice than in WT or Arrestin −/− controls. The paradigm used to investigate cone-driven response recovery is illustrated in Figure 5. A conditioning flash sufficiently intense to temporarily suppress all cone-driven responses was followed at different interstimulus intervals (ISIs) by a probe flash of the same intensity. Recovery of the cone-driven responses of the WT and Arrestin −/− mice was complete in about 1 sec (Fig.5A,C); in contrast, for the GRK1 −/− mouse, the first sign of recovery appeared only after 5 sec, and complete recovery required more than 50 sec (Fig. 5B).
Fig. 5.

Recovery of cone-driven ERGs after a conditioning flash in WT, GRK1 −/−, and Arrestin −/− mice. White probe flashes isomerizing ∼1.2% of the M cone pigment and ∼0.09% of the UV cone pigment were delivered after a conditioning flash isomerizing ∼1% of the M and ∼0.06% of the UV pigment at ISIs specified in seconds to the left of the traces. Responses obtained without immediately preceding conditioning flashes are marked as Control. The upward pointing arrowsfor the bottommost traces in each panel show the time of the flash (a time gap of 3–5 msec containing a flash artifact has been omitted from some traces); the downward pointing arrows on the topmost traces indicate the times when the first four test flashes were delivered for the WT and Arrestin −/− mice.A, Recovery in WT mouse. The control record was obtained with an orange (λ > 530 nm) steady background that produced ∼6000 photoisomerizations rod−1sec−1, suppressing rod signals (Lyubarsky et al., 1999); for all other recordings, rod activity was suppressed with the conditioning flash. Each trace is the average of 10 records. B, Recovery in GRK1 −/− mouse; eachtrace is the average of five measurements. C, Recovery in Arrestin −/− mouse; each trace is the average of 15 records.
Because the corneal-positive component of the cone-isolated ERG represents field potentials generated mostly by second-order neurons (Fig. 3B) and because GRK1 is expressed in cones, a question of special interest is whether the slowed recovery was caused by prolonged activation of the cones themselves or only by prolonged activity in neurons downstream from the photoreceptors. To address this question, we examined the behavior of the initial corneal-negative component of the ERG observed under cone-isolation conditions (Fig. 5); this small component exhibits properties, including its electrical sign, its magnitude, and its activation kinetics in response to intense flashes, consistent with origination in the suppression of cone photoreceptor circulating current (Lyubarsky et al., 1999). Indeed, in human subjects, the homologous but appropriately larger ERG component has been identified as originating in the suppression of cone circulating current (Hood and Birch, 1995; Cideciyan and Jacobson, 1996; Smith and Lamb, 1997). Thus, in Figure6, the initial portions of records from mice stimulated as in Figure 5 have been replotted on expanded scales to facilitate examination of the cone a-wave; here, the differences between the animals in the recoveries of the cone a-waves are seen to parallel the recoveries of the cone-driven b-wave (although the oscillatory potentials interfere with extraction of the cone a-wave from records of WT and Arrestin −/− mice in the first 200–300 msec after the conditioning flash).
Fig. 6.

Recovery of the a-wave component of the mouse ERG under cone isolation conditions for WT, GRK1 −/−, and Arrestin −/− mice. The format of presentation is the same as in Figure 5, except that the time base and amplitude scales have been expanded to reveal the initial 25 msec of the records. In each panel, the portion of the traces identified with the suppression of the cone circulating current has been emphasized bythickening of the trace. The traces of the WT and Arrestin −/− mice are the same as those shown in Figure 5; the data of the GRK1 −/− mouse were taken from a different animal than those of Figure 5B, obtained in an experiment engineered to minimize the flash artifact. Nonetheless, for the larger artifact, the slowed recovery of the cone a-wave of the GRK1 −/− mouse can also be seen in Figure 5B. (In this and in other figures, a 3.5 msec segment of the recorded traceimmediately after the flash trigger has been excised; this segment contains the flash artifact, which is caused largely by a difficult-to-eliminate magnetic interference effect.)
In Figure 7, we summarize the results of the experiment of Figures 5 and 6 and present data obtained with the same experimental protocol from additional WT, GRK1 −/−, and Arrestin −/− mice. Figure 7A plots the saturated amplitude of the cone-driven b-wave response relative to its baseline amplitude, as a function of time after the conditioning flash. Taking the average time to 50% b-wave recovery for WT and Arrestin −/− mice to be 0.4 sec, the cone b-wave of the GRK1 −/− mice is seen to recover 20–70 times more slowly. Figure 7B plots the recovery of the saturating cone a-wave on a common abscissa with A. Again, using the recovery to 50% amplitude as a benchmark, the cone a-waves of the GRK1 −/− mice are seen to recover 40–100 times more slowly than those of WT and Arrestin −/− mice.
Fig. 7.
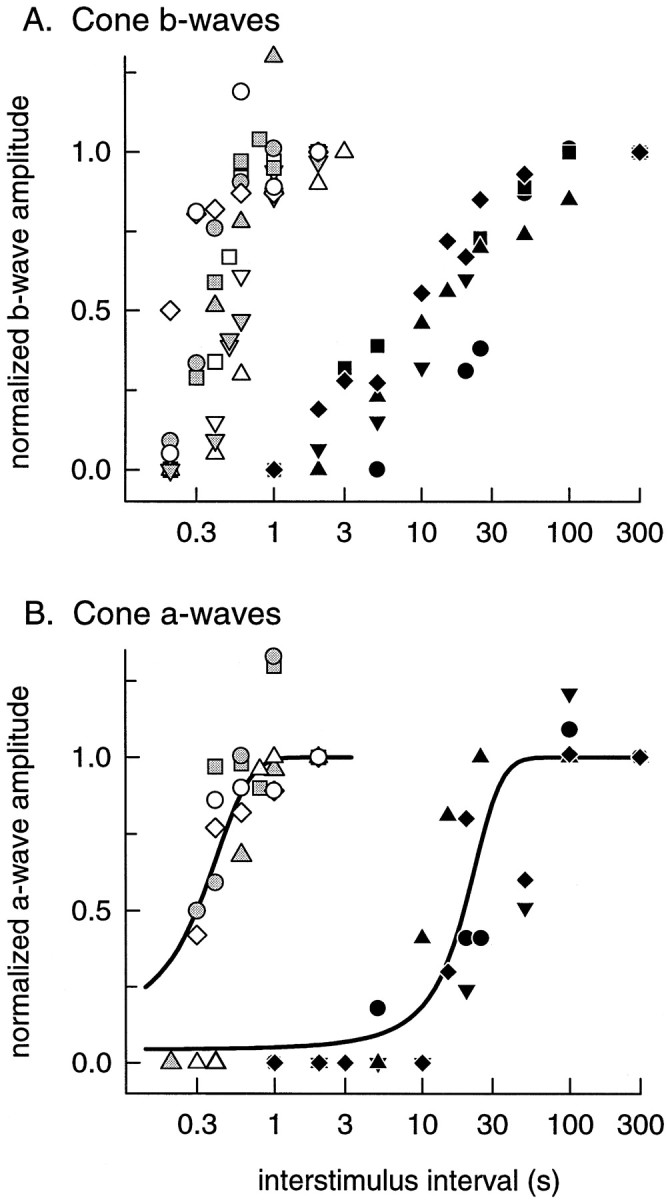
Time course of recovery of cone b-waves and cone a-waves from the conditioning flash. A, Normalized amplitudes of cone-driven b-waves for WT (open symbols, n = 5) , Arrestin −/− (gray-filled symbols, n = 4) , and GRK1 −/− mice (filled symbols, n = 5) plotted as function of the time (interstimulus interval) between the conditioning and probe flashes. B, Normalized amplitudes of cone a-waves plotted versus the interstimulus interval. The points extracted from the experiment of Figure 6 are plotted as circles in each case. All data were obtained with the experimental protocol illustrated in Figures 5 and 6; the same symbols are used inA and B for the data of the same animal. The curves plotted through the cone a-wave data have the form amax(t)/amax(∞) = 1/[1 + exp (−(t − t0)/τ], where amax is the saturated cone a-wave amplitude, and t0 = 0.3 sec and τ = 0.15 sec for the WT and Arrestin −/− data, and t0 = 19 sec and τ = 6.2 sec for the GRK1 −/− data; these curves are equivalent in form to that used by Thomas and Lamb (1999) to characterize the recovery of the human rod a-wave after a bleaching exposure. The normalization in each panel was based on the amplitude of the responses at ISIs of 2 or 3 sec for WT and Arrestin −/− and at 300 sec for GRK1 −/−. Note that the time axis is in logarithmic units.
DISCUSSION
Observations made with the rod a-wave in this investigation (Fig.3A) confirm previous findings (Yamamoto et al., 1997; Cideciyan et al., 1998; CK Chen et al., 1999) demonstrating that lack of GRK1 causes severe retardation of the recovery of retinal rod responses after intense stimulation, consistent with the hypothesis that GRK1 is essential for the normal inactivation of photoactivated rhodopsin (Wilden et al., 1986; Kuhn and Wilden, 1987;Palczewski et al., 1993; Chen et al., 1995; Ohguro et al., 1996; Xu et al., 1997; Yamamoto et al., 1997; Cideciyan et al., 1998; Zhao et al., 1998; CK Chen et al., 1999).
Our results make a strong case that GRK1 −/− mice have a specific defect in the inactivation of cone phototransduction, as follows. First, besides its expression in mammalian rods, GRK1 has only been found to be present in cones (Zhao et al., 1998); consistent with this general finding, the antisense probe generates no signal in the inner nuclear layer (Fig. 1), as would be expected, for example, were GRK1 present in cone ON bipolars and playing a role in their mGluR6 cascade (Masu et al., 1995). Thus, the cone b-wave recoveries of the GRK1 −/− mutants are not retarded because of a defect in the G-protein signaling cascade of the cone ON bipolars, but rather these slowed recoveries point to a defect in the cone photoreceptors that drive them. Second, the greatly slowed recoveries of the a-wave component recorded under cone isolation conditions provide specific functional evidence for a defect in the inactivation of cone transduction. The evidence is compelling because the ERG component isolated in Figure 6 (thickened portion of traces) is the murine cone a-wave, i.e., represents the suppression of the circulating current of the cones. Specifically, as presented previously (Lyubarsky et al., 1999), this component has the sign, magnitude, and activation kinetics expected from human ERG studies (Hood and Birch, 1995; Cideciyan and Jacobson, 1996; Smith and Lamb, 1997) and from microelectrode recordings from single mammalian cones (Schneeweis and Schnapf, 1995) for generation in the suppression of cone circulating current. Thus, the greatly slowed recovery of the cone-isolated a-wave in GRK1 −/− mutants (Figs. 6,7B) shows that the circulating currents of the cones of the mutant mice recover much more slowly from the conditioning flash than do the cone circulating currents of WT mice or those of mice lacking arrestin, which is necessary for normal rod inactivation (Xu et al., 1997) (Fig. 3A, c, f).
The very rapid recovery of the murine cone circulating current in WT and Arrestin −/− mice (Figs. 6, 7B) after a flash estimated to isomerize a few percent of the cone photopigments serves to underscore a feature of cones that differentiates them fundamentally from rods; assuming that the isomerized cone pigment is not regenerated in 1 sec, these results confirm that cones function well with amounts of bleached pigment that can suppress the circulating currents of rods. Moreover, the rapid recoveries in WT and Arrestin −/− mice also show that the flash intensities used in the experiments reported here are by no means nonphysiological for cones. Similarly rapid recoveries of human cone a-waves under comparable stimulation conditions have been reported recently (Mahroo et al., 1999).
Although the conclusion that GRK1 plays a critical role in the inactivation of murine cone phototransduction may seem surprising in view of evidence for cone-specific GRKs (Weiss et al., 1998), it bears emphasis that in only one previous investigation (Cideciyan et al., 1998) has any functional evidence been presented that any GRK plays a role in cone phototransduction. Although our results are thus not in qualitative conflict with the histological evidence for cone-specific GRKs, they nonetheless appear at odds quantitatively with the report ofCideciyan et al. (1998), who found a reliable, but only “slight slowing of cone deactivation kinetics” (measured with cone-isolated a-waves) in a human patient with a deletion of exon 5 of the GRK1 gene. What might account for the quantitative discrepancy between the human data and those reported here from mice? As possible explanations, we offer the following three hypotheses. First, the loss of exon 5 of human GRK1 gene may not constitute a functionally null mutation. Although it was demonstrated that GRK1 with an exon 5 deletion does not phosphorylate rhodopsin (Cideciyan et al., 1998), the data also show that a truncated GRK1 protein is expressed and that it remains in the cell without rapid degradation. Thus, it remains possible that a functional domain encoded in exons 1–4 can fold properly and participate in the recovery of cone phototransduction, perhaps by binding to cone-opsin rather than phosphorylating it. It has been shown in vitro that binding of GRK1 to photoactivated rhodopsin competitively blocks the activation of transducin (Pulvermuller et al., 1993). Second, it is possible that the loss of GRK1 in human cones can be compensated for by another GRK (Hisatomi et al., 1998; Weiss et al., 1998). Third, the recovery of cone phototransduction in human cones could depend primarily on the regeneration of cone pigments and not on interaction with GRK1. Clearly, our results reject the third hypothesis as it applies to mice, i.e., reject the hypothesis that independent of GRK1, regeneration of isomerized pigments in mouse cones under our stimulation conditions is sufficient to rapidly inactivate cone-opsin. Additional experiments are being planned to test the first and second hypotheses.
Footnotes
Correspondence should be addressed to Prof. Edward N. Pugh Jr., F. M. Kirby Center for Molecular Ophthalmology, Department of Ophthalmology, University of Pennsylvania School of Medicine, Stellar-Chance Building, Room 309B, 422 Curie Boulevard, Philadelphia, PA 19104-6069. E-mail: pugh@mail.med.upenn.edu.
Drs. Lyubarsky and Chen contributed equally to this work.
This work was supported by grants from the National Eye Institute.
REFERENCES
- 1.Baylor DA, Lamb TD, Yau K-W. The membrane current of single rod outer segments. J Physiol (Lond) 1979;288:589–611. [PMC free article] [PubMed] [Google Scholar]
- 2.Banin E, Cideciyan AV, Aleman TS, Petters RM, Wong F, Milam AH, Jacobson SG. Retinal rod photoreceptor-specific gene mutation perturbs cone pathway development. Neuron. 1999;23:549–557. doi: 10.1016/s0896-6273(00)80807-7. [DOI] [PubMed] [Google Scholar]
- 3.Calderone JB, Jacobs GH. Regional variations in the relative sensitivity to UV light in the mouse retina. Vis Neurosci. 1995;12:463–468. doi: 10.1017/s0952523800008361. [DOI] [PubMed] [Google Scholar]
- 4.Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 5.Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Simon MI, Matthes MT, Yasumura D, LaVail MM. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- 8.Cideciyan AV, Jacobson SG. An alternative phototransduction model for human rod and cone ERG a-waves: normal parameters and variation with age. Vision Res. 1996;36:2609–2621. doi: 10.1016/0042-6989(95)00327-4. [DOI] [PubMed] [Google Scholar]
- 9.Cideciyan AV, Zhao X, Nielsen L, Khani SC, Jacobson SG, Palczewski K. Null mutation in the rhodopsin kinase gene slows recovery kinetics of rod and cone phototransduction in man. Proc Natl Acad Sci USA. 1998;95:328–333. doi: 10.1073/pnas.95.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowling JE, Boycott BB. Organization of the primate retina: electron microscopy. Proc R Soc B Biol Sci. 1966;166:80–111. doi: 10.1098/rspb.1966.0086. [DOI] [PubMed] [Google Scholar]
- 11.Gloesman M, Ahnelt PK. Coexpression of M- and S-opsin extends over entire inferior mouse retina. Invest Ophthalmol Vis Sci. 1998;39:S1059. [Google Scholar]
- 12.Gorgels TGMF, Norren DV. Spectral transmittance of the rat lens. Vision Res. 1992;32:1509–1512. doi: 10.1016/0042-6989(92)90206-x. [DOI] [PubMed] [Google Scholar]
- 13.Hagins WA, Penn RD, Yoshikami S. Dark current and photocurrent in retinal rods. Biophys J. 1970;10:380–412. doi: 10.1016/S0006-3495(70)86308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harosi FI. Absorption spectra and linear dichroism of some amphibian photoreceptors. J Gen Physiol. 1975;66:357–382. doi: 10.1085/jgp.66.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hisatomi O, Matsuda S, Satoh T, Kotaka S, Imanishi Y, Tokunaga F. A novel subtype of G-protein-coupled receptor kinase, GRK7, in teleost cone photoreceptors. FEBS Lett. 1998;424:159–164. doi: 10.1016/s0014-5793(98)00162-8. [DOI] [PubMed] [Google Scholar]
- 16.Hood DC, Birch DG. Phototransduction in human cones measured using the alpha-wave of the ERG. Vision Res. 1995;35:2801–2810. doi: 10.1016/0042-6989(95)00034-w. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs GH, Neitz J, Deegan JF., II Retinal receptors in rodents maximally sensitive to ultraviolet light. Nature. 1991;353:655–656. doi: 10.1038/353655a0. [DOI] [PubMed] [Google Scholar]
- 18.Kuhn H, Wilden U. Deactivation of photoactivated rhodopsin by rhodopsin-kinase and arrestin. J Recept Res. 1987;7:283–298. doi: 10.3109/10799898709054990. [DOI] [PubMed] [Google Scholar]
- 19.Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN., Jr UV- and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahroo OAR, Paupoo AAV, Friedburg C, Lamb TD. Recovery of human cone photoreceptors following bleaching exposures. J Physiol (Lond) 1999;420:44P. [Google Scholar]
- 22.Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R, Takada M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995;80:757–765. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- 23.Ohguro H, Rudnicka-Nawrot M, Buczylko J, Zhao X, Taylor JA, Walsh KA, Palczewski K. Structural and enzymatic aspects of rhodopsin phosphorylation. J Biol Chem. 1996;271:5215–5224. doi: 10.1074/jbc.271.9.5215. [DOI] [PubMed] [Google Scholar]
- 24.Palczewski K, Buczylko J, Lebioda L, Crabb JW, Polans AS. Identification of the N-terminal region in rhodopsin kinase involved in its interaction with rhodopsin. J Biol Chem. 1993;268:6004–6013. [PubMed] [Google Scholar]
- 25.Peachey NS, Goto Y, al-Ubaidi MR, Naash MI. Properties of the mouse cone-mediated electroretinogram during light adaptation. Neurosci Lett. 1993;162:9–11. doi: 10.1016/0304-3940(93)90547-x. [DOI] [PubMed] [Google Scholar]
- 26.Pennisi ME, Lyubarsky AL, Pugh EN., Jr Extreme responsiveness of the pupil of the dark-adapted mouse to steady retinal illumination. Invest Ophthalmol Vis Sci. 1998;39:2148–2156. [PubMed] [Google Scholar]
- 27.Pugh EN, Jr, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram and the effect of age and light-rearing history on the magnitude of these components. In: Williams T, Thistle A, editors. Photostasis and related phenomena. Plenum; New York: 1998. pp. 93–128. [Google Scholar]
- 28.Pulvermuller A, Palczewski K, Hofmann KP. Interaction between photoactivated rhodopsin and its kinase: stability and kinetics of complex formation. Biochemistry. 1993;32:14082–14088. doi: 10.1021/bi00214a002. [DOI] [PubMed] [Google Scholar]
- 29.Remtulla S, Hallett PE. A schematic eye for the mouse, and comparisons with the rat. Vision Res. 1985;25:21–31. doi: 10.1016/0042-6989(85)90076-8. [DOI] [PubMed] [Google Scholar]
- 30.Schneeweis DM, Schnapf JL. Photovoltage of rods and cones in the macaque retina. Science. 1995;268:1053–1056. doi: 10.1126/science.7754386. [DOI] [PubMed] [Google Scholar]
- 31.Smith NP, Lamb TD. The a-wave of the human electroretinogram recorded with a minimally invasive technique. Vision Res. 1997;37:2943–2952. doi: 10.1016/s0042-6989(97)00094-1. [DOI] [PubMed] [Google Scholar]
- 32.Snyder AW, Pask C. The Stiles–Crawford effect: explanation and consequences. Vision Res. 1973;13:1115–1137. doi: 10.1016/0042-6989(73)90148-x. [DOI] [PubMed] [Google Scholar]
- 33.Stiles WS, Crawford BH. The luminous efficiency of rays entering the pupil at different points. Proc R Soc Lond B Biol Sci. 1933;112:428–450. [Google Scholar]
- 34.Szel A, Rohlich P, Caffe AR, Juliusson B, Aguirre G, van Veen T. Unique topographic separation of two spectral classes of cones in the mouse retina. J Comp Neurol. 1992;325:327–342. doi: 10.1002/cne.903250302. [DOI] [PubMed] [Google Scholar]
- 35.Thomas MM, Lamb TD. Light adaptation and dark adaptation of human rod photoreceptors measured from the a-wave of the electroretinogram. J Physiol (Lond) 1999;518:479–496. doi: 10.1111/j.1469-7793.1999.0479p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998;17:485–521. doi: 10.1016/s1350-9462(98)00006-8. [DOI] [PubMed] [Google Scholar]
- 37.Weiss ER, Rancourt D, Shirakava S, Ducceschi M, Bertram PT, Wong F, Kraft TW, Osawa S. The cloning of GRK7, a candidate cone opsin kinase, from cone- and rod-dominant mammalian retinas. Mol Vis. 1998;4:27. [PubMed] [Google Scholar]
- 38.Wilden U, Hall SW, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto S, Sippel K, Berson EL, Dryja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet. 1997;15:175–178. doi: 10.1038/ng0297-175. [DOI] [PubMed] [Google Scholar]
- 41.Zhao X, Huang J, Khani SC, Palczewski K. Molecular forms of human rhodopsin kinase (GRK1). J Biol Chem. 1998;273:5124–5131. doi: 10.1074/jbc.273.9.5124. [DOI] [PubMed] [Google Scholar]