

Abstract
The mesolimbic dopamine system is essential for reward-seeking behavior, and drugs of abuse are thought to usurp the normal functioning of this pathway. A growing body of evidence suggests that glutamatergic synapses on dopamine neurons in the ventral tegmental area (VTA) are modified during exposure to addictive drugs, producing sensitization, a progressive augmentation in the rewarding properties of psychostimulant drugs with repeated exposure. We have tested the hypothesis that psychostimulant exposure interferes with the synaptic plasticity of glutamatergic inputs to the VTA. We find that excitatory synapses onto VTA dopamine neurons exhibit long-term depression (LTD) in response to low-frequency stimulation and modest depolarization. LTD in the VTA is NMDA receptor-independent but is dependent on intracellular Ca2+ and can be induced by driving Ca2+ into the dopamine neuron. Brief exposure to amphetamine entirely blocks LTD at glutamatergic synapses in the VTA, by releasing endogenous dopamine that acts at D2 dopamine receptors. The block of LTD is selective, because amphetamine has no effect on hippocampal LTD. The LTD we have discovered in the VTA is likely to be an important component of excitatory control of the reward pathway; amphetamine will inhibit LTD, removing this normal brake on the glutamatergic drive to dopamine neurons. This effect of amphetamine represents an important mechanism by which normal function of the brain reward system may be impaired during substance abuse.
Keywords: long-term depression, VTA, dopamine, amphetamine, sensitization, psychostimulant, addiction
Daily administration of amphetamine, cocaine, or morphine results in a progressive enhancement or sensitization to many of the behavioral effects of the drug (Kalivas and Stewart, 1991; White, 1996). In rats, the enhancement is observed as progressive augmentation of spontaneous locomotor activity. In humans, the psychomotor stimulant effects of abused drugs are thought to relate to arousal and intense euphoria, and sensitization may underlie progressively more rewarding properties of drugs of abuse associated with craving (Robinson and Berridge, 1993; Kalivas et al., 1998; Self, 1998). Sensitization lasts for months in rats and perhaps many years in humans, suggesting that psychostimulants persistently modify brain function.
Despite the fact that amphetamine, cocaine, and morphine have distinct mechanisms of action, the ventral tegmental area of the midbrain (VTA) has consistently proven to be required for the development of sensitization to all three. The VTA contains dopamine neurons that project to cortical and limbic areas of the brain and is believed to be involved in reward-seeking behavior, drug abuse, and the initiation of sensitization (Wise and Bozarth, 1987; Wise and Rompre, 1989; Kalivas and Stewart, 1991; Koob, 1992; Wise, 1996; Schultz, 1998; White and Kalivas, 1998). Intra-VTA injections of amphetamine sensitize animals to peripherally delivered amphetamine and cocaine (Kalivas and Weber, 1988; Vezina and Stewart, 1993), whereas inhibitors of sensitization are effective when microinjected directly into the VTA. These data strongly suggest that the VTA is the initiation site for sensitization.
Considerable evidence suggests that sensitization is triggered by changes in glutamatergic transmission within the VTA (Wolf, 1998). Glutamate receptor antagonists block sensitization (Karler et al., 1989; Stewart and Druhan, 1993; Carlezone et al., 1999; Li and Wolf, 1999), and injection of an NMDA receptor antagonist directly into the VTA also blocks sensitization (Kalivas and Alesdatter, 1993). Lesions of the prefrontal cortex, which provides a major glutamatergic afferent input to the VTA, block sensitization to amphetamine (Wolf et al., 1995; Wolf and Xue, 1999). Moreover, repeated electrical stimulation of glutamatergic afferents to the VTA sensitizes animals to subsequent cocaine administration (Schenk and Snow, 1994). Finally, increased glutamate receptor expression and enhanced responsiveness to glutamate have been reported in the VTA in sensitized animals (Tong et al., 1995;White et al., 1995; Fitzgerald et al., 1996; Zhang et al., 1997). These data suggest the hypothesis that stimulant exposure causes plasticity at excitatory synapses on VTA dopamine neurons, leading to behavioral sensitization.
Long-term potentiation (LTP), a persistent increase in excitatory synaptic transmission, has been demonstrated recently at excitatory synapses on midbrain dopamine neurons (Bonci and Malenka, 1999; Overton et al., 1999). In the hippocampus and other brain regions expressing LTP, synaptic strength can also be downregulated by an opposing process, long-term depression (LTD) (Bear and Abraham, 1996). We report that in addition to exhibiting LTP, excitatory synapses on VTA neurons also exhibit LTD. LTD is entirely blocked by amphetamine, the first demonstration of modulation of synaptic plasticity in the dopaminergic reward pathway by a drug of abuse.
MATERIALS AND METHODS
Slice preparation. Sprague Dawley rats (16–28 d old) were decapitated, after deep halothane anesthesia. The brain was rapidly removed and placed in ice-cold artificial CSF (ACSF; in mm): 119 NaCl, 26 NaHCO3, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 glucose, saturated with 95% O2 and 5% CO2, pH 7.4. For VTA recordings, horizontal slices (250–350 μm) were prepared and stored with 1 mmkynurenic acid added to the ACSF (Jones and Kauer, 1999). For hippocampal recordings, coronal slices (400 μm) were prepared without kynurenate (Tecott et al., 1998). Slices were later transferred to a recording chamber where the slice was submerged in warmed ACSF without kynurenate (28–32°C). For VTA recordings, 100 μmpicrotoxin was added to block GABAA receptors to study excitatory synaptic transmission in isolation. Extracellular CaCl2 was raised to 5 mm in experiments using repetitive voltage steps (see Figs. 3,4).
Fig. 3.

Elevation of postsynaptic intracellular Ca2+ is sufficient to trigger LTD. A, A single example of a neuron recorded from for 40 min while collecting baseline EPSCs. At the arrow, synaptic stimulation was stopped, and the neuron was repetitively stepped 20 times to +10 mV for 3 sec, once every 5 sec. Synaptic stimulation was then resumed at 0.1 Hz. This protocol was sufficient to induce LTD. Inset,The averages of five EPSCs taken at the times indicated on the graph. Calibration: 200 pA, 10 msec. B, The average of 12 such experiments. All experiments in this figure were performed in ACSF containing 5 mm Ca2+. LTD elicited by repetitive depolarization is 79 ± 7% of control values (n = 11, 6 identified dopamine neurons).
Fig. 4.

Synaptically induced LTD occludes LTD produced by repetitive depolarization. A, Synaptic LTD was maximally induced by pairing 1 Hz stimulation with depolarization to −40 mV three times until no further LTD was elicited (openarrowheads). At this point, synaptic stimulation was stopped, and the neuron was repetitively stepped 20 times to +10 mV for 3 sec, once every 5 sec (arrow). No further LTD was apparent after repetitive depolarizations. Inset, The averages of five EPSCs taken at the times indicated on the graph are shown. Calibration: 200 pA, 10 msec. B, After maximally inducing LTD by pairing 1 Hz stimulation with depolarization to −40 mV two to four times until no further LTD was elicited, neurons were repetitively depolarized as described above (n = 7). LTD induced after repetitive depolarizations is 96 ± 8% of control values (n = 6). All experiments in this figure were performed in ACSF containing 5 mmCa2+.
Electrophysiology. Individual cells in the VTA were either recorded “blind” or were visualized using differential interference contrast on an upright microscope for whole-cell voltage-clamp recordings. Patch pipettes were filled with cesium gluconate-based internal solution (in mm): 117 cesium gluconate, 2.8 NaCl, 5 MgCl2, 0.5 CaCl2, 2 ATP-Na+, 1.7 GTP-Na+, 5 EGTA, and 20 HEPES. Experiments with intracellular BAPTA used a similar solution except that 90 mm cesium gluconate and 20 mm BAPTA were used, with no added CaCl2 or EGTA. Biocytin (0.4%) was added to allow post hoc identification of recorded cells by immunolabeling. Neurons were voltage-clamped at −60 mV except as noted. The cell input resistance and series resistance were monitored throughout the experiment, and experiments were discarded if these values changed by >10% during the experiment.
A bipolar stainless steel stimulating electrode was placed rostral to the recording site in the VTA to stimulate glutamatergic afferents at 0.1 Hz (stimulus intensities were typically 40–200 μA; 100 μsec). LTD was induced by stimulating excitatory afferents at 1 Hz for 6 min, while depolarizing the neuron to −40 mV. During this depolarization, the neurons often escaped the voltage clamp, firing action currents. It is likely that even when the cell did not fire, the synaptic regions were sufficiently unclamped to permit voltage fluctuations in response to synaptic currents.
Extracellular recordings from the CA1 region of hippocampal slices were performed as described previously, while stimulating CA3 afferents in the stratum radiatum (Tecott et al., 1998). Hippocampal LTD was induced by delivering paired pulses 50 msec apart for 7.5 min at 1 Hz. This stimulus train was then repeated 10 min later to maximize LTD.
Drugs were added directly to the ACSF perfusing the slice chamber at known concentrations at least 10 min before LTD induction. EPSC amplitudes and hippocampal field EPSP initial slopes were measured off-line using LabView software kindly donated by Dr. Daniel Madison. For statistical analysis, levels of LTD were assessed by averaging EPSC amplitudes or EPSP slopes for 10 min just before LTD induction and comparing this value with averaged values collected during the 10 min period from 10 to 20 min after LTD induction, except as noted. The mean values from this period for each test group were compared with the control using an unpaired, two-tailed t test; pvalues ≤ 0.05 were considered significant. Our data exhibit a normal distribution (there is no significant difference between the mean and median values for each data group, indicating that there is no skew in the distribution), and the variance for the different data sets is the same (the SDs of the data sets are not significantly different). Values are reported as means ± SEM.
Identification of dopamine neurons. Immediately after recording, VTA slices were fixed in 4% paraformaldehyde for at least 24 hr. Slices were then double-labeled for biocytin (to detect the recorded cell) and for tyrosine hydroxylase (TH) (to identify dopamine-synthesizing neurons). To classify cells as dopamine (DA) cells or nondopamine cells, we used a combination of physiology and immunohistochemistry as described previously (Jones and Kauer, 1999). Briefly, we tested for the presence of Ih, the pacemaker current prominent in dopamine neurons and absent in non-DA cells (Grace and Onn, 1989; Johnson and North, 1992). We also checked for TH immunoreactivity. When a cell was TH-positive, it was classed as a DA cell. When a cell was not recovered after fixation or was not apparently TH-positive (as happens frequently after lengthy whole-cell recordings), then if it had a prominentIh we also classed the cell as a DA cell. Other neurons were classed as non-DA cells.
Materials. Salts and BAPTA were obtained from Sigma (St. Louis, MO), and all other drugs were obtained from Research Biochemicals International (Natick, MA), with the exception of 7-(hydroxyimino)cycloprop[b]chroman-1a-carboxylate ethyl ester (CPCCOE), which was purchased from Tocris Cookson (Ballwin, MO). Stock solutions of amphetamine, eticlopride, sulpiride, spiperone, and CPCCOEt were made fresh daily; amphetamine and eticlopride were dissolved in water; sulpiride and spiperone were dissolved in 0.1N HCL; CPCCOEt was dissolved in DMSO. Ryanodine was dissolved in DMSO and stored at −20°C. Stock solutions were diluted 1:1000 for use in ACSF.
RESULTS
Stimulation of excitatory afferents to the VTA at 1 Hz for 6 min, during depolarization of cells to −40 mV, produced long-term depression of glutamatergic EPSCs recorded from VTA neurons (Fig.1A). The EPSC amplitude was depressed to 72 ± 3% of control values after LTD induction (n = 25; Fig. 1B). EPSCs from 22 of 25 cells were depressed by >10% 20 min after LTD induction (range, 101–41% of control EPSC amplitude;n = 25). These results are the first demonstration of LTD in the VTA and suggest that LTD is a mechanism normally present to dampen excitatory input.
Fig. 1.

Stimulation (1 Hz) elicits long-term depression at excitatory synapses in the VTA. A, A single experiment in which a VTA neuron was recorded from during excitatory afferent stimulation. At the arrow, electrical stimulation was delivered at 1 Hz while the neuron was held at −40 mV. Responses during the 1 Hz stimulation are omitted for clarity. Afterward, the stimulation frequency was returned to 0.1 Hz, and LTD was observed.Inset, The averages of five EPSCs taken just before (1) and after 1 Hz (2) stimulation. Calibration: 50 pA, 10 msec. B, Averaged results from 30 experiments eliciting LTD as described in A. Peak EPSC amplitudes were normalized and plotted over time. LTD could be induced in both dopamine and non-dopamine neurons within the VTA using this stimulation protocol (n = 22 dopamine neurons; n = 8 non-dopamine neurons). All cells tested were included in the ensemble average, regardless of whether or not they exhibited robust LTD.
We tested whether LTD in the VTA shares properties with LTD in other brain areas. NMDA receptors are required for LTD induction at many synapses, and evidence suggests that sensitization to psychostimulants and opiates requires activation of NMDA receptors in the VTA. However, blockade of NMDA receptors with APV did not prevent LTD in the VTA (Fig. 2A). At various synapses, including the CA3-to-CA1 synapse in the hippocampus, elevation of Ca2+ in the postsynaptic neuron is necessary for LTD induction (Linden and Connor, 1995;Cummings et al., 1996). In our experiments, inclusion of the Ca2+ chelator BAPTA (20 mm) in the whole-cell recording pipette for at least 30 min before LTD induction entirely blocked LTD (Fig.2B). The block of LTD was not caused by dialysis during long recordings, because control neurons recorded from for 30 min before LTD induction without BAPTA in the pipette solution did exhibit LTD after 1 Hz afferent stimulation (n = 7). These results show that LTD induction in the VTA requires an elevation of postsynaptic Ca2+ levels and that this Ca2+ must arise from sources other than the NMDA receptor.
Fig. 2.

NMDA receptors are not required, but intracellular Ca2+ elevation is required for LTD induction.A, Control data from Figure 1 (opencircles; n = 30) are shown; 50 μmd-APV (closedcircles) was present in the bathing medium throughout the experiment and for at least 10 min before inducing LTD.d-APV did not block LTD induced by 1 Hz stimulation during depolarization to −40 mV (n = 6, 5 identified dopamine neurons). This concentration of d-APV blocks NMDA receptors on VTA neurons (Jones and Kauer, 1999). LTD in the presence of APV is 79 ± 9% of control values (n = 5; not significantly different from control LTD). B, Control data from Figure 1(opencircles; n = 30) are shown; whole-cell recordings (closedcircles ) were made from neurons using pipettes containing 20 mm BAPTA (see Materials and Methods) for 30–60 min before delivering 1 Hz stimulation during depolarization to −40 mV. Intracellular BAPTA significantly attenuated LTD (n = 8, all identified dopamine neurons). LTD with intracellular BAPTA is 91 ± 6% of control values (n = 7).
The metabotropic glutamate receptor mGluR1 is present on dopamine neurons (Kosinski et al., 1998) and can increase intracellular Ca2+ levels by releasing Ca2+ from ryanodine-sensitive stores (Fiorillo and Williams, 1998). We therefore tested whether interfering with these processes also blocked LTD. We found that blocking mGluR1 using the selective antagonist CPCCOEt (100 μm) had no effect on LTD induction (EPSC amplitude 5–10 min after LTD induction, 70 ± 14% of baseline values; n = 8). Additionally, robust LTD was triggered in the presence of ryanodine (10 μm; EPSC amplitude, 64 ± 14% of baseline values; n = 4). These data suggest that activation of mGluR1 and subsequent release of Ca2+ from ryanodine-sensitive Ca2+ stores are not required for LTD induction.
To determine whether an increase in postsynaptic Ca2+ alone is sufficient to induce LTD without synaptic stimulation, we repetitively depolarized the dopamine neuron to activate voltage-gated Ca2+channels. After resuming synaptic stimulation, we found that LTD had been induced (Fig. 3). This result suggests the hypothesis that any event inducing a rise in postsynaptic Ca2+ can induce LTD. If the LTD induced artificially by driving Ca2+ into the postsynaptic neuron shares underlying mechanisms with synaptically induced LTD, then maximizing synaptic LTD should prevent further depression by the voltage-step protocol. This was indeed observed, indicating that synaptically induced LTD uses the same mechanisms used during LTD induced by repetitive depolarization (Fig.4). Taken together, our data indicate that elevation of intracellular Ca2+ is necessary and sufficient to trigger LTD at VTA excitatory synapses.
We hypothesized that psychostimulants might interfere with LTD, removing a normal brake on the excitation of dopamine neurons in the VTA. We therefore tested the effects of amphetamine on LTD by bathing VTA slices from drug-naïve animals in amphetamine for 15 min before an LTD-inducing protocol. LTD of VTA synapses was entirely blocked by 1 μm amphetamine (Fig.5; EPSC amplitude, 100 ± 6% of control values; p < 0.005). Long-term depression was first described at synapses in the hippocampus (Dudek and Bear, 1992; Mulkey and Malenka, 1992), and dopamine is known to modulate synaptic function in the hippocampus (Otmakhova and Lisman, 1996, 1998, 1999). To test the specificity of amphetamine's effects on LTD, we examined whether amphetamine can also block hippocampal LTD at the CA3-to-CA1 synapse. In contrast to its effects in the VTA, amphetamine (1 μm) had no effect on LTD at these hippocampal synapses (Fig. 6). These data indicate that amphetamine has selective effects on synaptic plasticity at excitatory synapses on midbrain dopamine neurons.
Fig. 5.

Amphetamine blocks LTD in VTA neurons.A, LTD was blocked in the presence of 1 μmamphetamine (horizontalbar) applied for 15 min before synaptic stimulation at 1 Hz for 6 min during depolarization to −40 mV. Insets, The averages of five EPSCs taken at the times indicated on the graph are shown. Calibration: 100 pA, 10 msec. B, The average of nine such experiments (6 identified dopamine neurons) is shown.
Fig. 6.

Amphetamine does not block LTD at CA3-to-CA1 synapses in the hippocampus. A, Two independent afferent pathways in st. radiatum were stimulated alternately at 0.1 Hz while field EPSPs were recorded. At the arrows (top graph), stimulation to Pathway 2 was stopped, and LTD was induced in Pathway 1, using two trains of 1 Hz stimulation (see Materials and Methods). After induction of LTD in Pathway 1, 1 μm amphetamine was added to the bathing medium. Fifteen minutes later, stimulation toPathway 1 was stopped, and LTD-inducing stimuli were delivered to Pathway 2 (arrows inbottomgraph). Amphetamine had no effect on LTD induction or maintenance. B, Averaged experiments show that identical LTD was induced in hippocampal slices whether or not 1 μm amphetamine was included in the bathing solution [closedcircles, control solution (n = 4); opencircles, 1 μm amphetamine present for at least 10 min before 1 Hz stimulation (n = 5)]. Responses during the 1 Hz stimulation are omitted for clarity.
Amphetamine blocks and reverses the action of the dopamine transporter, thereby increasing dopamine levels where the transporter is present. Dopamine depresses voltage-gated Ca2+channel function in a variety of peripheral and central neurons (Stack and Suprenant, 1991; Seabrook et al., 1994; Yan et al., 1997;Kuzhikandathil and Oxford, 1999), including midbrain dopamine cells (Cardozo and Bean, 1995). We hypothesized that dopamine released locally by amphetamine might block LTD by inhibiting Ca2+ entry into VTA neurons during the LTD induction protocol. Dopamine and the D2 receptor agonist quinpirole block VTA LTD, acting via D2 dopamine receptors (Thomas et al., 2000), indicating that amphetamine may block LTD at VTA synapses by releasing endogenous dopamine, which then acts at D2 receptors. We therefore examined whether D2 receptor antagonists interfere with the block of LTD by amphetamine. VTA slices were perfused with D2 receptor antagonists, and LTD was induced using 1 Hz stimulation and depolarization to −40 mV. The effect of amphetamine was attenuated by 30 nm eticlopride and was blocked by 100 nmeticlopride (Fig. 7). The less-selective D2 receptor antagonist spiperone (5 μm) also significantly blocked amphetamine's effects (EPSC amplitude after LTD induction, 75 ± 4% of control levels; n = 4). These data indicate that the block of LTD by amphetamine is mediated by D2 receptor activation.
Fig. 7.
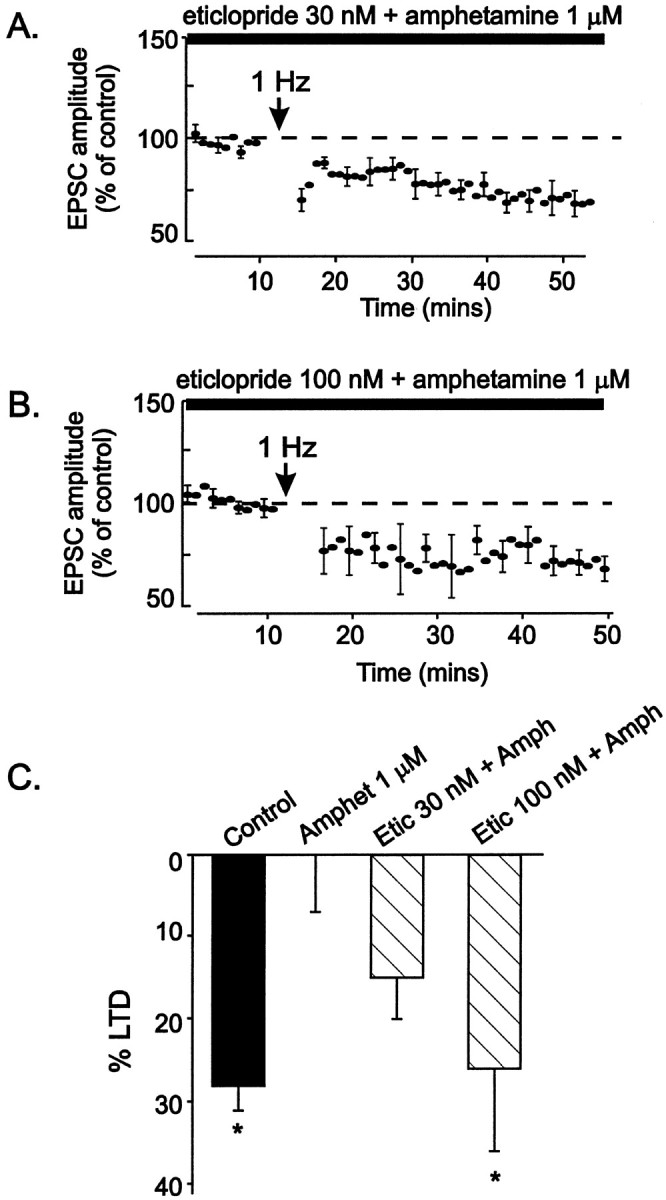
The D2 receptor antagonist eticlopride reverses the effects of amphetamine on LTD. Eticlopride was included in the bathing solution before 1 μm amphetamine was added. LTD was induced by depolarizing to −40 mV while stimulating synaptic afferents at 1 Hz for 6 min. A, Eticlopride (30 nm) attenuates the effects of amphetamine on LTD (n = 8, with 7 dopamine cells). B, Eticlopride (100 nm) attenuates the effects of amphetamine on LTD (n = 5, with all dopamine cells).C, The barchart shows the amount of LTD measured from 10 to 20 min after LTD induction in control cells (n = 25), amphetamine-treated cells (Amphet 1 μm; n = 8), cells treated with 30 nm eticlopride + 1 μmamphetamine (Etic 30 nm + Amph;n = 7), or cells treated with 100 nmeticlopride + 1 μm amphetamine (Etic 100 nm + Amph; n = 4).Asterisks indicate significant differences from cells treated with 1 μm amphetamine (p ≤ 0.05).
DISCUSSION
Our results are the first to show that excitatory synapses on VTA dopamine neurons can express LTD in addition to expressing LTP, exhibiting bidirectional modification depending on the pattern of afferent input and membrane potential. Under normal conditions, LTD will modulate information flow into the VTA, limiting excitation of dopamine neurons. The block of LTD by amphetamine is the first demonstration of an effect of an addictive drug on synaptic plasticity in the reward pathway. We speculate that the block of LTD, which occurs within minutes after a first exposure to amphetamine, is a rapid cellular consequence of amphetamine abuse.
Postsynaptic Ca2+ entry triggers LTD
A rise in intracellular Ca2+ is both necessary and sufficient to trigger LTD in VTA dopamine cells. Where does the Ca2+ come from during the protocol pairing modest depolarization with low-frequency synaptic stimulation? LTD at these synapses, unlike LTP (Bonci and Malenka, 1999; Overton et al., 1999), is not NMDA receptor-dependent, so Ca2+ entry through NMDA receptors is not necessary. Metabotropic glutamate receptors that release Ca2+ from ryanodine-sensitive intracellular stores and Ca2+-permeable AMPA channels are apparently not required for LTD (see also Thomas et al., 2000). Instead, the most likely hypothesis is that Ca2+ enters the cell through voltage-dependent Ca2+ channels, many types of which are expressed by VTA dopamine neurons (Grace and Onn, 1989; Kang and Kitai, 1993; Cardozo and Bean, 1995). Because Ca2+ channels are also essential for neurotransmitter release, it is difficult to test this hypothesis directly, and further work will be necessary to define precisely the channel types involved. Repetitive depolarization using voltage steps is likely to trigger Ca2+ entry through multiple voltage-gated Ca2+ channel types, particularly the high-voltage-activated L, N, P, and Q channels. The voltage-step protocol alone produces LTD, even without synaptic stimulation, demonstrating that LTD induction occurs postsynaptically and does not require synaptic activity or glutamate release. LTD elicited after voltage steps shares a common mechanism with LTD produced by pairing sustained modest depolarization with synaptic activity, because voltage steps have no further effect after synaptically triggered LTD is maximal. It is thus likely that whenever intracellular Ca2+ rises sufficiently, excitatory synapses onto VTA dopamine cells will be depressed.
We also observed LTD in nondopaminergic VTA neurons after pairing depolarization to −40 mV with synaptic stimulation at 1 Hz. Previous work showed that a protocol that triggers LTP in VTA dopamine neurons does not enhance synaptic transmission onto nondopamine neurons (Bonci and Malenka, 1999). Thus, although only dopamine cell synapses can be potentiated, excitatory synapses on both major types of VTA neurons can be depressed. Because our study focused primarily on dopamine neurons, further work will be required to determine whether the mechanisms underlying LTD at synapses on nondopamine cells are the same as those in dopamine cells and whether amphetamine can block LTD at synapses on nondopamine cells.
Amphetamine blocks LTD by activating D2 dopamine receptors
Treatment with 1 μm amphetamine entirely blocks LTD at synapses on dopamine neurons. Amphetamine interferes with LTD via a D2 receptor-mediated mechanism, because eticlopride, at 30–100 nm, attenuated amphetamine's effects. Dopamine and a D2 receptor agonist, quinpirole, also block LTD (Thomas et al., 2000). Several studies have shown that D2 receptor activation attenuates a variety of Ca2+ currents, including both low-voltage-activated T currents and high-voltage-activated L, N, P, and Q currents (Carbone and Lux, 1989; Stack and Suprenant, 1991;Seabrook et al., 1994; Cardozo and Bean, 1995; Yan et al., 1997;Kuzhikandathil and Oxford, 1999; Wolfe et al., 1999). We therefore hypothesize that amphetamine blocks and reverses the dopamine transporter to elevate extracellular dopamine, which activates D2 receptors and thereby depresses voltage-dependent Ca2+ currents. The inhibition of Ca2+ entry may reduce Ca2+ levels below the threshold needed to trigger LTD. Alternatively, other cellular sequelae of amphetamine-mediated dopamine receptor activation may interfere with LTD induction.
Modulation of LTD by dopamine and psychostimulants
VTA dopamine cells both in vitro and in vivohave unusually positive membrane potentials and action potential thresholds (Grace and Bunney, 1983; Grace, 1988), and it is not unlikely that their membrane potentials often remain near −40 mV for long periods of time. Because LTD induction in vitrorequires only relatively low-frequency synaptic activation, and because dopamine neurons usually fire at 1–8 Hz in vivo (Grace and Bunney, 1984), it is possible that in the brain under normal conditions VTA excitatory synapses often exist in a relatively depressed state. LTD may normally protect VTA dopamine neurons from excessive glutamatergic excitation. In the presence of amphetamine this protective brake is removed, permitting unrestricted excitation of dopamine neurons by glutamate afferents.
One important question raised by our results is whether endogenous dopamine blocks LTD under physiological conditions, as well as during psychostimulant-mediated dopamine elevation. Dopamine can be released from somatodendritic sites (Geffen et al., 1976; Cheramy et al., 1981), raising the possibility that LTD may be modulated by endogenous dopamine. This modulation is likely to operate on the seconds-to-minutes time scale, after local dopamine release and before removal of the neurotransmitter by the transporter. A transient block of LTD resulting from somatodendritic dopamine release could provide a window during which LTP is facilitated at synapses on dopamine cells, thus acting as a gate for strengthening afferent excitatory input. In contrast, after psychostimulant administration, psychostimulant levels remain high for hours, and levels of dopamine in the VTA may consequently also remain elevated for prolonged periods. The resulting persistent blockade of LTD would be expected to promote glutamatergic activity from cortical and subcortical regions and to increase the likelihood of NMDA receptor-dependent LTP.
Overactivity of dopaminergic neurotransmission in the VTA is postulated to contribute to the development of psychotic symptoms, both in schizophrenia and in amphetamine psychosis. Antipsychotic drugs effective in treating both disorders have D2 receptor antagonist properties. Somatodendritic dopamine release does not have a dramatic effect on the firing rate of most dopamine neurons (Bunney and Aghajanian, 1975; Pucak and Grace, 1996), but our findings suggest that, acutely, D2 receptor antagonists may promote the development of LTD in the VTA, reducing dopamine neuron activity in the short-term. The effects of longer-term exposure to these drugs have yet to be tested.
Amphetamine effects in the VTA promote behavioral sensitization
NMDA receptor-dependent LTP has been proposed as a possible mechanism involved in the onset of behavioral sensitization (Tong et al., 1995; White, 1996; Wolf, 1998). Three mechanisms have been identified by which amphetamine will promote the abnormal excitation of VTA dopamine neurons by glutamatergic afferents. First, although a rapid effect of psychostimulants is hyperpolarization of dopamine neurons via autoreceptors, this effect desensitizes in the presence of amphetamine, removing one source of inhibition (Seutin et al., 1991). Second, psychostimulants potently elevate extracellular glutamate in the VTA, perhaps as a result of decreased glutamate reuptake (Xue et al., 1996; Kalivas and Duffy, 1998; Wolf and Xue, 1999). Finally, our results indicate that amphetamine strongly modulates excitatory synaptic transmission by blocking LTD. We propose that these three mechanisms will act in concert to promote NMDA receptor-dependent LTP, resulting in an abnormal strengthening of excitatory synapses on dopamine neurons in the VTA. This hypothesis can account for the block of sensitization by intra-VTA administration of an NMDA receptor antagonist (Vezina and Stewart, 1993) and the ability of high-frequency electrical stimulation of glutamate afferents to the VTA by itself to produce sensitization to peripherally administered psychostimulants (Schenk and Snow, 1994). The resulting glutamatergic excitation of dopamine neurons may represent an early trigger in the development of sensitization and addiction.
Footnotes
This work was supported by National Institutes of Health Grant DA 11289. We wish to thank Andrew Pittman and Katherine Papastephanou for excellent technical assistance and Drs. Lawrence Katz, Donald Lo, Richard Mooney, and Yong Li for helpful comments on this manuscript.
S.J. and J.L.K. contributed equally to this work.
Correspondence should be addressed to Dr. Julie Kauer at her present address: Brown University, Department of Molecular Pharmacology, Physiology, and Biotechnology, P.O. Box G-B4, Providence, RI 02912.
REFERENCES
- 1.Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 2.Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunney BS, Aghajanian GK. Evidence for drug actions on both pre- and postsynaptic catecholamine receptors in the CNS. In: Usdin E, Bunney WE, editors. Pre- and postsynaptic receptors. Dekker; New York: 1975. pp. 89–122. [PubMed] [Google Scholar]
- 4.Carbone E, Lux HD. Modulation of Ca channels in peripheral neurons. Ann NY Acad Sci. 1989;1560:346–357. doi: 10.1111/j.1749-6632.1989.tb24114.x. [DOI] [PubMed] [Google Scholar]
- 5.Cardozo DL, Bean BP. Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol. 1995;74:1137–1148. doi: 10.1152/jn.1995.74.3.1137. [DOI] [PubMed] [Google Scholar]
- 6.Carlezone WA, Jr, Rasmussen K, Nestler EJ. AMPA antagonist LY293558 blocks the development, without blocking the expression, of behavioral sensitization to morphine. Synapse. 1999;31:256–262. doi: 10.1002/(SICI)1098-2396(19990315)31:4<256::AID-SYN3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 7.Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- 8.Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 9.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geffen LB, Jessell TM, Cuello AC, Iversen LL. Release of dopamine from dendrites in rat substantia nigra. Nature. 1976;260:258–260. doi: 10.1038/260258a0. [DOI] [PubMed] [Google Scholar]
- 13.Grace AA. In vivo and in vitro intracellular recordings from rat midbrain dopamine neurons. Ann NY Acad Sci. 1988;537:51–76. doi: 10.1111/j.1749-6632.1988.tb42096.x. [DOI] [PubMed] [Google Scholar]
- 14.Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons. 1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- 15.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grace AA, Onn S-P. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission in the ventral tegmental area via serotonin receptors. J Neurosci. 1999;19:9780–9787. doi: 10.1523/JNEUROSCI.19-22-09780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalivas PW, Alesdatter JE. Involvement of NMDA receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;267:486–495. [PubMed] [Google Scholar]
- 20.Kalivas PW, Duffy P. Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J Neurochem. 1998;70:1497–1502. doi: 10.1046/j.1471-4159.1998.70041497.x. [DOI] [PubMed] [Google Scholar]
- 21.Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- 22.Kalivas PW, Weber B. Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther. 1988;245:1095–1101. [PubMed] [Google Scholar]
- 23.Kalivas PW, Pierce RC, Cornish J, Sorg BA. A role for sensitization in craving and relapse in cocaine addiction. J Psychopharmacol. 1998;12:49–53. doi: 10.1177/026988119801200107. [DOI] [PubMed] [Google Scholar]
- 24.Kang Y, Kitai ST. A whole cell patch-clamp study on the pacemaker potential in dopaminergic neurons of rat substantia nigra compacta. Neurosci Res. 1993;18:209–221. doi: 10.1016/0168-0102(93)90056-v. [DOI] [PubMed] [Google Scholar]
- 25.Karler R, Calder LD, Chaudhry IA, Turkanis SA. Blockade of “reverse tolerance” to cocaine and amphetamine by MK-801. Life Sci. 1989;45:599–606. doi: 10.1016/0024-3205(89)90045-3. [DOI] [PubMed] [Google Scholar]
- 26.Koob GF. Drugs of abuse: anatomy, pharmacology, and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 27.Kosinski CM, Standaert DG, Testa CM, Penney JB, Jr, Young AB. Expression of metabotropic glutamate receptor 1 isoforms in the substantia nigra pars compacta of the rat. Neuroscience. 1998;86:783–798. doi: 10.1016/s0306-4522(97)00654-4. [DOI] [PubMed] [Google Scholar]
- 28.Kuzhikandathil EV, Oxford GS. Activation of human D3 dopamine receptor inhibits P/Q-type calcium channels and secretory activity in AtT-20 cells. J Neurosci. 1999;19:1698–1707. doi: 10.1523/JNEUROSCI.19-05-01698.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Wolf ME. Can the “state-dependency” hypothesis explain prevention of amphetamine sensitization in rats by NMDA receptor antagonists? Psychopharmacology (Berl) 1999;141:351–361. doi: 10.1007/s002130050844. [DOI] [PubMed] [Google Scholar]
- 30.Linden DJ, Connor JA. Long-term synaptic depression. Annu Rev Neurosci. 1995;18:319–357. doi: 10.1146/annurev.ne.18.030195.001535. [DOI] [PubMed] [Google Scholar]
- 31.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 32.Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otmakhova NA, Lisman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via a cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otmakhova NA, Lisman JE. Dopamine selectively inhibits the direct cortical pathway to the CA1 hippocampal region. J Neurosci. 1999;19:1437–1445. doi: 10.1523/JNEUROSCI.19-04-01437.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Overton PG, Richards CD, Berry MS, Clark D. Long-term potentiation at excitatory amino acid synapses on midbrain dopamine neurons. NeuroReport. 1999;10:221–226. doi: 10.1097/00001756-199902050-00004. [DOI] [PubMed] [Google Scholar]
- 36.Pucak ML, Grace AA. Effects of haloperidol on the activity and membrane physiology of substantia nigra dopamine neurons recorded in vitro. Brain Res. 1996;713:44–52. doi: 10.1016/0006-8993(95)01460-8. [DOI] [PubMed] [Google Scholar]
- 37.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 38.Schenk S, Snow S. Sensitization to cocaine's motor activating properties produced by electrical kindling of the medial prefrontal cortex but not of the hippocampus. Brain Res. 1994;659:17–22. doi: 10.1016/0006-8993(94)90858-3. [DOI] [PubMed] [Google Scholar]
- 39.Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- 40.Seabrook GR, Knowles M, Brown N, Myers J, Sinclair H, Patel S, Freedman SB, McAllister G. Pharmacology of high-threshold calcium currents in GH4C1 pituitary cells and their regulation by activation of human D2 and D4 dopamine receptors. Br J Pharmacol. 1994;112:728–734. doi: 10.1111/j.1476-5381.1994.tb13138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Self DW. Neural substrates of drug craving and relapse in drug addiction. Ann Med. 1998;30:379–389. doi: 10.3109/07853899809029938. [DOI] [PubMed] [Google Scholar]
- 42.Seutin V, Verbanck P, Massotte L, Dresse A. Acute amphetamine-induced subsensitivity of A10 dopamine autoreceptors in vitro. Brain Res. 1991;558:141–144. doi: 10.1016/0006-8993(91)90731-a. [DOI] [PubMed] [Google Scholar]
- 43.Stack J, Suprenant A. Dopamine actions on calcium currents, potassium currents and hormone release in rat melanotrophs. J Physiol (Lond) 1991;439:37–58. doi: 10.1113/jphysiol.1991.sp018655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart J, Druhan JP. Development of both conditioning and sensitization to the behavioral activating effects of amphetamine is blocked by the non-competitive NMDA receptor antagonist, MK-801. Psychopharmacology (Berl) 1993;110:125–132. doi: 10.1007/BF02246961. [DOI] [PubMed] [Google Scholar]
- 45.Tecott LH, Logue SF, Wehner JM, Kauer JA. Perturbed dentate gyrus function in serotonin 5-HT2C receptor mutant mice. Proc Natl Acad Sci USA. 1998;95:15026–15031. doi: 10.1073/pnas.95.25.15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas MJ, Malenka RC, Bonci A. Modulation of long-term depression by dopamine in the mesolimbic system. J Neurosci. 2000;20:5581–5586. doi: 10.1523/JNEUROSCI.20-15-05581.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tong Z-Y, Overton PG, Clark D. Chronic administration of (+)-amphetamine alters the reactivity of midbrain dopaminergic neurons to prefrontal cortex stimulation in the rat. Brain Res. 1995;674:63–74. doi: 10.1016/0006-8993(94)01439-o. [DOI] [PubMed] [Google Scholar]
- 48.Vezina P, Stewart J. Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Res. 1993;516:99–106. doi: 10.1016/0006-8993(90)90902-n. [DOI] [PubMed] [Google Scholar]
- 49.White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;16:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 50.White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- 51.White FJ, Hu X-T, Zhang X-F, Wolf ME. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther. 1995;273:445–454. [PubMed] [Google Scholar]
- 52.Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996;19:319–340. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- 53.Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- 54.Wise RA, Rompre P-P. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- 55.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:1–42. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 56.Wolf ME, Xue CJ. Amphetamine-induced glutamate efflux in the rat ventral tegmental area is prevented by MK-801, SCH 23390, and ibotenic acid lesions of the prefrontal cortex. J Neurochem. 1999;73:1529–1538. doi: 10.1046/j.1471-4159.1999.0731529.x. [DOI] [PubMed] [Google Scholar]
- 57.Wolf ME, Dahlin SL, Hu X-T, Xue C-J, White K. Effects of lesions of prefrontal cortex, amygdala, or fornix on behavioral sensitization to amphetamine: comparison with NMDA antagonists. Neuroscience. 1995;69:417–439. doi: 10.1016/0306-4522(95)00248-h. [DOI] [PubMed] [Google Scholar]
- 58.Wolfe SE, Howard DE, Schetz JA, Cheng CJ, Webber R, Beatty DM, Chronwall BM, Morris SJ. Dopamine D2-receptor isoforms expressed in AtT20 cells inhibit Q-type high-voltage-activated Ca2+ channels via a membrane-delimited pathway. J Neurochem. 1999;72:479–490. doi: 10.1046/j.1471-4159.1999.0720479.x. [DOI] [PubMed] [Google Scholar]
- 59.Xue C-J, Ng JP, Li Y, Wolf ME. Acute and repeated systemic amphetamine administration: effects on extracellular glutamate, aspartate, and serine levels in rat ventral tegmental area and nucleus accumbens. J Neurochem. 1996;67:352–363. doi: 10.1046/j.1471-4159.1996.67010352.x. [DOI] [PubMed] [Google Scholar]
- 60.Yan Z, Song W-J, Surmeier DJ. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X-F, Hu X-T, White FJ, Wolf ME. Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves AMPA receptors. J Pharmacol Exp Ther. 1997;281:699–706. [PubMed] [Google Scholar]