

Abstract
To date, delivery of neurotrophic factors has only allowed to transiently protect axotomized facial motoneurons against cell death. In the present report, long-term protection of these neurons was evaluated by continuously expressing the neurotrophic factor glial cell line-derived neurotrophic factor (GDNF) within the facial nucleus using a lentiviral vector system. The viral vector was injected unilaterally into the facial nucleus of 4-month-old Balb/C mice. In contrast to axotomy in other adult rodents, facial nerve lesion in these animals leads to a progressive and sustained loss and/or atrophy of >50% of the motoneurons. This model thus represents an attractive model to evaluate potential protective effects of neurotrophic factors for adult-onset motoneuron diseases, such as amyotrophic lateral sclerosis. One month after unilateral lentiviral vector injection, the facial nerve was sectioned, and the animals were killed 3 months later. Viral delivery of the GDNF gene led to long-term expression and extensive diffusion of GDNF within the brainstem. In addition, axotomized motoneurons were completely protected against cell death, because 95% of the motoneurons were present as demonstrated by both Nissl staining and choline acetyltransferase immunoreactivity. Furthermore, GDNF prevented lesion-induced neuronal atrophy and maintained proximal motoneuron axons, despite the absence of target cell reinnervation. This is the first evidence that viral-mediated delivery of GDNF close to the motoneuron cell bodies of the facial nucleus of adult mice can lead to complete and long-term protection against lesion-induced cell death.
Keywords: axotomy, facial nerve, GDNF, gene expression, gene therapy, gene transfer, lentiviral vector, motoneuron, neuroprotection, neurotrophic factor, Balb/C mice
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disorder characterized by the loss of motoneurons leading to progressive paralysis and death within 2–5 years. With the exception of mutations in the gene for superoxide dismutase I (SOD) found in ∼2% of all cases (Rosen, 1993), the etiology of the disease is still unknown and no treatment is currently available. A number of animal models, such as facial or sciatic nerve axotomy in neonatal rodents, have been developed to evaluate potential protective effects of neurotrophic factors (Sendtner et al., 1990;Koliatsos et al., 1993; Henderson et al., 1994; Zurn et al., 1994; Yan et al., 1995). However, axotomy in neonates leads to a rapid cell death (4–5 d) (Snider et al., 1992), whereas degeneration is progressive and extends over several months in ALS. Second, ALS is an adult-onset motoneuron disease, and mature motoneurons may be less dependent on trophic support than developing neurons (Pollin et al., 1991). In the present report, motoneuron axotomy was performed in adult Balb/C mice because, in contrast to axotomy in other adult rodents, facial nerve lesion in these animals leads to a progressive and long-term loss of motoneurons (Kou et al., 1995; Ferri et al., 1998). This relatively slow cell death more closely resembles the degeneration observed in motoneuron diseases because it extends over a period of several weeks (Kou et al., 1995). It thus represents a better model than axotomy in neonatal animals to evaluate potential protective effects of neurotrophic factors for adult-onset motoneuron disorders. Spinal root avulsion, another adult lesion model, is less appropriate because not only motoneuron loss but also complete loss of associated Schwann cells occurs (Koliatsos et al., 1994).
To date, delivery of recombinant neurotrophic factors close to the cut nerve in neonatal rats and mice has only allowed to transiently protect axotomized motoneurons against cell death (Schmalbruch and Rosenthal, 1995; Vejsada et al., 1995). Long-term protection against degeneration is therefore still a major unresolved issue. To obviate this problem, adenoviral vectors have been used to deliver neurotrophic factors to the cut nerve stump of lesioned motoneurons. However, <10% of the motoneurons retrogradely transported and expressed the transgenes, and only a partial rescue was observed (Baumgartner and Shine, 1997;Gimenez y Ribotta et al., 1997; Gravel et al., 1997). We therefore assessed whether continuous expression of glial cell line-derived neurotrophic factor (GDNF) in the vicinity of the motoneuron cell bodies of the facial nucleus would allow efficient and sustained protection against cell death. GDNF was chosen because it is the most potent neurotrophic factor for cultured embryonic motoneurons described to date (Henderson et al., 1994), as well as the most efficient factor to rescue axotomized neonatal motoneurons when delivered close to the nerve stump (Yan et al., 1995; Vejsada et al., 1998). A lentiviral (human immunodeficiency virus)-based vector system was used to deliver GDNF directly into the brain. The core particles of this vector system are pseudotyped with vesicular stomatitis virus glycoprotein G, which confers a broad tropism and allows to infect a large variety of different cell types (Naldini et al., 1996b). Moreover, the vector has been shown to transduce both dividing and nondividing cells such as neurons and to integrate transgenes into the chromosomes of their targets, resulting in the long-term expression of the transgene. This efficient gene delivery system has been applied previously in vivo in the brain of rats and monkeys (Naldini et al., 1996a;Zufferey et al., 1997; Kordower et al., 1999; Déglon et al., 2000).
MATERIALS AND METHODS
Production of recombinant lentiviral vectors. The cDNA coding for a nuclear-localized β-galactosidase (LacZ), the human GDNF containing a Kozak consensus sequence (a 636 bp fragment, position 1–151 and 1–485; GenBank accession numbers L19062 and L19063), or a mutated GDNF (deletion of amino acids 74–85 of the mature GDNF, leading to the absence of secretion of the trophic factor) (Choi-Lundberg et al., 1997) were cloned in the SIN-W-PGK transfer vector (Déglon et al., 2000). The packaging construct used in this study was the pCMVDR-8.92 (derived from the pCMVDR-8.91plasmid: destruction of the BamHI restriction site in the coding region of the rev gene). To further decrease the risk of recombination and production of replication-competent retroviruses, the Rev gene was inserted in the pRSV-Rev plasmid. The viral particles were pseudotyped with the vesicular stomatitis virus G-protein encoded by the pMD.G plasmids described previously (Naldini et al., 1996b). The viral particles were produced by transient transfection of 293T cells (Naldini et al., 1996b). Forty-eight hours later, the supernatant was collected and filtered, and the particle content was determined by ELISA assay (NEN, Boston, MA). High-titer stocks were obtained by ultracentrifugation. The pellet was resuspended in PBS and 1% BSA and stored frozen at −80°C. The batches of virus were tested for the absence of replication-competent viral vectors (Naldini et al., 1996b). The titers of the lentiviral vector encoding LacZ (LV-LacZ) stocks were determined on 293T cells. The cells were plated at a density of 2 × 105 cells per well on six-well tissue culture dishes (Costar, Cambridge, MA). Serial dilutions of the viral stocks were added, and the number of LacZ-infected cells was analyzed 48 hr later. Titers were calculated by counting the number of blue foci per well and dividing it by the dilution factor. LacZ-expressing viruses with ranges from 3 to 7 × 108 transfection units/ml were obtained. In all in vivo experiments, LacZ, GDNF, or mutated GDNF-expressing virsuses (LV-LacZ, LV-GDNF, and LV-mGDNF, respectively) were matched for particle content (200,000 ng p24 antigen/ml as measured by ELISA assay). These experiments were performed in biosafety level 2 laboratories.
Animals and surgery. Adult BalB/C mice (4 months old; IFFA Credo, Reims, France) were anesthetized with an intraperitoneal injection of 60 mg/kg pentobarbital and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). The experiments were performed in accordance with the European Community Council Directive (86/609/EEC) for care and use of laboratory animals. A midline incision was performed on the skin covering the skull, and the bregma point was determined. The skull was opened with a dental drill at a location 6.2 mm caudal and 1.2 mm lateral to the bregma, the coordinates for the facial nucleus. One microliter of lentiviral vector expressing either GDNF, mutated GDNF, or the reporter gene LacZ was injected at the rate of 0.2 μl/min down to a depth of 5.6 mm. Other control groups consisted of animals that received either a 1 μl PBS solution at the same coordinates or no treatment. Thirty days later, the animals were anesthetized with pentobarbital. The skin was incised below the right ear. The right facial nerve was exposed, freed of connective tissue, and transected at its exit from the stylomastoïd foramen. A small segment of the nerve was removed to prevent regeneration. The skin incision was sutured with a Prolene 6–0 filament (Johnson & Johnson). The success of the procedure was confirmed by the absence of movement of the whiskers on the side of the lesion. The animals were then returned to their cages and maintained in a P2 facility. They were killed 30 or 90 d later by an overdose of pentobarbital and perfused under RNase-free condition with PBS, followed by a 4% paraformaldehyde solution. Thereafter, the brainstem was removed and post-fixed in the same fixative overnight and cryoprotected in PBS containing 25% sucrose. Frozen sections of the brainstem (20-μm-thick) were cut on a Reichert Jung Cryocut 1800 at −24°C.
Immunohistochemistry. Brainstem sections from animals injected with LV-LacZ were processed for β-galactosidase immunoreactivity and double stained for glial fibrillary acidic protein (GFAP) or SMI-32, an antibody recognizing nonphosphorylated neurofilament protein used to identify motoneurons and their central processes. Free-floating sections were blocked in a solution containing 10% normal goat serum and 0.1% Triton X-100 in PBS for 3 hr. The following primary antibodies were used: rabbit polyclonal anti-β-galactosidase, 1:500 (5 Prime → 3 Prime Inc., Boulder, CO), mouse monoclonal anti-GFAP, 1:50 (Boehringer Mannheim, Mannheim, Germany), and mouse anti-SMI-32, 1:4000 (Sternberger Monoclonals, Lutherville, MD). Primary antibodies were diluted in the blocking solution, and sections were incubated overnight at 4°C. Sections were then incubated for 3 hr with the secondary goat anti-mouse Cy3-conjugated antibody at a 1:400 dilution and goat anti-rabbit FITC-conjugated antibody at a 1:100 dilution in PBS (Jackson ImmunoResearch, West Grove, PA). Finally, sections were mounted onto glass slides and coverslipped using Fluorosave (Calbiochem, La Jolla, CA). For choline acetyltransferase (ChAT) immunohistochemistry, sections were washed in PBS and incubated in 10% normal goat serum for 2 hr, followed by an overnight incubation at 4°C with a monoclonal ChAT antibody at a 1:400 dilution (generous gift of Dr. Cozzari, Istituto di Biologia Cellulare, Roma, Italy) (Umbriaco et al., 1994). Control sections were incubated without the primary antibody. After incubation with the secondary antibody, binding was detected using an Elite avidin-biotin-peroxidase kit (Vector Laboratories, Burlingame, CA). To evaluate diffusion of GDNF in the brainstem, immunohistochemistry using an anti-human GDNF antibody was performed. Briefly, 20-μm-thick sections were first quenched for 20 min in 0.1m sodium periodate, followed by a 1 hr blocking in 100 mm Tris/150 mm NaCl (TBS) containing 2% BSA, 3% normal horse serum (NHS), and 0.5% Triton X-100. They were then incubated for 48 hr at room temperature in TBS containing biotinylated goat anti-GDNF antibody (R & D Systems, Wiesbaden, Germany) at a 1:250 dilution, 1% BSA, 1% NHS, and 0.04%Triton X-100. The sections were then incubated for 1 hr at room temperature in the same buffer containing biotinylated horse anti-goat IgG (1:200) (Vector Laboratories), 1% NHS, and 1% BSA. Antibody binding was revealed according to standard procedures using an Elite avidin-biotin-peroxidase kit (Vector Laboratories) and DAB.
In situ hybridization. To confirm the expression of the transgene close to the facial nucleus, in situ hybridization was performed with a woodchuck hepatitis virus (WHV) digoxigenin (DIG) riboprobe. The WHV regulatory element is present on all lentiviral constructs used in this study (Déglon et al., 2000). A 591 bp fragment of the WHV post-transcriptional element (position 1093–1684 of the WHV complete genome; GenBank accession number J04514) was cloned in the pBluescript SK+ plasmid (Stratagene, La Jolla, CA). The antisense riboprobe was synthesized from a BamHI linearized pBluescript-WHV vector according to the supplier's manual (DIG RNA labeling kit; Roche Products, Rotkreuz, Switzerland). The concentration of the digoxigenin-11 UTP- labeled probe was measured by dot blot using a DIG-labeled control RNA as internal standard. Frozen brain tissue obtained as described above was embedded in RNase-free Tissue Tek OCT Compound (Bayer, Zurich, Switzerland) and 20 μm sections were cut at −24°C using a cryostat. Free-floating sections were washed in 0.1% DEPC in PBS twice for 15 min, treated with HCl 0.02 N for 10 min, and equilibrated in 5× SSC (0.75 mm NaCl and 0.075 mm Na3 citrate) for 15 min. The sections were then prehybridized in 50% formamide, 5× SSC, 5% dextran sulfate, 1× Denhardt's solution (1% bovine serum albumin, 1% polyvinylpyrrolidone, and 1% Ficoll 400), 250 μg/ml heat-denatured sheared salmon sperm DNA, and 100 μg/ml yeast t-RNA for 2 hr at 48°C. Hybridization was performed overnight at 48°C with the DIG riboprobe in the prehybridization buffer. Sections were then washed in 2× SSC for 30 min at room temperature, followed by 2× SSC for 1 hr and 0.1× SSC for 1 hr at 48°C. The hybridized DIG riboprobe was revealed as described by the manufacturer (Boehringer Mannheim).
GDNF ELISA assay. To evaluate the level of GDNF expression in the brainstem, 4-month-old Balb/C mice received intrafacial nucleus injections of either 1 μl of lentivirus carrying the GDNF or LacZ gene, or the carrier solution PBS (see below). Sixty days later, the animals were killed with an overdose of pentobarbital and perfused with an ice-cold solution of PBS containing 0.2% ascorbic acid. The brainstem was immediately removed and frozen at −70°C on dry ice. It was then cut in the midline and resuspended in 500 μl of PBS containing 0.5% Triton X-100 and protease inhibitors [chymotrypsin, papain, pronase, thermolysin, and trypsin (Complete Protease Inhibitor; Boehringer Mannheim)]. The proteins were extracted by sonication and centrifuged at 10,000 rpm for 10 min. The supernatants were frozen to −70°C before further use. The amount of human GDNF present in the sample was determined using an ELISA assay (Promega, Madison, WI). Because of the low recovery rate of GDNF from mouse brain, an internal standard curve was performed by adding increasing doses of recombinant human GDNF to brainstem tissue of control mice before extraction.
Motoneuron counts. To evaluate the number of motoneurons in the facial nucleus, adjacent sections stained for Nissl, and ChAT and SMI-32 immunoreactivity, were counted in every fifth section on both the lesioned and control sides. Motoneuron survival was expressed as the percentage of cells surviving on the lesioned side compared with the nonlesioned side. With Nissl staining, only neurons with clearly visible nucleoli and a large soma diameter (∼20 μm) were counted. All ChAT-immunopositive cells were counted. SMI-32-positive cells were counted as motoneurons when they presented a large soma diameter. The number of cells expressing the GDNF construct was evaluated by in situ hybridization for the WHV post-transcriptional enhancer element in randomly selected animals.
Motoneuron soma size. Motoneuron soma size was measured by digitizing the most central section of the facial nucleus of all animals in each group (∼350 cells per group) with a Panasonic CCD camera and by drawing the perimeter of each motoneuron at a magnification of 200×. Results were analyzed using NIH Image software (version 1.47).
Statistical analysis. All data were expressed as the mean ± SEM. For statistical evaluation of motoneuron numbers identified by either Nissl staining or ChAT immunoreactivity, the number of cells present in each facial nucleus was counted, multiplied by five (every fifth section was counted), and then multiplied by the Abercrombie correction factor, which compensates for double counting in adjacent sections (Abercrombie, 1946). Standard one-way ANOVA followed by a Scheffe's PLSD post hoc test was performed using the statistical software Statview 4.0. Statistical analyses of the square areas of the cell bodies were performed using one-way ANOVA followed by a Scheffe's PLSD post hoctest.
RESULTS
Transgene expression in unlesioned animals
β-galactosidase expression
Two months after LV-LacZ injection, β-galactosidase was found to be expressed in 20,595 ± 2737 cells (n = 4) in brainstem sections of nonlesioned animals. Labeling was observed over a rostrocaudal and lateral distance of 2.5 ± 0.13 and 0.9 ± 0.11 mm (n = 4), respectively, including the whole extent of the facial nucleus (Fig.1). Long-term expression of the transgene was confirmed by the presence of 24,213 ± 3130 (n = 4) β-galactosidase-positive cells 4 months after the injection. Because of the small size and the location of the facial nucleus, the injection needle could be placed within the facial nucleus itself in approximately half of the cases, as evidenced by the localization of the needle tract by Nissl staining. In the latter case, 6293 ± 753 (n = 2) cells were infected within the facial nucleus itself. Double-labeling with anti-SMI-32 antibodies (used as a marker for motoneurons) showed that 35 ± 7.7% (n = 2) of the motoneurons expressed β-galactosidase. When the injection was performed above the nucleus, 10.9 ± 5.1% (n = 2) of the SMI-32-positive motoneurons were labeled. Using the glial cell marker GFAP, 17.27 ± 1.2% (n = 4) of the infected cells were found to be astrocytes.
Fig. 1.

β-galactosidase expression in the brainstem of nonlesioned mice 60 d after the injection of LV-LacZ. The number of β-galactosidase-positive cells per 20 μm section was evaluated every 100 μm over a rostrocaudal distance of 3 mm. The center of the facial nucleus was determined and used as a reference point in the four animals (number of cells per section ± SEM).
Localization of the GDNF construct
The GDNF construct itself was localized by in situhybridization for the post-transcriptional enhancer element of the WHV (Fig. 2). Sixty days after viral infection, the mRNA was found to be present in 17,045 ± 981 (n = 4) cells covering a distance of 2.4 ± 0.2 mm rostrocaudally. In the cases in which the injection was directly within the facial nucleus, 41.6 ± 2.1% (n = 3) of the motoneurons expressed the transgene, as verified on adjacent Nissl-stained sections.
Fig. 2.
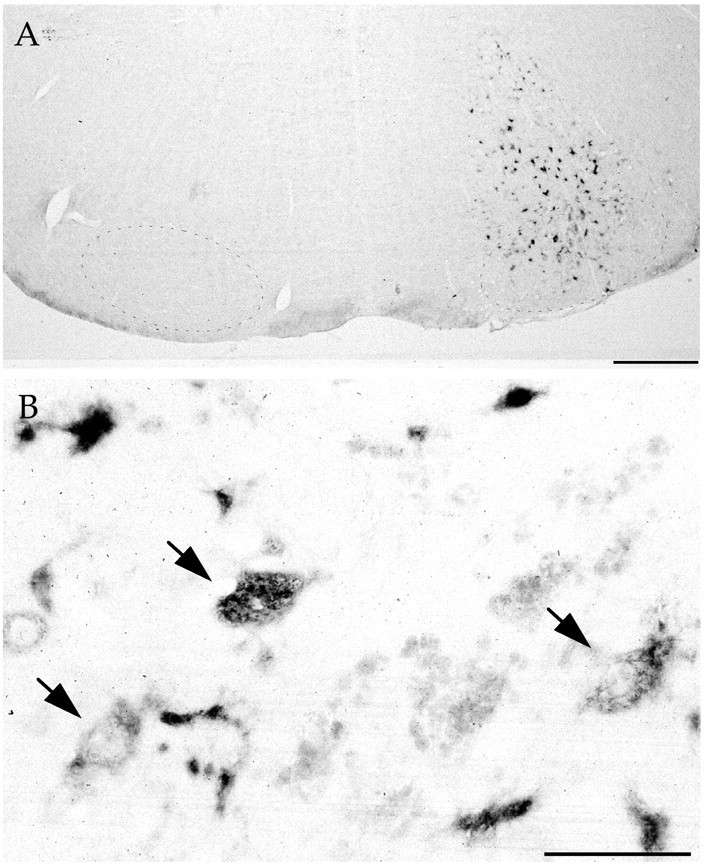
In situ hybridization for the WHV element. A, Nonlesioned mice 60 d after LV-GDNF injection in the facial nucleus. The region of the facial nucleus is indicated by small dots. Scale bar, 500 μm.B, Close-up view showing motoneurons in the facial nucleus (arrows) and other infected cells. Scale bar, 100 μm.
GDNF expression and diffusion
In vitro infection of 293T cells with LV-GDNF allows the synthesis and secretion of 319 ± 46 ng GDNF/106 cells per day, as assayed by ELISA (Déglon et al., 2000). In addition, the released GDNF is bioactive because it increases ChAT activity twofold to threefold in embryonic day 14 spinal cord cultures (Déglon et al., 2000). Infection with LV-mGDNF resulted in no detectable GDNF production. An ELISA assay for GDNF was performed 60 d after viral infection on brainstem tissue punches of 0.8 mm diameter and 2 mm depth. Mice that had received injections of LV-GDNF expressed 1.69 ± 0.51 ng of GDNF per punch (n = 4), whereas GDNF levels were below the detection limit on the uninjected side or in control animals that had received LV-LacZ (n = 4). To assess long-term expression of GDNF in and around the facial nucleus, GDNF production and diffusion were evaluated by immunohistochemistry. The micrograph in Figure 3 reveals that GDNF diffused over long distances, beyond the location of the transduced cells (Fig.2).
Fig. 3.
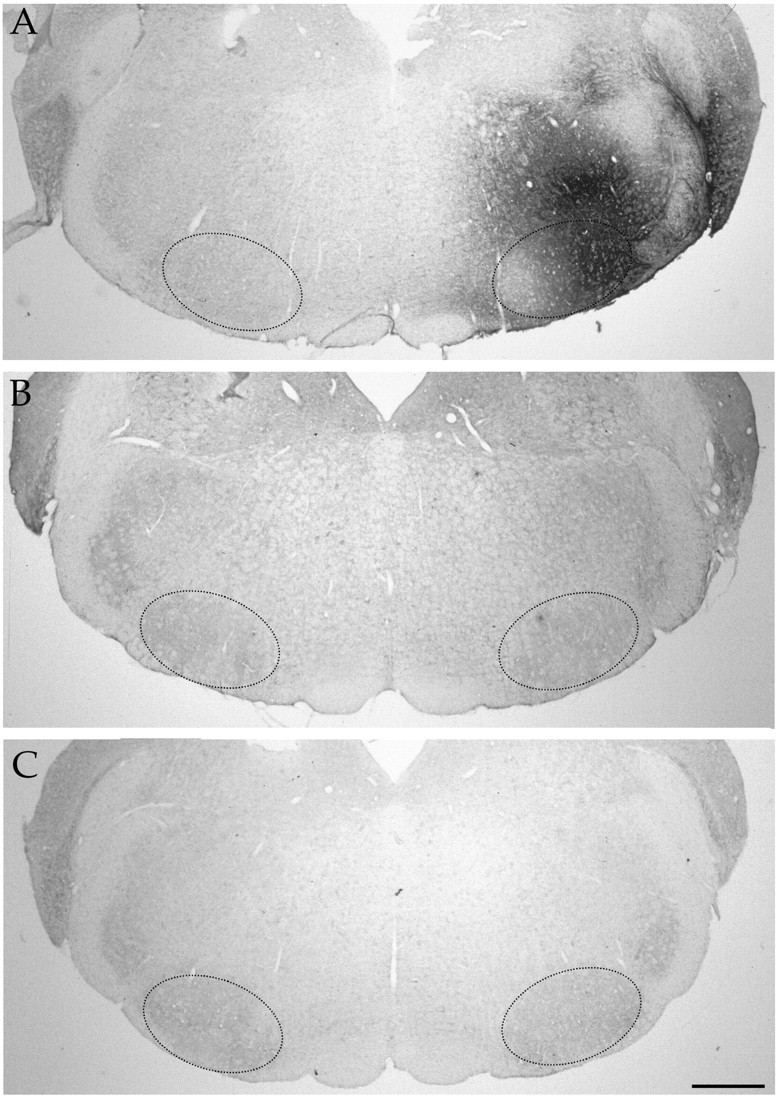
Immunodetection of GDNF in the brainstem.A, GDNF immunoreactivity 4 months after injection of LV-GDNF close to the facial nucleus. The region of the facial nucleus is indicated by small dots. Animals injected with LV-LacZ (B) or LV-mGDNF (C). Scale bar, 500 μm.
Protective effects of GDNF in lesioned animals
Facial nerve axotomy in 5-month-old Balb/C mice led to a severe loss of motoneurons, with only 40.2 ± 2.1 (n = 4) and 46.3 ± 2.5% (n = 6) of the neurons surviving compared with the nonlesioned side 30 and 90 d after the lesion, respectively (no statistical difference between these two groups;p > 0.1) (Fig.4A,B, Nissl staining). Note that motoneuron loss does not seem to occur naturally in these mice with age, because 2947 ± 30 and 2929 ± 200 (n = 4) facial motoneurons were present at 2 and 8 months of age, respectively, and because ChAT levels were also stable with age (data not shown). Injection of PBS, LV-LacZ, or LV-mGDNF had no significant effect on cell loss, with 44.95 ± 0.8 (n = 4), 51.8 ± 7.2 (n = 4), and 44 ± 6.6% (n = 6) of the motoneurons remaining after 30 d, respectively (Fig. 4A) (no statistical difference between these groups and untreated animals). Similar results were obtained in the different control groups 90 d after the lesion (Fig. 4B). In contrast, animals pretreated with LV-GDNF showed a complete rescue of the axotomized motoneurons, with 109.4 ± 4.3 (n = 7,p < 0.0001) and 95 ± 4.9% (n = 8, p < 0.0001) of the cells remaining after 30 and 90 d, respectively (Fig.4A,B). Evaluation of the number of ChAT-positive cells 30 d after the lesion showed that only 26.2 ± 4.8 (n = 4) and 23.7 ± 8.9% (n = 4) of the cells were present in PBS- and LV-mGDNF-treated animals, respectively, whereas in the presence of GDNF, 77.9 ± 6.1% of the cells expressed ChAT (n= 4, p < 0.001) (Fig. 4C). Protective effects were sustained for several months, with 95.3 ± 2.5% (n = 8) of the cells expressing ChAT at 3 months in the presence of GDNF but only 29.9 ± 5.2 (n = 6), 35.4 ± 5.2 (n = 6), 38.37 ± 6.7 (n = 6), and 45.5 ± 4.5% (n = 6) in animals treated with LV-LacZ, LV-mGDNF, PBS, or no treatment, respectively (Fig. 4D). Photomicrographs of Nissl- and ChAT-stained sections 3 months after the lesion are shown in Figures 5 and6, respectively. Importantly, not only cell bodies but also central processes were maintained in the brainstem in the presence of GDNF, as evidenced by immunostaining with anti-neurofilament antibody (Fig. 7). (Note that SMI-32 does not distinguish between axons and dendrites.) Thus, motoneuron cell bodies as well as proximal processes were present with GDNF despite the absence of target cell reinnervation, as evidenced by the absence of whisker movement.
Fig. 4.

Number of motoneurons in mice injected with PBS, LV-LacZ, LV-mGDNF, or LV-GDNF 30 (A, C) and 90 (B, D) d after transection of the facial nerve. A, B, Nissl staining.C, D, ChAT immunostaining. Motoneuron survival is expressed as the percentage of cells surviving on the lesioned compared with the nonlesioned side. Only GDNF is statistically different from PBS, LacZ, and mGDNF or untreated mice; ***p < 0.001.
Fig. 5.
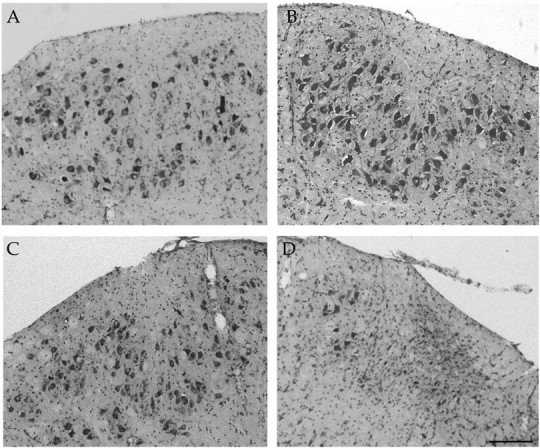
Nissl stains of cross-sections through the facial nucleus from lesioned animals injected with LV-GDNF (A,B) or LV-mGDNF (C, D) 90 d after nerve transection (Nissl staining). A,C, Nonlesioned side. B, D, Lesioned side. Scale bar, 200 μm.
Fig. 6.

ChAT immunostaining of cross-sections through the facial nucleus from lesioned animals injected with LV-GDNF (A) and LV-LacZ (B) 90 d after nerve transection. Left, Nonlesioned side.Right, Lesioned side. Scale bar, 500 μm.
Fig. 7.

Central processes of facial motoneurons stained with anti-neurofilament antibody (SMI-32). A, Nonlesioned, noninjected side. B, Lesioned side injected with lenti-mutated GDNF. C, Lesioned side injected with lenti-GDNF. Scale bar, 40 μm.
Assessment of the distribution of motoneuron soma sizes on the lesioned and the nonlesioned side showed that there was a decrease in the percentage of cells in the 400–500 μm2size and an increase in the 250–350 μm2size 3 months after axotomy (Fig.8A–E). This indicates that, in addition to cell loss, there is also atrophy in neurons remaining after the lesion. Furthermore, there is an increase in the percentage of cells in the 400–700 μm2size in the presence of GDNF on the lesioned but also on the nonlesioned side (Fig. 8C,F), indicating hypertrophy induced by the neurotrophic factor. Similar changes were observed at 1 month. Continuous expression of GDNF in the facial nucleus region induced a sustained weight loss starting day 3 after injection and reaching 31.2 ± 2.4% (n = 8) loss at 40 d, which it maintained for 4 months. Despite this important loss and a 27.3 ± 3.1% (n = 8) decrease in food intake, these animals have unchanged activity levels as evaluated using a monitoring box (data not shown).
Fig. 8.
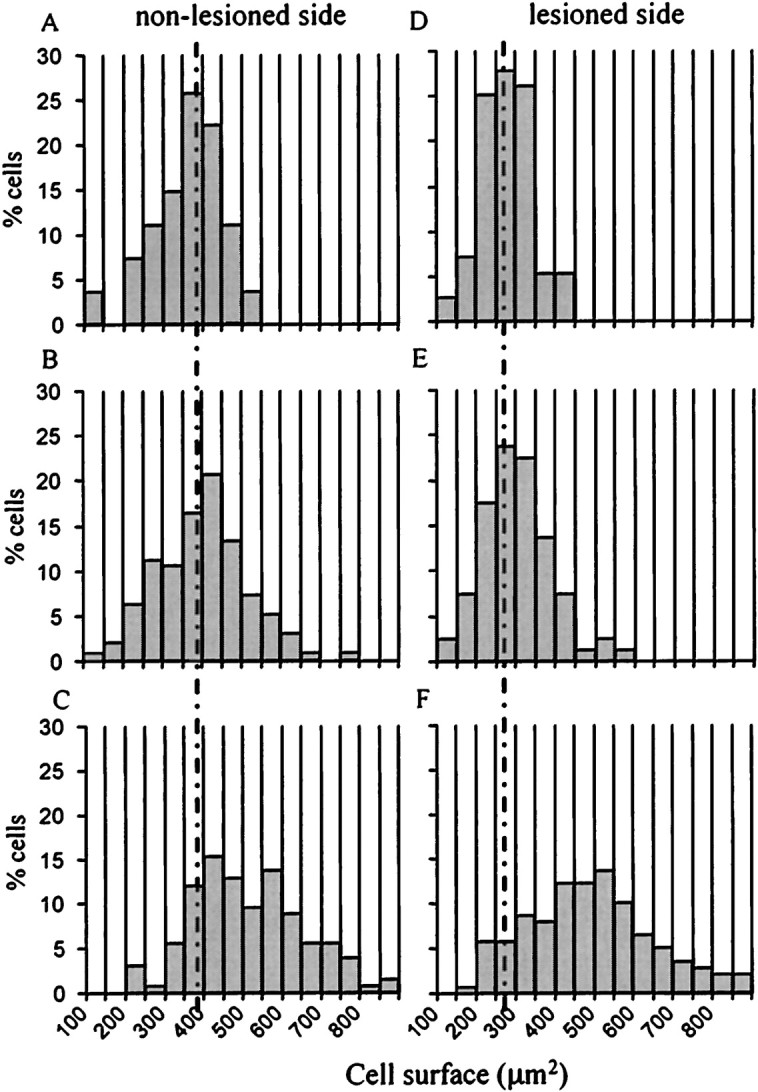
Effect of GDNF on the motoneuron size distribution 3 months after the lesion. A–C, Size distribution (in square micrometers) of facial motoneurons on the nonlesioned side:A, untreated; B, LacZ; andC, GDNF. D–F, Soma size distribution on the lesioned side: D, Noninfected; E, LacZ; and F, GDNF-infected animals.
DISCUSSION
The present report demonstrates that, using facial nerve lesion in adult Balb/C mice, expression of the neurotrophic factor GDNF close to the motoneuron cell bodies of the facial nucleus using a lentiviral vector system leads to extensive diffusion of GDNF in the brainstem and to a complete and sustained protection against lesion-induced cell death.
Infection of the brainstem with a lentiviral vector encoding the reporter gene LacZ leads to β-galactosidase expression in >20,000 cells within and surrounding the facial nucleus. A comparable number of transduced cells has been obtained previously in rat striatum using a similar vector (Zufferey et al., 1997). Viral infection allows to stably express the transgene for up to at least 4 months. Only a mild inflammation consisting in minor perivascular cuffing and in aggregation of lymphocytes around the injection tract is observed. Both neurons and astrocytes are transduced, with up to 45% of the SMI-32-positive facial motoneurons expressing the reporter gene in the best experiments. In comparison, only 5–10% of the axotomized facial motoneurons express β-galactosidase after injection of recombinant adenovirus into lesioned nerves or facial muscles of neonatal rats (Baumgartner and Shine, 1997; Gravel et al., 1997).
In contrast to nerve axotomy in other strains of adult mice and in rats, which only results in the downregulation of a number of proteins, including ChAT, the enzyme responsible for the synthesis of the neurotransmitter acetylcholine (Lams et al., 1988; Yan et al., 1994), facial nerve axotomy in 2-month-old Balb/C mice has been shown to lead to 20–30% cell loss (Kou et al., 1995; Ferri et al., 1998). In our hands, facial nerve lesion leads to the sustained loss of as much as 60% of the motoneurons, probably because of the older age of the mice at the time of the lesion (5 months). However, the difference may also in part be attributable to our counting method because we considered only clearly identifiable motoneurons with a large soma diameter (∼20 μm). Therefore, part of the cells not included in our counts may have been counted by others. In addition, some of the motoneurons that were not counted were probably atrophic and not lost (Fig. 8).
Intraparenchymal delivery of the GDNF gene in adult Balb/C mice leads to sustained expression and extensive diffusion of GDNF within the brainstem. This viral gene delivery system thus allows much better diffusion of the neurotrophic factor within the brain parenchyma than intraventricular bolus injections of GDNF (Lapchak et al., 1998). We also demonstrate that GDNF expression within and close to the facial nucleus allows to completely protect facial motoneurons against axotomy-induced cell death and/or atrophy, for as long as 3 months. Recombinant GDNF or GDNF released by encapsulated genetically engineered cells applied in the vicinity of the cut nerve in neonatal rodents has also been shown to be protective (Henderson et al., 1994;Zurn et al., 1994; Oppenheim et al., 1995; Yan et al., 1995; Houenou et al., 1996). However, only a transient, 1–2 week rescue could be obtained (Vejsada et al., 1998). Protection lasting for 28 d was obtained in neonatal rats by repeated application of GDNF at days 0 and 14 after axotomy. However, although rescue was complete at 14 d, it was only partial (40%) at 28 d (Matheson et al., 1997). In an attempt to prolong the rescue period, adenoviral vectors have been used recently to deliver neurotrophic factors either to denervated muscles (Baumgartner and Shine, 1997; Gimenez y Ribotta et al., 1997) or directly into the lesioned nerves (Gravel et al., 1997). Although survival of axotomized motoneurons was maintained for at least 5 weeks under those conditions (Gravel et al., 1997), only a 40–60% rescue was observed (Baumgartner and Shine, 1997; Gimenez y Ribotta et al., 1997; Gravel et al., 1997). This may be attributable to the fact that only 5–10% of the motoneurons were transduced. In contrast, direct delivery and continuous expression of GDNF via viral gene transfer within and surrounding the facial motoneuron cell bodies leads to sustained and complete neuroprotection as evaluated both by Nissl staining and ChAT immunohistochemistry. It is interesting to note that, despite the variation in the number of motoneurons infected by the lentiviral vector, we observe no correlation between the percentage of motoneurons infected by LV-GDNF and the extent of rescue. Thus, GDNF released by neurons and astrocytes at a distance can reach motoneuron cell bodies of the facial nucleus and protect them against degeneration. This indicates that both autocrine and paracrine mechanisms are involved. Evidence for a paracrine mechanism of action of GDNF has been obtained previously in neonatal rats in which a larger number of motoneurons survived after facial nerve axotomy than were transduced (Baumgartner and Shine, 1997; Gravel et al., 1997). Upregulation of the signaling component of the GDNF receptorc-ret and the ligand binding subunit GDNFR-α has been described to occur after facial nerve lesion in the rat (Burazin and Gundlach, 1998). Such increased expression, combined with long-distance diffusion of GDNF and action both via an autocrine and paracrine mechanism, may explain the efficient neuroprotection obtained in adult Balb/C mice.
GDNF does not only protect facial motoneuron cell bodies against cell death, but it also maintains the proximal processes of these cells. This occurs despite the lack of target cell reinnervation as evidenced by the absence of whisker movement on the lesioned side. In addition, GDNF also prevents atrophy and/or induces hypertrophy in the lesioned motoneurons. Motoneuron hypertrophy has also been described to occur with GDNF after neonatal motor nerve axotomy and ventral root avulsion (Henderson et al., 1994; Li et al., 1995; Oppenheim et al., 1995). The mechanism for this hypertrophy is unknown but may resemble the mechanism observed with nerve growth factor on sympathetic and sensory neurons (Levi-Montalcini, 1987).
Mice treated with GDNF show a sustained weight loss. This effect appears to be specific to GDNF delivery close to the facial nucleus, because intrastriatal and intranigral injection of identical amounts of LV-GDNF in rat and mouse does not induce weight loss (Déglon et al., 2000; data not shown). A 10–15% weight reduction has been described previously to occur after intraventricular injection of microgram quantities of GDNF (Lapchak et al., 1997; Giehl et al., 1998). The mechanisms responsible for this weight loss are unknown. It is possible that indirect activation of higher brain centers involved in the control of feeding occurs in the present study (Kupfermann, 1994).
In conclusion, the present report provides the first evidence that continuous delivery of nanogram quantities of GDNF close to the facial nucleus of adult mice via a lentiviral vector system can lead to (1) sustained expression of the transgene, (2) extensive diffusion of the molecule, and (3) complete and long-term protection of motoneurons and their central processes against lesion-induced cell death and atrophy. Delivery of trophic factors close to the degenerating motoneuron cell bodies may be a prerequisite to obtain significant neuroprotection in SOD transgenic mice, currently the best model of ALS (Gurney et al., 1994), as well as in ALS patients, because application in the periphery has not given any positive results so far.
Footnotes
This work was supported by the Swiss National Science Foundation. A.F.H. is supported by the Roche Research Foundation MD-PhD program. We thank Anne Maillard, Fabienne Pidoux, Maria Rey, and Dana Hornfeld for their technical assistance and helpful comments.
Correspondence should be addressed to Dr. Anne D. Zurn, Division of Surgical Research and Gene Therapy Center, Pavillon 4, Centre Hospitalier Universitaire Vaudois, 1011 Lausanne, Switzerland. E-mail:anne.zurn@chuv.hospvd.ch.
REFERENCES
- 1.Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- 2.Baumgartner BJ, Shine HD. Targeted transduction of CNS neurons with adenoviral vectors carrying neurotrophic factor genes confers neuroprotection that exceeds the transduced population. J Neurosci. 1997;17:6504–6511. doi: 10.1523/JNEUROSCI.17-17-06504.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burazin TA, Gundlach AL. Up-regulation of GDNFR-alpha and c-ret mRNA in facial motoneurons following facial nerve injury in the rat. Mol Brain Res. 1998;55:331–336. doi: 10.1016/s0169-328x(98)00017-5. [DOI] [PubMed] [Google Scholar]
- 4.Choi-Lundberg DL, Lin Q, Chang YN, Chiang YL, Hay CM, Mohajeri H, Davidson BL, Bohn MC. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science. 1997;275:838–841. doi: 10.1126/science.275.5301.838. [DOI] [PubMed] [Google Scholar]
- 5.Déglon N, Tseng JL, Bensadoun J-C, Zurn AD, Arsenijevic Y, Pereira de Almeida L, Zufferey R, Trono D, Aebischer P. Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson's disease. Hum Gene Ther. 2000;11:179–190. doi: 10.1089/10430340050016256. [DOI] [PubMed] [Google Scholar]
- 6.Ferri CC, Moore FA, Bisby MA. Effects of facial nerve injury on mouse motoneurons lacking the p75 low-affinity neurotrophin receptor. J Neurobiol. 1998;34:1–9. [PubMed] [Google Scholar]
- 7.Giehl KM, Schacht CM, Yan Q, Mestres P. Infusion of GDNF into the cerebrospinal fluid through two different routes: effects on body weight and corticospinal neuron survival. NeuroReport. 1998;9:2809–2813. doi: 10.1097/00001756-199808240-00023. [DOI] [PubMed] [Google Scholar]
- 8.Gimenez y Ribotta M, Revah F, Pradier L, Loquet I, Mallet J, Privat A. Prevention of motoneuron death by adenovirus-mediated neurotrophic factors. J Neurosci Res. 1997;48:281–285. doi: 10.1002/(sici)1097-4547(19970501)48:3<281::aid-jnr11>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.Gravel C, Gotz R, Lorrain A, Sendtner M. Adenoviral gene transfer of ciliary neurotrophic factor and brain-derived neurotrophic factor leads to long-term survival of axotomized motor neurons. Nat Med. 1997;3:765–770. doi: 10.1038/nm0797-765. [DOI] [PubMed] [Google Scholar]
- 10.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddigue T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 11.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 12.Houenou LJ, Oppenheim RW, Li L, Lo AC, Prevette D. Regulation of spinal motoneuron survival by GDNF during development and following injury. Cell Tissue Res. 1996;286:219–223. doi: 10.1007/s004410050690. [DOI] [PubMed] [Google Scholar]
- 13.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 14.Koliatsos VE, Price WL, Pardo CA, Price DL. Ventral root avulsion: an experimental model of death of adult motor neurons. J Comp Neurol. 1994;342:35–44. doi: 10.1002/cne.903420105. [DOI] [PubMed] [Google Scholar]
- 15.Kordower P, Bloch J, Ma SY, Chu Y, Palfi S, Roitberg BZ, Emborg M, Hantraye P, Déglon N, Aebischer P. Lentiviral gene transfer to the nonhuman primate brain. Exp Neurol. 1999;160:1–16. doi: 10.1006/exnr.1999.7178. [DOI] [PubMed] [Google Scholar]
- 16.Kou SY, Chiu AY, Patterson PH. Differential regulation of motor neuron survival and choline acetyltransferase expression following axotomy. J Neurobiol. 1995;27:561–572. doi: 10.1002/neu.480270410. [DOI] [PubMed] [Google Scholar]
- 17.Kupfermann I. Neural control of feeding. Curr Opin Neurobiol. 1994;4:869–876. doi: 10.1016/0959-4388(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 18.Lams BE, Isacson O, Sofroniew MV. Loss of transmitter-associated enzyme staining following axotomy does not indicate death of brainstem cholinergic neurons. Brain Res. 1988;475:401–406. doi: 10.1016/0006-8993(88)90635-x. [DOI] [PubMed] [Google Scholar]
- 19.Lapchak PA, Miller PJ, Collins F, Jiao S. GDNF attenuates behavioural deficits and regulates nigrostriatal dopaminergic and peptidergic markers in 6-hydroxydopamine-lesioned adult rats: comparison of intraventricular and intranigral delivery. Neuroscience. 1997;78:61–72. doi: 10.1016/s0306-4522(97)83045-x. [DOI] [PubMed] [Google Scholar]
- 20.Lapchak PA, Araujo DM, Hilt DC, Jiao S, Collin F, Miyoshi Y, Yi A, Zhang Z, Gash DM. Topographical distribution of [125I]-glial cell line-derived neurotrophic factor in unlesioned and MPTP-lesioned rhesus monkey brain following a bolus intraventricular injection. Brain Res. 1998;789:9–22. doi: 10.1016/s0006-8993(97)01495-9. [DOI] [PubMed] [Google Scholar]
- 21.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Wu W, Lin LF, Lei M, Oppenheim RW, Houenou LJ. Rescue of adult mouse motoneurons from injury-induced cell death by glial cell line-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:9771–9775. doi: 10.1073/pnas.92.21.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matheson CR, Wang J, Collins FD, Yan Q. Long-term survival effects of GDNF on neonatal rat facial motoneurons after axotomy. NeuroReport. 1997;8:1739–1742. doi: 10.1097/00001756-199705060-00034. [DOI] [PubMed] [Google Scholar]
- 24.Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996a;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996b;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 26.Oppenheim RW, Houenou LJ, Johnson JE, Lin LF, Li L, Lo AC, Newsome AL, Prevette DM, Wang S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- 27.Pollin MM, McHanwell S, Slater CR. The effect of age on motor neurone death following axotomy in the mouse. Development. 1991;112:83–89. doi: 10.1242/dev.112.1.83. [DOI] [PubMed] [Google Scholar]
- 28.Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364:362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- 29.Schmalbruch H, Rosenthal A. Neurotrophin-4/5 postpones the death of injured spinal motoneurons in newborn rats. Brain Res. 1995;700:254–260. doi: 10.1016/0006-8993(95)00982-v. [DOI] [PubMed] [Google Scholar]
- 30.Sendtner M, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature. 1990;345:440–441. doi: 10.1038/345440a0. [DOI] [PubMed] [Google Scholar]
- 31.Snider WD, Elliott JL, Yan Q. Axotomy-induced neuronal death during development. J Neurobiol. 1992;23:1231–1246. doi: 10.1002/neu.480230913. [DOI] [PubMed] [Google Scholar]
- 32.Umbriaco D, Watkins KC, Descarries L, Cozzari C, Hartman BK. Ultrastructural and morphometric features of the acetylcholine innervation in adult rat parietal cortex: an electron microscopic study in serial sections. J Comp Neurol. 1994;348:351–373. doi: 10.1002/cne.903480304. [DOI] [PubMed] [Google Scholar]
- 33.Vejsada R, Sagot Y, Kato AC. Quantitative comparison of the transient rescue effects of neurotrophic factors on axotomized motoneurons in vivo. Eur J Neurosci. 1995;7:108–115. doi: 10.1111/j.1460-9568.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- 34.Vejsada R, Tseng JL, Lindsay RM, Acheson A, Aebischer P, Kato AC. Synergistic but transient rescue effects of BDNF and GDNF on axotomized neonatal motoneurons. Neuroscience. 1998;84:129–139. doi: 10.1016/s0306-4522(97)00497-1. [DOI] [PubMed] [Google Scholar]
- 35.Yan Q, Matheson C, Lopez OT, Miller JA. The biological responses of axotomized adult motoneurons to brain-derived neurotrophic factor. J Neurosci. 1994;14:5281–5291. doi: 10.1523/JNEUROSCI.14-09-05281.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 37.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotech. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 38.Zurn AD, Baetge EE, Hammang JP, Tan SA, Aebischer P. Glial cell line-derived neurotrophic factor (GDNF), a new neurotrophic factor for motoneurones. NeuroReport. 1994;6:113–118. doi: 10.1097/00001756-199412300-00030. [DOI] [PubMed] [Google Scholar]