

Abstract
Mutations in the L1 neural cell adhesion molecule, a transmembrane glycoprotein, cause a spectrum of congenital neurological syndromes, ranging from hydrocephalus to mental retardation. Many of these mutations are single amino acid changes that are distributed throughout the various domains of the protein. Defective herpes simplex virus vectors were used to express L1 protein with the clinical missense mutations R184Q and D598N in the Ig2 and Ig6 extracellular domains, respectively, and S1194L in the cytoplasmic domain. All three mutant proteins were expressed at similar levels in infected cells. Neurite outgrowth of cerebellar granule cells was stimulated on astrocytes expressing wild-type or S1194L L1, whereas those expressing R184Q and D598N L1 failed to increase neurite length. Live cell immunofluorescent staining of L1 demonstrated that most defective vector-infected cells did not express R184Q or D598N L1 on their cell surface. This greatly diminished cell-surface expression occurred in astrocytes, neurons, and non-neural cells. In contrast to wild-type or S1194L L1, the R184Q and D598N L1 proteins had altered apparent molecular weights and remained completely endoglycosidase H (endoH)-sensitive, suggesting incomplete post-translational processing. We propose that some missense mutations in human L1 impede correct protein trafficking, with functional consequences independent of protein activity. This provides a rationale for how expressed, full-length proteins with single amino acid changes could cause clinical phenotypes similar in severity to knock-out mutants.
Keywords: gene transfer, herpes simplex virus, protein trafficking, membrane glycoproteins, mental retardation, neurite outgrowth
Mutations in the gene for L1 (L1CAM) are linked to a number of human CNS syndromes, including X-linked hydrocephalus (XLH or HSAS), MASA syndrome (mental retardation, aphasia, shuffling gait, and adducted thumbs with dysgenesis of the corticospinal tract), and X-linked complicated spastic paraparesis (Jouet et al., 1994;Vits et al., 1994). L1CAM knock-out mice exhibit a range of similar defects that vary depending on genetic background (Dahme et al., 1997; Cohen et al., 1998; Fransen et al., 1998a; Demyanenko et al., 1999). Missense mutations encoding single amino acid substitutions are the most common type among the 93 clinical mutations in human L1 (http://dnalab-www.uia.ac.be/dnalab/l1/) (Van Camp et al., 1996;Yamasaki et al., 1997). This broad spectrum of pathological mutations with associated clinical phenotypes provides a basis to study structure–function relationships. Some laboratories have investigated the functional consequences of clinical mutations (Zhao and Siu, 1996;Zhao et al., 1998; De Angelis et al., 1999), but none have examined the fate of full-length L1 mutant proteins expressed in cells of the nervous system.
L1 is a member of the immunoglobulin superfamily (Moos et al., 1988), containing six immunoglobulin-like (Ig) repeats and five fibronectin type III repeats in its extracellular domain (see Fig.1A). The short cytoplasmic tail interacts with a number of kinases, such as calmodulin kinase II (Wong et al., 1996a), p90rsk (Wong et al., 1996b), Src (Ignelzi et al., 1994), and EphB2 (Zisch et al., 1997), as well as ankyrin and the cytoskeleton (Davis and Bennett, 1994). L1 functions through both homophilic interactions with L1 expressed on an opposing cell-surface (Lemmon et al., 1989; Miura et al., 1992) and heterophilic interactions with other binding partners, including Ig superfamily member axonin-1/TAG-1 (Malhotra et al., 1998), integrins (Yip et al., 1998), and others (for review, see Brummendorf and Rathjen, 1996). In the developing nervous system, L1 protein is thought to participate in cell adhesion, neurite outgrowth, axon fasciculation, and neuronal migration (Fischer et al., 1986; Lagenaur and Lemmon, 1987; Cervello et al., 1991; Asou et al., 1992; Miura et al., 1992; Hankin and Lagenaur, 1994; Zhao et al., 1998).
Fig. 1.
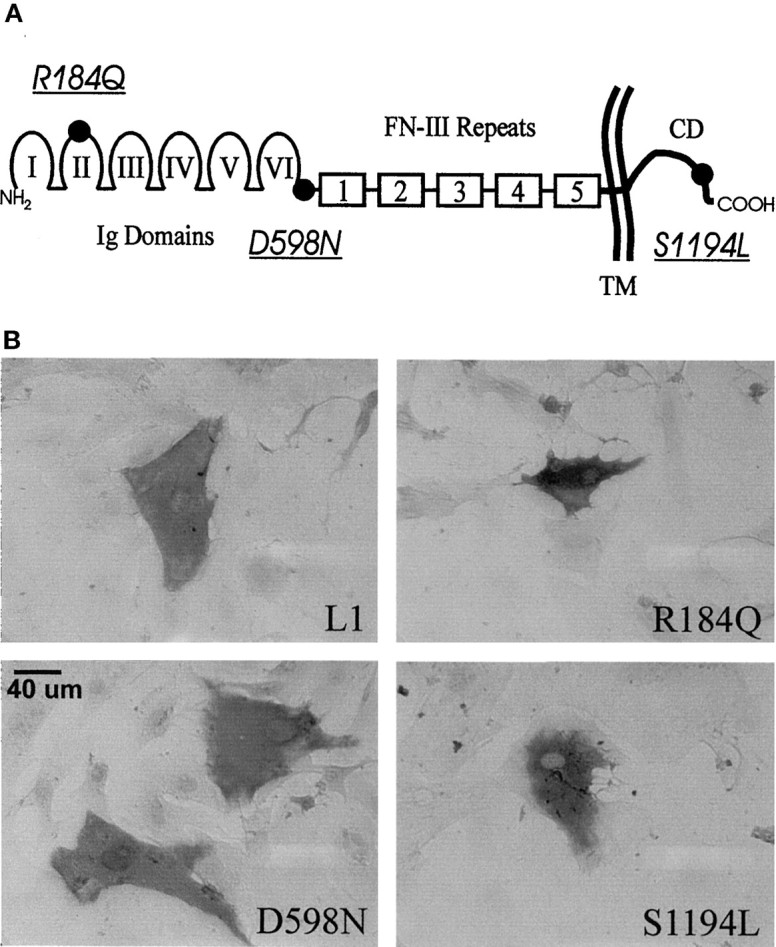
A, Schematic of the L1 cell adhesion molecule showing domain structure: Ig-like domains,I–VI; fibronectin-type III repeats 1–5,FN-III; transmembrane region, TM; and cytoplasmic domain, CD. The location of the mutations (underlined and italicized) are indicated (●). B, Immunohistochemical staining of L1 protein in fixed-permeabilized astrocytes after infection with dvL1.
We have studied three clinical missense mutations affecting different regions of the L1 molecule and producing a spectrum of clinical severities. Patients with the R184Q amino acid substitution (see Fig.1A) diagnosed with XLH present with severe hydrocephalus and a conglomeration of MASA symptoms and often die within 1 year of birth (Jouet et al., 1994). Patients with the D598N mutation present with MASA syndrome with no overt hydrocephalus, a less severe phenotype than R184Q (Vits et al., 1994; De Angelis et al., 1999). S1194L mutation patients have been diagnosed with MASA or XLH (Fransen et al., 1994), showing a midrange severity. To understand how these single base substitutions affect the function of L1 protein and cause such a spectrum of developmental disorders, we expressed the mutant proteins in primary cells of the nervous system using defective herpes simplex virus (HSV) vectors (dv) (Yazaki et al., 1996).
MATERIALS AND METHODS
Construction of mutant L1 amplicon plasmids. Amplicon plasmid pHC-hL1 encoding human L1 (Yazaki et al., 1996) and pHCL encoding lacZ (Kaplitt et al., 1991) have been described previously. pHC-AP, encoding human placental alkaline phosphatase (AP) (Berger et al., 1987), was constructed from pHCL-CAP (New and Rabkin, 1996) by digestion with SpeI and ligation of the large fragment. pHC-hL1 plasmids encoding the R184Q (G551A), D598N (G1792A), and S1194L (C3581T) mutant forms of L1 were generated using the GeneEditor site-directed mutagenesis kit (Promega, Madison, WI) as directed. The following 5′ phosphorylated HPLC purified antisense primers (Bio-Synthesis Inc., Lewisville, TX) were used for mutagenesis: G551A, 5′-ATC GTC ACC TGC TCG TCC TGC TTG ATG-3′; G1792A, 5′-CTC CAC CAC ATT CAG TTC GGT ACT GGC CAC-3′; and C3581T, 5′-CGT TGA GCA ATG GCT GGC TGC-3′. Mutations were confirmed by changes in restriction endonuclease sites (loss of BsrBI and gain of BspMI for G551A, loss of XcmI for G1792A, and gain of BsrDI for C3581T) and automated fluorescent dye-labeled terminator sequencing on a Perkin-Elmer (Emeryville, CA) Applied Biosystem 377 DNA sequencer. The L1 transmembrane domain and signal sequence from each plasmid were also sequenced to rule out additional errant mutations affecting protein trafficking.
Defective HSV vector generation. Purified amplicon plasmid DNA was cotransfected with HSV tsK DNA into Vero cells using Lipofectamine (Life Technologies, Gaithersburg, MD) as described by the manufacturer, and incubated at permissive temperature for tsK (31.5°C) until total cytopathic effect was observed. Virus was passaged at high multiplicity until the best defective vector to helper virus ratio was reached. Virus stocks were prepared by a freeze-thaw–sonication regimen, followed by low-speed centrifugation (2000 × g for 10 min at 4°C) to remove cell debris, and stored in 3% Ficoll. TsK helper virus (obtained from J. Subak-Sharpe Institute of Virology, Glasgow, Scotland) was titered on Vero cells by plaque assay [plaque forming units (pfu)] at the permissive temperature (31.5°C). Defective vector was titered on Vero cells under nonpermissive conditions for the helper virus (39.5°C) by counting individual transgene-expressing cells [defective particle units (dpu)] after immunohistochemistry for L1, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) histochemistry for β-galactosidase, or 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazoliumchloride histochemistry for AP (New and Rabkin, 1996). The titers of the defective vector stocks used were as follows: dvHCL (encodinglacZ), 1.5 × 107 dpu/ml: 9.5 × 107 pfu/ml; dvHC-AP (encoding human placental alkaline phosphatase), 2 × 107 dpu/ml: 2 × 108 pfu/ml; dvHC-hL1, 1.3 × 106 dpu/ml: 1.1 × 107 pfu/ml; dvHC-hL1(R184Q), 2.5 × 106 dpu/ml: 3 × 107 pfu/ml; dvHC-hL1(D598N), 8.8 × 105 dpu/ml: 9.6 × 106 pfu/ml; and dvHC-hL1(S1194L), 2.8 × 106 dpu/ml: 2.1 × 107 pfu/ml. Titers of L1 defectives are likely undercounted because of the sensitivity of L1 immunohistochemistry.
Cell cultures. Vero (African green monkey kidney) and COS-7 [Vero cells transformed with SV40 T-Ag (Cosman and Tevethia, 1981)] cells were obtained from American Type Culture Collection and cultured in DMEM (Life Technologies) supplemented with 10% calf serum (CS) (HyClone, Logan, UT). L3–1 cells, mouse L fibroblasts stably transfected with rat L1, kindly provided by Dr. K. Uyemura (Keio University, Tokyo, Japan) (Miura et al., 1992), were cultured in DMEM–10% CS with 500 μg/ml G-418 (Life Technologies).
Primary astrocytes were prepared from postnatal day 6 (P6) rat cerebella using a modification of the procedure of Levi et al. (1989). Cerebellum was dissected out, and meninges were removed. Cells were mechanically dispersed, treated with trypsin and DNase–soybean trypsin inhibitor (Sigma, St. Louis, MO), plated, and passaged in 10% fetal calf serum (FCS) (HyClone)–DMEM and antibiotic–antimycotic (BioFluids, Rockville, MD) to purify astrocytes. Passage 4 to 5 astrocyte cultures were used in experiments (Yazaki et al., 1996). Cells stained positively for glial fibrillary acidic protein using immunofluorescence [monoclonal antibody (mAb); Sigma].
Cerebellar granule cell (CGC) neurons were isolated from P6 rat cerebella (Levi et al., 1989). Long-term cultures were resuspended in Complete Media (Basal Media Eagle, 10% fetal calf serum, 5 mm glutamine, 1% antibiotics, and 25 mm KCl), plated at 2.5 × 105cells/cm2 in poly-d-lysine-coated four-well chamber slides (Nunc, Naperville, IL), treated 1 d later with AraC (2.5 μg/ml), and infected after 6 d in vitro. CGCs (5 × 106) for neurite outgrowth assays were washed with sterile PBS without Ca2+ or Mg2+ (PBS), incubated in 4 mg/ml PKH26 fluorescent dye (Sigma) for 5 min, washed twice in Complete Media, and plated on astrocyte monolayers as described below.
Cells were infected with dv diluted in PBS with 1 gm/l glucose and 1% heat-inactivated FCS (PBS-GS) and incubated at 37.5°C for 1.5–2 hr. The inoculum was removed, replaced with DMEM with 1% heat-inactivated FCS, 5 mm glutamine, and 1% antibiotics, and the infected cultures were incubated at 39.5°C to minimize helper virus (tsK) toxicity.
Neurite outgrowth assay. Astrocytes in two-well chamber slides were infected at a 0.25 multiplicity of infection (number of dv particles per cell, dv titer determined in Vero cells), which resulted in ∼15–20% transgene-positive cells as determined by L1 immunofluorescence of fixed-permeabilized duplicate cultures. Two hours after infection, PKH26-labeled CGCs (2 × 104 per well) were plated on the infected astrocytes and incubated in DMEM with 25 mm HEPES and N2 media supplement (Life Technologies) at 39.5°C for 18 hr. Cultures were washed with PBS, fixed with 4% paraformaldehyde in PBS for 20 min, washed, and coverslipped with Fluormount-G (Southern Biotechnology, Birmingham, AL).
Fluorescent neurons and neurites were visualized at 400× magnification on an Olympus Optical (Tokyo, Japan) AH2 Vanox-S microscope using fluorescence filters for rhodamine. Images were digitally captured using a Toshiba three-chip color CCD low-light level camera, and neurite lengths were measured using Optimas 5.2 software (Media Cybernetics). Neurites were defined as cell projections longer than one cell body width, with no contact on other labeled CGCs (to avoid the tropic effect of endogenous L1 and other neuronally expressed surface molecules). Normalized neurite lengths were computed within each experiment using neurite lengths of CGCs on mock-infected astrocytes. These normalized lengths were pooled from four separate experiments and analyzed using one-way ANOVA. To determine which groups were different from one another, Tukey's studentized range tests were used.
Immunostaining for L1. Cells were infected with 102-103 dpu of indicated dv, and incubated for 16.5 hr at 39.5°C. For immunohistochemical staining, cells were fixed in 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, incubated with 1:1000 5G3 anti-human L1 mAb (generous gift of Ralph Reisfeld, Scripps Research Institute, La Jolla, CA), washed, blocked with 10% horse serum, and incubated with biotinylated horse anti-mouse antibody (Ab) (1:1000; Vector Laboratories, Burlingame, CA). Bound Ab was detected with horseradish peroxide-conjugated avidin (Vectastain Elite kit; Vector) and visualized by incubation with 3,3′-diaminobenzidine, 0.05% NH4Cl, 1% glucose, and glucose oxidase (1.5 U/ml; Sigma). Pictures were taken on a Nikon (Tokyo, Japan) Diaphot microscope.
For surface human L1 staining, cells were washed with chilled PBS and incubated with 5G3 anti-human L1 mAb (1:1000) in DMEM–10% FCS at 4°C for 1.5 hr. Cells were washed, fixed with 4% paraformaldehyde in PBS, washed, blocked in PBS with 10% normal goat serum for 1 hr, and incubated with rhodamine red-conjugated goat anti-mouse antibody (1:500; Jackson ImmunoResearch, West Grove, PA) at room temperature for 1 hr. Fixed-permeabilized cell labeling was performed as above, except cells were fixed and then permeabilized with 0.25% Triton X-100 in PBS before incubation with primary Ab. Live/fixed-permeabilized double immunofluorescence was performed as above for surface L1 except Oregon green-conjugated goat anti-mouse Ab (1:500; Molecular Probes, Eugene, OR) was used as the secondary Ab. Excess primary Ab was blocked with goat anti-mouse IgG (1:20; Sigma) in 10% goat serum in PBS for 1 hr at room temperature. Cells were then permeabilized, incubated with 5G3 mAb for 1.5 hr, and finally with rhodamine red-labeled goat anti-mouse Ab. Cells were coverslipped with Fluormount-G.
Blinded quantification was performed by counting positive cells in multiple rows per treatment well using either FITC or rhodamine bandpass filters on a Nikon Optiphot microscope. At least five random rows of a two- or four-well chamber slide were examined for each treatment. Average numbers of positively stained cells were determined per row. Matched surface/permeabilized immunofluorescence staining ratios were analyzed with or without logarithmic transformation using one-way ANOVA followed by Tukey's studentized range test. Images (single 0.1 μm sections) were taken on an Olympus confocal laser-scanning microscope with Fluoview 2.0 software.
Deglycosylation and immunoblotting of L1. Astrocytes were infected at a multiplicity of infection of 0.5 and incubated overnight at 39.5°C. Cells were washed and then suspended in chilled 20 mm Tris–1 mm EDTA using a cell scraper, pelleted, and resuspended in cracking buffer (130 mm Tris-HCl, 20% glycerol, 3% β-mercaptoethanol, 3% SDS, and 0.03% bromphenol blue). Lysates (5 μg of R184Q and D598N or 12 μg of L1, S1194L, lacZ, AP, mock, and L3–1) were denatured for 5 min at 95°C and incubated overnight in 75 mm sodium citrate, pH 5.5, and 0.05% PMSF with or without 0.5 mU of endoglycosidase H (Boehringer Mannheim, Indianapolis, IN) at 37°C. Samples were denatured, separated by 5% SDS-PAGE, and transferred to a polyvinylidene difluoride membrane (Immobilon; Millipore, Bedford, MA). The membrane was blocked overnight with Detector Block (KPL, Gaithersburg, MD), incubated for 2 hr at 4°C with rabbit L1-C anti-L1 polyclonal Ab (1:1000 in Detector Block), which recognizes L1 from all mammalian species (kindly provided by Dr. K. Uyemura), and washed with TBS with Tween-20 (50 mm Tris, 0.9% NaCl, and 0.1% Tween-20). Bound primary antibody was detected with HRP-conjugated donkey anti-rabbit secondary antibody (Amersham Pharmacia Biotech, Arlington Heights, IL) and visualized with Supersignal ECL (Pierce, Rockford, IL), as described by the manufacturer, using Amersham ECL hyperfilm. Film was scanned, and figures were prepared using CorelGraphics. Protein concentration was determined using Bradford assay (Bio-Rad, Hercules, CA) on cell lysates prepared by sonication and freeze–thawing.
RESULTS
Defective HSV vectors expressing mutant L1
Defective HSV vectors are generated from amplicon plasmids (containing bacterial plasmid sequences, HSV cis-acting elements, and foreign transcription units, including promoter and cDNA for transgene) that are replicated and packaged in cells infected with helper HSV. Amplicon plasmids containing human L1 cDNA were mutagenized using oligonucleotides containing single-base changes, G551A, G1792A, and C3581T, encoding clinical L1 mutations R184Q, D598N, and S1194L, respectively (Fig. 1A). Defective vectors encoding these mutant forms of L1 expressed similar levels of L1 as confirmed by immunohistochemistry of fixed-permeabilized cells (Fig. 1B). Defective vectors encoding lacZ (dvHCL), with the cytosolic protein product β-galactosidase, and AP (dvHC-AP), a membrane-bound protein, were used as controls.
Neurite extension of cerebellar neurons is enhanced on normal and S1194L L1-expressing astrocytes
To determine the functional activity of mutant L1 protein, we examined the ability of cerebellar astrocytes expressing mutant L1 to stimulate the elongation of CGC neurites. Purified CGCs from P6 rats were plated on astrocytes expressing wild-type or mutant L1, or as controls AP or β-galactosidase. Infection of astrocytes with dvHCL or dvHC-AP did not alter the inherent ability of astrocytes to promote a limited degree of neurite outgrowth (Fig.2). Only astrocytes expressing wild-type or S1194L L1 were able to promote neurite outgrowth compared with mock-infected astrocytes, with a greater than 50% increase in mean neurite length after 18 hr. R184Q- and D598N-expressing astrocytes did not elicit any increase in neurite length compared with mock-infected AP- or β-galactosidase-expressing astrocytes.
Fig. 2.

Wild-type and S1194L mutant L1 stimulate neurite outgrowth. Relative change in mean neurite lengths of CGCs plated on astrocytes infected with dvL1 (wild-type or indicated mutant), dvHCL (LacZ), or dvHC-AP (AP). Data pooled from four separate experiments after normalization to neurite lengths on mock-infected astrocytes. *p < 0.05 by Tukey's studentized range tests conducted after one-way ANOVA (p = 0.0001).
L1 proteins with mutations in the extracellular region are not efficiently expressed on the cell surface
One possible reason for the inability of R184Q or D598N L1 to stimulate neurite outgrowth could be the lack of cell-surface expression. Therefore, we determined the localization of mutant L1 protein using indirect immunofluorescence 16.5 hr after infection of cells with dvL1. In the first instance, we compared live cell with fixed or fixed-permeabilized staining of duplicate-infected cerebellar astrocyte cell cultures using a human L1-specific antibody (Table1, Astrocytes Live/perm duplicates). We found that, even in the absence of permeabilization, there was slight staining of intracellular L1 that did not occur with live-cell staining (data not shown). Second, we sequentially double-stained live and then fixed-permeabilized cells in the same culture. Cell-surface L1 is labeled with one fluorophore (Fig.3A,C,E,G), and total L1 is labeled with a second (Fig.3B,D,F,H). The number of immunofluorescent cells was quantified (Fig. 4), and the ratio of surface to total labeling indicates the proportion of cells expressing L1 protein on the cell surface (Table 1, Double labeling).
Table 1.
Proportion of normal or mutant L1 that is surface expressed in defective vector-infected cells
| Astrocytes | COS-7 | Vero | ||||
|---|---|---|---|---|---|---|
| Live/perm duplicates | Double labeling | Live/perm duplicates | Double labeling | Live/perm duplicates | Double labeling | |
| R184Q | 0.07 ± 0.03 | 0.11 ± 0.01 | 0.15 ± 0.02 | 0.04 ± 0.01 | 0.18 ± 0.07 | 0.07 ± 0.02 |
| D598N | 0.07 ± 0.02 | 0.11 ± 0.02 | 0.12 ± 0.02 | 0.10 ± 0.01 | 0.28 ± 0.03 | 0.58 ± 0.03 |
| S1194L | 0.57 ± 0.07 | 0.47 ± 0.03 | 0.86 ± 0.05 | 0.87 ± 0.03 | 1.00 ± 0.08 | 0.93 ± 0.01 |
| L1 | 0.88 ± 0.15 | 0.71 ± 0.16 | 0.81 ± 0.07 | 0.65 ± 0.02 | 1.06 ± 0.09 | 0.85 ± 0.02 |
Live/perm duplicate ratios were determined from duplicate infected cultures (16.5 hr after infection) that were either live/surface stained or stained after fixation–permeabilization. Double-labeling ratios were determined from surface/permeabilized staining as in Figures 3 and 6. Means of the ratios of surface labeled to permeabilized-labeled cells are reported ± SE. Matched surface/permeabilized immunofluorescence staining ratios were analyzed using one-way ANOVA, followed by Tukey's Studentized range test. R184Q and D598N versus L1 and S1194L ratios differed significantly in all cell types (one-way ANOVA, p = 0.0001; Tukey's,p < 0.05), but neither R184Q versus D598N nor L1 versus S1194L differed significantly from each other, except L1 versus S1194L in double-labeled astrocytes and R184Q versus D598N in double-labeled Veros. There was no significant difference between L1 versus S1194L surface expression in double-labeled astrocytes when the ratios were analyzed after logarithmic transformation; all other significant differences remained.
Fig. 3.
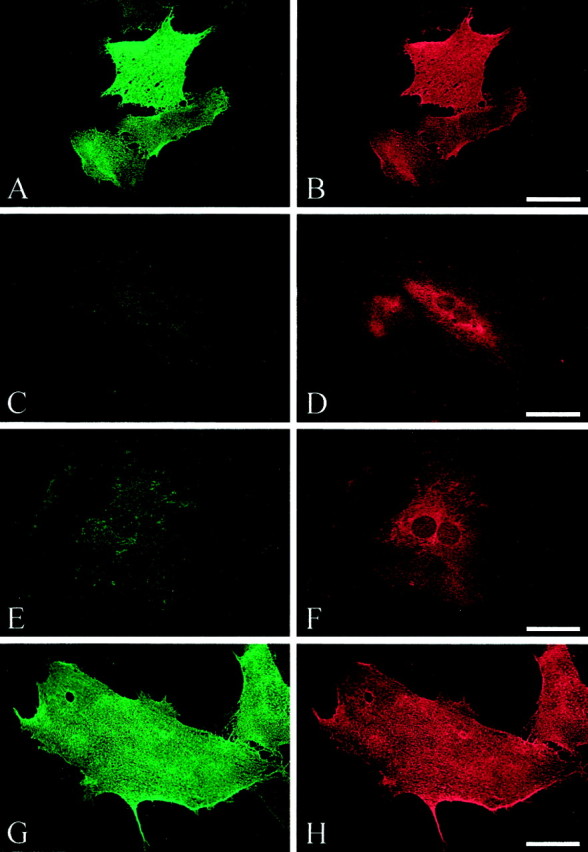
Surface (A, C,E, G) and permeabilized (B, D, F,H) staining of L1. Double immunofluorescence was performed on rat cerebellar astrocytes infected with dvL1 (16.5 hr after infection): wild type, A, B; R184Q,C, D; D598N, E,F; and S1194L, G, H. In most cells expressing R184Q and D598N mutant L1, L1 protein failed to reach the cell surface. Scale bar, 50 μm.
Fig. 4.

Quantification of surface versus permeabilized L1 double labeling on dvL1-infected astrocytes (16.5 hr after infection). Mean ± SE number of positive cells for surface (open bars) or permeabilized (filled bars) staining per row using 20× objective.
All forms of L1 protein were highly expressed in infected astrocytes (Fig.3B,D,F,H); however, only wild-type and S1194L L1 proteins were strongly expressed on the cell surface of most infected astrocytes (Fig.3A,G; Table 1). The two L1 proteins with mutations in the extracellular region, R184Q and D598N, were not typically detected by live-cell staining (Fig.3C,E). In general, <15% of infected astrocytes expressed R184Q or D598N L1 on their cell surface (Table 1). The low proportion of cells expressing mutant L1 on the their cell surface did not change when examined at earlier (12 hr) or later (24 hr) times after infection (data not shown), suggesting that this was not attributable to more rapid turnover of the mutant proteins or insufficient time to reach the surface. No dv-infected astrocytes expressing either AP or β-galactosidase stained positive for surface or cytosolic L1 (data not shown).
To demonstrate that this protein-trafficking defect was not unique to astrocytes, we examined dvL1-infected CGCs. These cells express endogenous rat L1 (Fig.5A). As seen with the infected astrocytes, dvL1-infected CGC neurons expressed human L1 as detected after fixation–permeabilization (Fig.6B,D,F,H). The human-specific monoclonal antibody (5G3) only detects the exogenous vector-derived human L1, not endogenous L1 or vector-derived rat L1 (data not shown). Again, only wild-type and S1194L L1 proteins were consistently detected on the cell surface. This surface expression covered the entire neuron and was not limited to axons (Fig.6A,G), a distribution similar to that seen for endogenous L1 in these cells (Fig. 5). The R184Q and D598N mutant L1 proteins were not usually expressed on the cell surface but were distributed in the cytosol and within nerve processes (Fig.6D,F).
Fig. 5.

Endogenous L1 expression in rat CGC neurons. L1 was detected by immunofluorescence (A) using a species nonspecific anti-L1 antibody in fixed-permeabilized CGC neurons (B, differential-interface contrast microscopy). Scale bar, 50 μm.
Fig. 6.
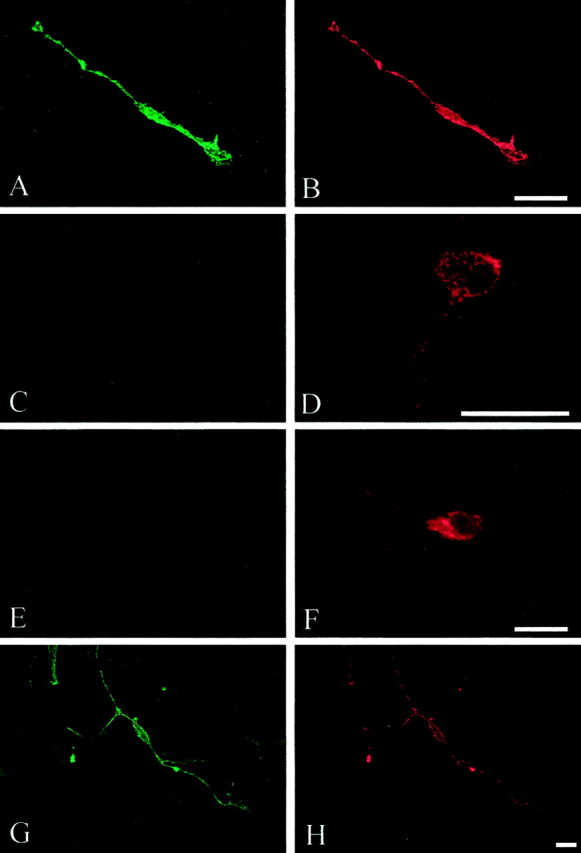
Surface (A, C,E, G) and permeabilized (B, D, F,H) staining of exogenous L1. Double immunofluorescence was performed on CGC neurons infected with dvL1 (16.5 hr after infection): wild type, A,B; R184Q, C, D; D598N,E, F; and S1194L, G,H. In most cells expressing R184Q and D598N mutant L1, L1 protein failed to reach the cell surface but was detected within the neuronal cytosol, including processes. Scale bar, 20 μm.
We then tested non-neural cells to determine whether this effect was nervous system-specific. Again, similar results were seen when Vero (African green monkey kidney cells) and COS-7 (SV40 immortalized Vero cells) were infected with dvL1 (Table 1). The only difference was an increased proportion of cell-surface labeling for D598N L1 protein in Vero cells; however, the intensity of the surface staining was very low compared with wild-type or S1194L L1.
L1 proteins with mutations in the extracellular region are underglycosylated
To evaluate whether vector-derived wild-type and mutant human L1 proteins were full-length, expressed at similar levels, and correctly modified post-translationally, cell lysates from dvL1-infected astrocytes were separated by SDS-PAGE and immunoblotted for L1 using a species nonspecific polyclonal antibody to L1 (Fig. 7). Primary rat astrocytes do not endogenously express L1, because no protein was detected in mock-, lacZ-, or AP vector-infected astrocytes (Fig. 7, Mock, LacZ, AP). All L1 mutants expressed similar levels of full-length protein; however, the R184Q and D598N L1 proteins were not fully modified post-translationally. Normal and S1194L L1 protein had a characteristic doublet pattern for L1 (Fig. 7, L1−,S1194L−), with apparent molecular weights (MW) of ∼210 and 200 kDa (Miura et al., 1992). The R184Q and D598N dv-infected cell lysates only contained the bottom band of the L1 doublet (Fig. 7, R184Q−, D598N−). The top band of the L1 doublet has been reported to be the cell-surface form of L1(Zisch et al., 1997). This suggests that any R184Q or D598N L1 protein that reaches the cell surface, as seen with immunofluorescence, is not fully modified.
Fig. 7.

Western blot of lysates from vector-infected astrocytes. Cell lysates were treated with (+) or without (−) endoH to remove unmodified N-linked glycosylations. Five (R184Q or D598N) or 12 (all others, so lower MW band at 200 kDa would be visible) μg of protein were loaded per lane. The upper MW band, the fully glycosylated form of L1 (∼210 kDa), is resistant to endoH (compare − with + in L1, S1194L, and L3–1), whereas the lower MW band of L1 is sensitive.
During normal glycoprotein processing, translated proteins are glycosylated in stages, which increases their MW. For N-linked glycosylations, asparagine residues are initially glycosylated in the endoplasmic reticulum (ER). These glycosylations are later modified and then extended in sequential compartments of the Golgi (for review, see (Dennis et al., 1999). To test for L1 glycosylation, dv-infected astrocyte lysates were treated with endoH, which removes unmodified N-linked glycosylations. The top band of the wild-type and S1194L L1 doublet was resistant to endoH treatment, whereas thebottom band was susceptible, as indicated by the shift in MW after endoH treatment (Fig. 7, L1+,S1194+). This ∼150 kDa shifted band is the same MW as deglycosylated L1 reported previously (Faissner et al., 1985). The single, bottom band present for R184Q or D598N mutant L1 protein was completely shifted after endoH treatment (Fig. 7,R184Q+, D598N+). The top band from L3–1 cells, mouse L fibroblasts stably transfected with rat L1, ran at a slightly higher position than human L1, and this band is also endoH-resistant (Fig. 7, L3–1+). This may be because of differences between rat and human L1 protein or processing differences in mouse L cells versus rat cerebellar astrocytes. The endoH-sensitive band of rat L1 from the L3–1 cells runs at the same size as human L1 (Fig. 7, L3–1 vs L1).
Treatment of dvL1-infected astrocytes with N-acetylgalactosamine, which cleaves O-linked glycosylations, had no effect on L1 banding pattern (data not shown). This confirms an earlier report that L1 has no significant O-linked glycosylations (Faissner et al., 1985).
DISCUSSION
To begin to understand how single amino acid substitutions in human L1 protein lead to such devastating clinical syndromes, we used defective HSV vectors to express mutated L1 proteins in cells of the CNS. We found that R184Q or D598N mutant L1 proteins, which have extracellular region amino acid substitutions, are not present on the cell surface of most dvL1-infected cells of CNS and non-CNS origin. The reason some cells in a population express mutant L1 on the cell surface whereas the majority do not remains to be determined. This does not seem to be a phenotype of a particular cell type, because the proportion was similar in astrocytes, neurons (preliminary data), and COS-7 cells. The lack of cell-surface expression would result in the inability of L1 to mediate binding and signaling in cells that express these mutant L1 proteins in vivo, as illustrated by the inability of these mutants to promote neurite outgrowth in vitro. The effect of these single amino acid changes on protein trafficking rather than protein function also suggests a new model of how such mutations could cause clinical phenotypes similar to those seen with mouse null mutations. The degree of cell-surface expression could affect the severity of disease and account for the large variability in clinical phenotypes. If these two extracellular domain mutations are representative, it may also indicate how mutations throughout different domains of L1 could account for similar phenotypes.
In contrast, the cytoplasmic domain mutant S1194L was expressed on the cell surface of all these cell types at near normal levels and appeared to function as well as wild-type L1 as a substrate for neurite outgrowth when expressed by astrocytes. Western blot analysis showed that vector-derived normal and S1194L L1 proteins had a characteristic MW doublet pattern, whereas the R184Q and D598N mutant proteins were only present as the lower MW form. Zisch et al. (1997) have shown that the upper MW band of L1 is the cell surface-expressed form of the protein (Zisch et al., 1997), supporting our live cell-surface L1 labeling. A low proportion of cells expressed R184Q and D598N mutant proteins on their cell surface, yet no upper MW band was detected. This suggests that the lower MW form of L1 protein is able to the reach the cell surface, either through the same trafficking pathway as wild-type L1 protein or an entirely different one. The lack of neurite outgrowth activity of the R184Q and D598N mutant proteins could be attributable to the small number of astrocytes expressing L1 on their cell surface (i.e., a dose–response effect), or that the mutant, incompletely processed forms of L1 at the cell surface are unable to mediate neurite outgrowth.
The lower MW band likely represents newly synthesized, not yet surface-expressed L1 that is moving through the protein post-translational processing machinery. R184Q and D598N L1 proteins were endoH-sensitive and therefore received N-linked glycosylations in the ER but were not further modified in the Golgi apparatus (making them endoH-resistant) (Dennis et al., 1999). Normally a protein is not trafficked to the plasma membrane until its carbohydrate modifications are complete, i.e., glycosylations in the ER and modifications–changes to these glycosylations in the Golgi.
Possible fates of mutant L1 proteins
One explanation for R184Q and D598N mistargeting would be misfolding of the protein. Modeling studies suggest this possibility for the R184Q mutant but not the D598N mutant (Bateman et al., 1996). Misfolded proteins are often targeted for degradation, yet both the R184Q and D598N mutant proteins were expressed at similar levels as wild-type L1 protein. We did not find evidence for ubiquination of these mutant proteins (data not shown). Molecular modeling suggests that R184Q is part of a buried salt bridge (Bateman et al., 1996), whereas peptide inhibition studies suggest it is present on an exposed surface (Zhao et al., 1998). This region of the L1 Ig2 domain has been shown to be important in adhesion and neurite outgrowth (Zhao et al., 1998), and chimeric L1 protein containing the R184Q mutation has <20% of wild-type L1 homophilic binding activity (De Angelis et al., 1999). The combination of limited cell-surface expression of the R184Q mutant protein and its decreased binding activity could generate the severe phenotype seen in patients.
The D598N change resides within the Ig6 domain, which induces neurite outgrowth, supposedly because of the RGD sequence that does not include position 598 (Felding-Habermann et al., 1997; Yip et al., 1998). Molecular modeling of the L1 extracellular domains identified position 598 as a “conserved-surface” residue; i.e., not critical in protein folding but nevertheless conserved among species (Bateman et al., 1996). Because this residue is unlikely to disrupt the integrity the domain, it likely affects an interaction with a ligand of L1 (Brummendorf et al., 1998). In contrast to an extracellular ligand, it may interact with an intracellular chaperone or other ligand during post-translational processing. Substitution of asparagine at this site introduces a possible substrate for additional glycosylation, but analysis using Motif (http://www.motif.genome.ad.jp/) did not identify this sequence as a candidate for additional glycosylation. A recombinant protein containing the extracellular region of L1 with the D598N mutation had only a small reduction in homophilic binding (De Angelis et al., 1999). Therefore, the limited surface expression of this mutant is likely the root of its physiological defect and provides a rationale for the conservation of this residue across species.
In contrast to our results, De Angelis et al. (1999) reported recently that transiently transfected COS cells expressed R184Q and D598N mutant proteins on their cell surface. We have found that paraformaldehyde fixation can permeabilize cells enough to detect cytosolic antigens, which does not occur with live-cell staining. Others have shown that dv-derived gene products are trafficked correctly to various subcellular compartments in neurons (Craig et al., 1995; West et al., 1997; Jareb and Banker, 1998; Stowell and Craig, 1999), suggesting that dv infection does not disrupt normal protein processing and trafficking.
Contrary to the extracellular domain substitutions, the S1194L intracellular domain mutant protein behaved like wild-type L1. The S1194L mutant protein was expressed on the cell surface at levels similar to wild-type L1, induced neurite outgrowth as a substrate as well as wild-type, and had the same post-translational modifications. However, our studies did not examine the intracellular signaling properties of L1. S1194 is neither within the ankyrin binding domain (Davis and Bennett, 1994; Dahlin-Huppe et al., 1997; Garver et al., 1997; Hortsch et al., 1998; Zhang et al., 1998), nor is this serine normally phosphorylated (Heiland et al., 1996; Wong et al., 1996a,b). Still, it is conserved among human, rat, mouse, chick, and evenDrosophila homologs of L1 (Zisch et al., 1997), and patients with this mutation can be quite severely affected, being diagnosed with either XLH or MASA syndrome.
Clinical parallels in other systems
There are a few examples of clinically relevant proteins being mistargeted because of single amino acid changes. M467T and M467K substitutions in the rBAT protein result in cystinuria; the protein shows delayed trafficking to the surface and underglycosylation (Calonge et al., 1994; Chillaron et al., 1997). We did not observe any change in surface expression of either R184Q or D598N L1 protein at up to 24 hr after infection (data not shown). The R281W substitution in the Kell glycoprotein, found on red blood cells, results in decreased cell-surface expression and altered glycosylation (Yazdanbakhsh et al., 1999). As with the L1 mutants, the mutant Kell glycoprotein is endoH-sensitive. Additional clinical examples occur in the cystic fibrosis transmembrane conductance regulator (Smit et al., 1995), low density lipoprotein receptor (Pathak et al., 1988), intestinal surcrase-isomaltase (Fransen et al., 1991), and αIIbβ3 integrin (Wilcox et al., 1994). These and other examples of altered processing of glycoproteins, together with our results, suggest this to be an important mechanism by which mutant surface proteins fail to achieve normal function, regardless of their tissue or cell of origin. To our knowledge, the results presented here are the first evidence suggesting such a mechanism in the developing CNS.
Alterations in ER processing of proteins have been divided into two classes: those that lead to degradation of the protein and those that accumulate in the ER (Aridor et al., 1999). We do not believe the mutant L1 proteins are rapidly degraded because their levels are similar to normal L1 protein, and inhibitors that block the proteasome degradation pathway do not alter the levels of wild-type or mutant L1 protein (our unpublished observations). However, L1 also fails to exclusively colocalize with the ER markers concanavalin A and calnexin (our unpublished observations).
Implications for the classification of L1 CAM mutations
L1 mutations have been divided into three classes: nonsense mutations generating truncated and presumably secreted protein, thereby leaving the cell surface devoid of L1 and having a more profound and severe effect on the patient; missense mutations, divided into extracellular and intracellular locales, theoretically interfering with L1 binding, signaling, or both, but usually having a more mild phenotype (Yamasaki et al., 1997; Fransen et al., 1998b). In light of our results, it might be useful to reclassify mistrafficked mutants, such as R184Q and D598N, which would result in little or no functional L1 on the cell surface. This may clarify why some of the extracellular substitution mutations have been difficult to classify, because they can result in either severe or milder phenotypes (Yamasaki et al., 1997; Fransen et al., 1998b). It remains to be determined whether these mistrafficked mutant proteins interact or interfere with intracellular signaling pathways.
Footnotes
This work was supported by National Institutes of Health Training Grant T32 HD07459 (H.D.M.) and an ARCS Foundation scholarship. We thank Ralph Reisfeld for the 5G3 antibody, Keiichi Uyemura for the L1-C antibody and L3–1 cells, Takahito Yazaki for constructing plasmid pHC-hL1, and Matthew Kelley, Barbara Bregman, Geoff Goodhill, Rodolfo Rivas, and Pablo Hernaiz Driever for helpful discussions and critical comments. Also instrumental were Henry Yang in the Lombardi Sequencing Core Facility, Mariella C. Tefft of the Lombardi Biostatistics Unit, and Dr. Suzette Mueller, Director of the Lombardi Microscopy Core Facility. We thank the members of the Rabkin and Martuza laboratories for their valuable advice and encouragement throughout the course of this work.
Correspondence should be addressed to Samuel D. Rabkin at his present address: Massachusetts General Hospital East, Building 149, Room 2510, Box 17, Charlestown, MA 02129. E-mail:rabkin@helix.mgh.harvard.edu.
Dr. Martuza's present address: Neurosurgery Service, Massachusetts General Hospital, WHT 502, 55 Fruit Street, Boston, MA 02114.
REFERENCES
- 1.Aridor M, Bannykh SI, Rowe T, Balch WE. Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J Biol Chem. 1999;274:4389–4399. doi: 10.1074/jbc.274.7.4389. [DOI] [PubMed] [Google Scholar]
- 2.Asou H, Miura M, Kobayashi M, Uyemura K. The cell adhesion molecule L1 has a specific role in neural cell migration. NeuroReport. 1992;3:481–484. doi: 10.1097/00001756-199206000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Bateman A, Jouet M, MacFarlane J, Du JS, Kenwrick S, Chothia C. Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J. 1996;15:6050–6059. [PMC free article] [PubMed] [Google Scholar]
- 4.Berger J, Garattini E, Hua JC, Udenfriend S. Cloning and sequencing of human intestinal alkaline phosphatase cDNA. Proc Natl Acad Sci USA. 1987;84:695–698. doi: 10.1073/pnas.84.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brummendorf T, Rathjen FG. Structure/function relationships of axon-associated adhesion receptors of the immunoglobulin superfamily. Curr Opin Neurobiol. 1996;6:584–593. doi: 10.1016/s0959-4388(96)80089-4. [DOI] [PubMed] [Google Scholar]
- 6.Brummendorf T, Kenwrick S, Rathjen FG. Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr Opin Neurobiol. 1998;8:87–97. doi: 10.1016/s0959-4388(98)80012-3. [DOI] [PubMed] [Google Scholar]
- 7.Calonge MJ, Gasparini P, Chillaron J, Chillon M, Gallucci M, Rousaud F, Zelante L, Testar X, Dallapiccola B, Di Silverio F, Barcelo P, Estivill X, Zorzano A, Nunes V, Papacín M. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet. 1994;6:420–425. doi: 10.1038/ng0494-420. [DOI] [PubMed] [Google Scholar]
- 8.Cervello M, Lemmon V, Landreth G, Rutishauser U. Phosphorylation-dependent regulation of axon fasciculation. Proc Natl Acad Sci USA. 1991;88:10548–10552. doi: 10.1073/pnas.88.23.10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chillaron J, Estevez R, Samarzija I, Waldegger S, Testar X, Lang F, Zorzano A, Busch A, Palacin M. An intracellular trafficking defect in type I cystinuria rBAT mutants M467T and M467K. J Biol Chem. 1997;272:9543–9549. doi: 10.1074/jbc.272.14.9543. [DOI] [PubMed] [Google Scholar]
- 10.Cohen NR, Taylor JS, Scott LB, Guillery RW, Soriano P, Furley AJ. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1998;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- 11.Cosman DJ, Tevethia MJ. Characterization of a temperature-sensitive, DNA-positive, nontransforming mutant of simian virus 40. Virology. 1981;112:605–624. doi: 10.1016/0042-6822(81)90306-8. [DOI] [PubMed] [Google Scholar]
- 12.Craig AM, Wyborski RJ, Banker G. Preferential addition of newly synthesized membrane protein at axonal growth cones. Nature. 1995;375:592–594. doi: 10.1038/375592a0. [DOI] [PubMed] [Google Scholar]
- 13.Dahlin-Huppe K, Berglund E, Ranscht B, Stallcup W. Mutational analysis of the L1 neuronal cell adhesion molecule identifies membrane-proxomal amino acids of the cytoplasmic domain that are required for cytoskeletal anchorage. Mol Cell Neurosci. 1997;9:144–156. doi: 10.1006/mcne.1997.0608. [DOI] [PubMed] [Google Scholar]
- 14.Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- 15.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- 16.De Angelis E, MacFarlane J, Du JS, Yeo G, Hicks R, Rathjen FG, Kenwrick S, Brummendorf T. Pathological missense mutations of neural cell adhesion molecule L1 affect homophilic and heterophilic binding activities. EMBO J. 1999;18:4744–4753. doi: 10.1093/emboj/18.17.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demyanenko GP, Tsai AY, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dennis JW, Granovsky M, Warren CE. Protein glycosylation in development and disease. BioEssays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 19.Faissner A, Teplow DB, Kubler D, Keilhauer G, Kinzel V, Schachner M. Biosynthesis and membrane topography of the neural cell adhesion molecule L1. EMBO J. 1985;4:3105–3113. doi: 10.1002/j.1460-2075.1985.tb04052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felding-Habermann B, Silletti S, Mei F, Siu CH, Yip PM, Brooks PC, Cheresh DA, TE OT, Ginsberg MH, Montgomery AM. A single immunoglobulin-like domain of the human neural cell adhesion molecule L1 supports adhesion by multiple vascular and platelet integrins. J Cell Biol. 1997;139:1567–1581. doi: 10.1083/jcb.139.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer G, Kunemund V, Schachner M. Neurite outgrowth patterns in cerebellar microexplant cultures are affected by antibodies to the cell surface glycoprotein L1. J Neurosci. 1986;6:605–612. doi: 10.1523/JNEUROSCI.06-02-00605.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fransen JA, Hauri HP, Ginsel LA, Naim HY. Naturally occurring mutations in intestinal sucrase-isomaltase provide evidence for the existence of an intracellular sorting signal in the isomaltase subunit. J Cell Biol 115 1991. 45 57[Erratum (1991) 115: 1473]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fransen E, Schrander-Stumpel C, Vits L, Coucke P, Van Camp G, Willems PJ. X-linked hydrocephalus and MASA syndrome present in one family are due to a single missense mutation in exon 28 of the L1CAM gene. Hum Mol Genet. 1994;3:2255–2256. doi: 10.1093/hmg/3.12.2255. [DOI] [PubMed] [Google Scholar]
- 24.Fransen E, R DH, Van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SK, De Zeeuw CI, De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum Mol Genet. 1998a;7:999–1009. doi: 10.1093/hmg/7.6.999. [DOI] [PubMed] [Google Scholar]
- 25.Fransen E, Van Camp G, R DH, Vits L, Willems PJ. Genotype-phenotype correlation in L1 associated diseases. J Med Genet. 1998b;35:399–404. doi: 10.1136/jmg.35.5.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hankin M, Lagenaur C. Cell adhesion molecules in the early developing mouse retina: retinal neurons show preferential outgrowth in vitro on L1 but not N-CAM. J Neurobiol. 1994;35:472–487. doi: 10.1002/neu.480250503. [DOI] [PubMed] [Google Scholar]
- 28.Heiland PC, Hertlein B, Traub O, Griffith LS, Schmitz B. The neural adhesion molecule L1 is phosphorylated on tyrosine and serine residues. NeuroReport. 1996;7:2675–2678. doi: 10.1097/00001756-199611040-00053. [DOI] [PubMed] [Google Scholar]
- 29.Hortsch M, Homer D, Malhotra JD, Chang S, Frankel J, Jefford G, Dubreuil RR. Structural requirements for outside-in and inside-out signaling by Drosophila neuroglian, a member of the L1 family of cell adhesion molecules. J Cell Biol. 1998;142:251–261. doi: 10.1083/jcb.142.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ignelzi MA, Jr, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- 31.Jareb M, Banker G. The polarized sorting of membrane proteins expressed in cultured hippocampal neurons using viral vectors. Neuron. 1998;20:855–867. doi: 10.1016/s0896-6273(00)80468-7. [DOI] [PubMed] [Google Scholar]
- 32.Jouet M, Rosenthal A, Armstrong G, MacFarlane J, Stevenson R, Paterson J, Metzenberg A, Ionasescu V, Temple K, Kenwrick S. X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat Genet. 1994;7:402–407. doi: 10.1038/ng0794-402. [DOI] [PubMed] [Google Scholar]
- 33.Kaplitt M, Pfaus J, Kleopolos S, Hanlon B, Rabkin S, Pfaff D. Expression of a functional foreign gene in adult mammalian brain following in vivo transfer via herpes simplex virus type 1 defective vector. Mol Cell Neurosci. 1991;2:320–330. doi: 10.1016/1044-7431(91)90062-s. [DOI] [PubMed] [Google Scholar]
- 34.Lagenaur C, Lemmon V. An L1-like molecule, the 8D9 antigen, is a potent substrate for neurite extension. Proc Natl Acad Sci USA. 1987;84:7753–7757. doi: 10.1073/pnas.84.21.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemmon V, Farr KL, Lagenaur C. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 1989;2:1597–1603. doi: 10.1016/0896-6273(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 36.Levi G, Francesca A, Ciotti MT, Thangnipon W, Kingsbury A, Balazs R. Preparation of 98% pure cerebellar granule cell cultures. In: Shahar A, de Vellis J, Vernadakis A, Haber B, editors. A dissection and tissue culture manual of the nervous system. Liss; New York: 1989. pp. 211–214. [Google Scholar]
- 37.Malhotra JD, Tsiotra P, Karagogeos D, Hortsch M. Cis-activation of L1-mediated ankyrin recruitment by TAG-1 homophilic cell adhesion. J Biol Chem. 1998;273:33354–33359. doi: 10.1074/jbc.273.50.33354. [DOI] [PubMed] [Google Scholar]
- 38.Miura M, Asou H, Kobayashi M, Uyemura K. Functional expression of a full-length cDNA coding for rat neural cell adhesion molecule L1 mediates homophilic intercellular adhesion and migration of cerebellar neurons. J Biol Chem. 1992;267:10752–10758. [PubMed] [Google Scholar]
- 39.Moos M, Tacke R, Scherer H, Teplow D, Fruh K, Schachner M. Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature. 1988;334:701–703. doi: 10.1038/334701a0. [DOI] [PubMed] [Google Scholar]
- 40.New K, Rabkin S. Co-expression of two gene products in the CNS using double-cassette defective herpes simplex virus vectors. Mol Brain Res. 1996;37:317–323. doi: 10.1016/0169-328x(95)00262-q. [DOI] [PubMed] [Google Scholar]
- 41.Pathak RK, Merkle RK, Cummings RD, Goldstein JL, Brown MS, Anderson RG. Immunocytochemical localization of mutant low density lipoprotein receptors that fail to reach the Golgi complex. J Cell Biol. 1988;106:1831–1841. doi: 10.1083/jcb.106.6.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smit LS, Strong TV, Wilkinson DJ, Macek M, Mansoura MK, Jr, Wood DL, Cole JL, Cutting GR, Cohn JA, Dawson DC. Missense mutation (G480C) in the CFTR gene associated with protein mislocalization but normal chloride channel activity. Hum Mol Genet. 1995;4:269–273. doi: 10.1093/hmg/4.2.269. [DOI] [PubMed] [Google Scholar]
- 43.Stowell JN, Craig AM. Axon/dendrite targeting of metabotropic glutamate receptors by their cytoplasmic carboxy-terminal domains. Neuron. 1999;22:525–536. doi: 10.1016/s0896-6273(00)80707-2. [DOI] [PubMed] [Google Scholar]
- 44.Van Camp G, Fransen E, Vits L, Raes G, Willems PJ. A locus-specific mutation database for the neural cell adhesion molecule L1CAM (Xq28). Hum Mutat. 1996;8:391. doi: 10.1002/(SICI)1098-1004(1996)8:4<391::AID-HUMU19>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 45.Vits L, Van Camp G, Coucke P, Fransen E, De Boulle K, Reyniers E, Korn B, Poustka A, Wilson G, Schrander-Stumpel C, Winter RM, Schwartz CE, Willems PJ. MASA syndrome is due to mutations in the neural cell adhesion gene L1CAM. Nat Genet. 1994;7:408–413. doi: 10.1038/ng0794-408. [DOI] [PubMed] [Google Scholar]
- 46.West AE, Neve RL, Buckley KM. Targeting of the synaptic vesicle protein synaptobrevin in the axon of cultured hippocampal neurons: evidence for two distinct sorting steps. J Cell Biol. 1997;139:917–927. doi: 10.1083/jcb.139.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilcox DA, Wautier JL, Pidard D, Newman PJ. A single amino acid substitution flanking the fourth calcium binding domain of alpha IIb prevents maturation of the alpha IIb beta 3 integrin complex. J Biol Chem. 1994;269:4450–4457. [PubMed] [Google Scholar]
- 48.Wong EV, Schaefer AW, Landreth G, Lemmon V. Casein kinase II phosphorylates the neural cell adhesion molecule L1. J Neurochem. 1996a;66:779–786. doi: 10.1046/j.1471-4159.1996.66020779.x. [DOI] [PubMed] [Google Scholar]
- 49.Wong EV, Schaefer AW, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J Biol Chem. 1996b;271:18217–18223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]
- 50.Yamasaki M, Thompson P, Lemmon V. CRASH syndrome: mutations in L1CAM correlate with severity of the disease. Neuropediatrics. 1997;28:175–178. doi: 10.1055/s-2007-973696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yazaki T, Martuza RL, Rabkin SD. Expression of L1 in primary astrocytes via a defective herpes simplex virus vector promotes neurite outgrowth and neural cell migration. Mol Brain Res. 1996;43:311–320. doi: 10.1016/s0169-328x(96)00186-6. [DOI] [PubMed] [Google Scholar]
- 52.Yazdanbakhsh K, Lee S, Yu Q, Reid ME. Identification of a defect in the intracellular trafficking of a kell blood group variant. Blood. 1999;94:310–318. [PubMed] [Google Scholar]
- 53.Yip PM, Zhao X, Montgomery AM, Siu CH. The Arg-Gly-Asp motif in the cell adhesion molecule L1 promotes neurite outgrowth via interaction with the alphavbeta3 integrin. Mol Biol Cell. 1998;9:277–290. doi: 10.1091/mbc.9.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X, Davis JQ, Carpenter S, Bennett V. Structural requirements for association of neurofascin with ankyrin. J Biol Chem. 1998;273:30785–30794. doi: 10.1074/jbc.273.46.30785. [DOI] [PubMed] [Google Scholar]
- 55.Zhao X, Siu CH. Differential effects of two hydrocephalus/MASA syndrome-related mutations on the homophilic binding and neuritogenic activities of the cell adhesion molecule L1. J Biol Chem. 1996;271:6563–6566. doi: 10.1074/jbc.271.12.6563. [DOI] [PubMed] [Google Scholar]
- 56.Zhao X, Yip PM, Siu CH. Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J Neurochem. 1998;71:960–971. doi: 10.1046/j.1471-4159.1998.71030960.x. [DOI] [PubMed] [Google Scholar]
- 57.Zisch AH, Stallcup WB, Chong LD, Dahlin-Huppe K, Voshol J, Schachner M, Pasquale EB. Tyrosine phosphorylation of L1 family adhesion molecules: implication of the Eph kinase Cek5. J Neurosci Res. 1997;47:655–665. doi: 10.1002/(sici)1097-4547(19970315)47:6<655::aid-jnr12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]