

Abstract
Neurotrophins activate several different intracellular signaling pathways that in some way exert neuroprotective effects. In vitro studies of sympathetic and cerebellar granule neurons have demonstrated that the survival effects of neurotrophins can be mediated via activation of the phosphatidylinositol 3-kinase (PI3-kinase) pathway. Neurotrophin-mediated protection of other neuronal types in vitro can be mediated via the extracellular signal-related protein kinase (ERK) pathway. Whether either of these pathways contributes to the neuroprotective effects of neurotrophins in the brain in vivo has not been determined. Brain-derived neurotrophic factor (BDNF) is markedly neuroprotective against neonatal hypoxic-ischemic (H-I) brain injuryin vivo. We assessed the role of the ERK and PI3-kinase pathways in neonatal H-I brain injury in the presence and absence of BDNF. Intracerebroventricular administration of BDNF to postnatal day 7 rats resulted in phosphorylation of ERK1/2 and the PI3-kinase substrate AKT within minutes. Effects were greater on ERK activation and occurred in neurons. Pharmacological inhibition of ERK, but not the PI3-kinase pathway, inhibited the ability of BDNF to block H-I-induced caspase-3 activation and tissue loss. These findings suggest that neuronal ERK activation in the neonatal brain mediates neuroprotection against H-I brain injury, a model of cerebral palsy.
Keywords: cerebral palsy, neurotrophin, MAP kinase, PI3-kinase, ischemia, apoptosis
Neuronal dysfunction and loss contribute to a variety of acute as well as chronic diseases of the brain. Understanding the mechanisms underlying neuronal cell death and the means by which it can be prevented may lead to better treatments. The neurotrophins (NTs), including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), NT-3, and NT-4/5, make up a family of neurotrophic factors that are important regulators of naturally occurring cell death in the peripheral nervous system (PNS) (Chao, 1992; Snider, 1994) and also regulate neuronal development and maturation in the CNS (Chen et al., 1997; Fagan et al., 1997;Martinez et al., 1998). Survival-promoting effects of neurotrophins are elicited by activation of different intracellular signaling cascades including the phosphatidylinositol 3-kinase (PI3-kinase) and the extracellular signal-related kinase (ERK) pathways. In several neuronal cell types in vitro, the PI3-kinase pathway appears to mediate the protective effects of growth factors. NGF-mediated activation of the PI3-kinase pathway appears to be required for survival effects in pheochromocytoma 12 (PC12) (Yao and Cooper, 1995) and superior cervical ganglion neurons (Crowder and Freeman, 1998), whereas activation of PI3-kinase by BDNF in motoneurons (Dolcet et al., 1999) and insulin-like growth factor-1 in cerebellar granule neurons, oligodendrocytes, and PC12 cells also appears to be important for survival (Vemuri and McMorris, 1996; D'Mello et al., 1997; Miller et al., 1997; Parrizas et al., 1997).
Although PI3-kinase is clearly important for growth factor-mediated neuronal survival in certain cells and conditions, in other neuronal cell types and under different conditions, growth factor-mediated activation of the ERK-signaling pathway appears to mediate survival effects. For example, ERK activation can promote PC12 survival (Xia et al., 1995) and is involved in survival-promoting effects of BDNF on retinal ganglion and cerebellar granule neurons (Meyer-Franke et al., 1998; Bonni et al., 1999). A study also demonstrates that BDNF neuroprotection of cortical neurons can be mediated via the ERK or PI3-kinase pathway depending on the injurious stimulus (Hetman et al., 1999). Thus, growth factor-mediated protection of neurons may be via different pathways depending on factors such as cell type, culture conditions, and the injurious stimulus. Whether growth factor-stimulated activation of the ERK or the PI3-kinase pathways is required for neuronal protection in the CNS in vivo has not been studied.
The hypoxic-ischemic (H-I) encephalopathy in survivors of perinatal asphyxia is a major contributor to morbidity and mortality (Vanucci, 1990; Volpe, 1995; Taylor et al., 1999). A well characterized model of neonatal H-I injury is one in which unilateral carotid ligation in postnatal day 7 (P7) rats is followed by exposure to hypoxia (Levine, 1960). This results in reproducible brain injury ipsilateral but not contralateral to the carotid ligation (Rice et al., 1981; Johnston, 1983). Using this model, our previous studies have demonstrated that BDNF is markedly neuroprotective (Cheng et al., 1997; Han et al., 2000). Herein, we address whether the ERK and PI3-kinase pathways are activated by BDNF in vivo and whether either is required for BDNF's neuroprotective effects.
MATERIALS AND METHODS
Animals and surgical procedures. Newborn Sprague Dawley rats (dam plus 12 pups per litter) were obtained from Sasco Breeders when the pups were 3–4 d of age. The pups were housed with the dam in the home cage under a 12:12 hr light/dark cycle, with food and water available ad libitum throughout the study. The neonatal H-I brain injury is based on the Levine procedure (Levine, 1960; Rice et al., 1981; Gidday et al., 1994; Cheng et al., 1997). Briefly, pups at P7 were anesthetized with 2.5% halothane (balance, room air), and the left common carotid artery was surgically exposed and permanently ligated. The incisions were sutured, and the pups were returned to the dam for a 2 hr recovery and feeding period. Pups were then placed in individual containers (37°C water bath to maintain normothermia) through which humidified 8% oxygen (balance, nitrogen) flowed for the next 2.5 hr. After completion of the H-I, the pups were returned to their home cage with dam and littermates.
Intracerebroventricular injection of BDNF and protein kinase inhibitors. To determine whether intracerebroventricular (ICV) injection of BDNF activates the ERK and PI3-kinase pathways in the neonatal brain, P7 rats received an ICV injection of a 5 μl solution containing either vehicle (VEH; PBS, pH 7.4) or BDNF (5 μg in vehicle) into the left hemisphere as described previously (Cheng et al., 1997). Injections were performed with a Hamilton syringe with a 27 gauge needle. The location of each injection was 2 mm rostral, 1.5 mm lateral to bregma, and 2 mm deep to the skull surface. Recombinant human BDNF was a gift of Dr. Qiao Yan (Amgen, Thousand Oaks, CA).
An MEK1/2 inhibitor, U0126, and a PI3-kinase inhibitor, wortmannin, were purchased from Calbiochem (La Jolla, CA) and dissolved in DMSO. To determine whether these protein kinase inhibitors were able to block the activation of the ERK1/2 and AKT pathways induced by BDNFin vivo, P7 pups were ICV injected with a 5 μl solution containing VEH (1 μl of DMSO and 4 μl of PBS, pH 7.4), U0126, or wortmannin, 2 mm rostral, 1.5 mm lateral, and 2 mm deep to the skull surface. Thirty minutes later, pups received a second ICV injection of VEH (5 μl of PBS, pH 7.4) or BDNF (5 μg/animal in 5 μl of PBS). At different time points after BDNF treatment, pups were anesthetized with 150 mg/kg pentobarbital intraperitoneally, perfused through the left ventricle with PBS, pH 7.4, and brain tissues from the hippocampus and parietal cortex were dissected. Brain tissues were then immediately frozen in dry ice and stored at −70°C.
Immunofluorescent labeling. Rat pups were anesthetized with 150 mg/kg pentobarbital intraperitoneally and then perfused through the left ventricle with PBS, pH 7.4. Brains were then post-fixed overnight in 4% paraformaldehyde in 0.1 m phosphate buffer at 4°C, cryoprotected in 30% (w/v) sucrose in 0.1 m phosphate buffer, pH 7.4 (4°C), frozen in powdered dry ice, and stored at −70°C. Serial (40 μm) coronal sections were cut on a freezing sliding microtome.
Forty micrometer free-floating sections through the forebrain were processed for immunofluorescent labeling as described previously (Han et al., 2000). Tissue sections were blocked with 3% goat serum in TBS and incubated overnight with mouse anti-neuronal nuclei (1:100) antibody (NeuN; Chemicon, Temecula, CA) and anti-phospho-ERK1/2 antibody (1:250; New England Biolabs, Beverly, MA). After washing, secondary antibodies conjugated to the fluorescent markers Alexa-488 and Alexa-568 (Molecular Probes, Eugene, OR) were applied to sections for 1 hr. Sections were then washed, mounted on slides, coverslipped with Vectashield mounting media (Vector Laboratories, Burlingame, CA), and examined with a Nikon (Melville, NY) FXL fluorescence microscope.
Western blotting. Tissue samples from the hippocampus and cortex were lysed by homogenizing in 300 μl of lysis buffer containing 10 mm HEPES, pH 7.4, 5 mmMgCl2, 1 mm DTT, 1% Triton X-100, 2 mm EGTA, 2 mm EDTA, 25 mmβ-glycerophosphate, 0.1 mm okadaic acid, 0.5 mm sodium orthovanadate, 1 mm PMSF, and protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN). Lysates were centrifuged at 12,000 × g for 10 min at 4°C, and the protein concentration was determined by a BCA protein assay kit (Pierce, Rockford, IL). Protein samples (50 μg per lane) diluted in SDS-PAGE sample buffer (50 mmTris-HCl, pH 6.8, 100 mm DTT, 2% SDS, 0.1% bromphenol blue, and 10% glycerol) were boiled for 10 min, electrophoresed on a 12.5% SDS-polyacrylamide gel, and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Blots were blocked with 3% dried milk in TBS containing 0.05% Tween 20 overnight. Blots were then incubated for 2–3 hr with the following antibodies: anti-phospho-ERK1/2 antibody (1:2000), anti-ERK1/2 antibody (1:3000), anti-phospho-AKT antibody (1:1000), and anti-AKT antibody (1:1000) that were purchased from New England Biolabs. Proteins were visualized with enhanced chemiluminescence (Amersham, Arlington Heights, IL) using a previously described procedure (Han et al., 2000).
Asp-Glu-Val-Asp-(7-amino-4-methylcoumarin) cleavage assay. P7 pups were ICV injected with protein kinase inhibitors with or without BDNF as described above before the carotid artery ligation and exposure to 8% oxygen for 2.5 hr. Twenty-four hours after H-I, tissues from the hippocampus and cortex in both lesioned and unlesioned hemispheres were rapidly dissected and frozen in dry ice. The Asp-Glu-Val-Asp-(7-amino-4-methylcoumarin) (DEVD-AMC) cleavage assay was performed as described previously (Han et al., 2000). Tissue samples were homogenized in lysis buffer (10 mm HEPES, pH 7.4, 5 mm MgCl2, 1 mmDTT, 1% Triton X-100, 2 mm EGTA, 2 mm EDTA, 1 mm PMSF, and protease inhibitor cocktail) and centrifuged at 12,000 × g for 10 min at 4°C. Tissue lysates (10 μl) were incubated in a 96-well plate with 90 μl of assay buffer (10 mm HEPES, pH 7.4, 42 mmKCl, 5 mm MgCl2, 1 mm DTT, and 10% sucrose) containing 30 μm acetyl-DEVD-AMC (Calbiochem). The emitted fluorescence was measured every 5 min for 30 min at room temperature at an excitation wavelength of 360 nm and an emission wavelength of 460 nm using a microplate fluorescence reader (Bio-Tek Instruments, Winooski, VT). DEVD-AMC cleavage activity was obtained from the slope by plotting fluorescence units against time. Acetyl-AMC (Calbiochem) was used to obtain a standard curve, and the enzyme activity was calculated as picomoles of AMC per milligram of protein per minute.
Assessment of brain damage caused by H-I. One week after H-I, brain sections were prepared as described above, and damage caused by H-I was determined by calculating the amount of surviving tissue in coronal sections as described previously (Cheng et al., 1998; Han et al., 2000). Briefly, coronal sections from the genu of the corpus callosum to the end of the dorsal hippocampus were stained with cresyl violet. The cross-sectional areas of the striatum, cortex, and hippocampus in each of eight equally spaced reference planes were assessed with the NIH Image analysis system (version 1.57) linked to a Nikon microscope. The sections corresponded approximately to plates 12, 15, 17, 20, 23, 28, 31, and 34 in a rat brain atlas (Paxinos and Watson, 1986). The total cross-sectional area in each brain region was then calculated in all sections assessed, and the percent area loss in the lesioned versus the unlesioned hemisphere was determined for each animal.
RESULTS
BDNF activates the ERK and PI3-kinase pathways in the neonatal brain
Previous in vitro studies have shown that neurotrophins such as BDNF can activate both the ERK and PI3-kinase pathways in responsive cells (Dudek et al., 1997; Qiu et al., 1998; Bonni et al., 1999; Dolcet et al., 1999; Encinas et al., 1999; Hetman et al., 1999). We asked whether BDNF activated these pathways in the neonatal rat brain in vivo. ICV injection of BDNF (5 μg) resulted in activation of both the ERK and PI3-kinase pathways as assessed in the hemisphere ipsilateral to the injection (Fig.1). The phosphorylation of both ERK1/2 (p-ERK1/2) as well as the serine–threonine protein kinase AKT (p-AKT) was increased by 30 min after ICV injection, with prolonged activation lasting up to at least 12 hr after a single ICV injection. Effects of the ERK pathway are mediated via phosphorylation of ERK1/2, and those of the PI3-kinase activation are often mediated by phosphorylation of AKT (also known as PKB or RAC), one of its immediately downstream protein kinase effectors (Dudek et al., 1997; Kaufmann-Zeh et al., 1997). Similar effects of BDNF on p-ERK1/2 and p-AKT were seen in both the cortex (Fig. 1B) and hippocampus (Fig.1A) with a greater percent increase over baseline for p-ERK1/2 than for p-AKT. Despite the BDNF-stimulated increases in p-ERK1/2 and p-AKT, there was no change in total ERK or AKT (Fig. 1). Because the BDNF receptor trkB has been shown to be localized to neurons (Klein et al., 1989, 1990), we suspected that the effect of BDNF on these intracellular signaling pathways was caused by a direct effect on cortical and hippocampal neurons. Indeed BDNF treatment increased p-ERK1/2-immunoreactivity (-IR) in neurons (Fig.2). p-ERK1/2-IR was detectable at low levels in neuronal cell processes at baseline with no obvious staining of neuronal cell bodies or nuclei. However, 2 hr after ICV BDNF treatment, p-ERK1/2-IR was present in neuronal processes, cell bodies, and nuclei within neurons (NeuN-positive cells) throughout all layers of the cortex and hippocampus (Fig. 2). There was no apparent change in p-ERK1/2 staining of astrocytes or oligodendrocytes after BDNF treatment (data not shown).
Fig. 1.

BDNF increases phosphorylation of ERK1/2 and AKT in the neonatal brain. P7 rat pups received an ICV injection of vehicle (PBS) or BDNF (5 μg in PBS). Brain tissues from the hippocampus (A) and cortex (B) were dissected at the various times indicated and lysed. Proteins were separated by 12.5% SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was first performed with an anti-phospho-ERK1/2 antibody; blots were stripped and reprobed with anti-phospho-AKT, anti-ERK1/2, and anti-AKT antibodies. On theright in A and B,p44 corresponds to ERK1, and p42corresponds to ERK2. Similar results were found in four independent experiments.
Fig. 2.

BDNF induces phosphorylation of ERK1/2 in neurons. Brain sections were prepared from P7 rats 30 min after an ICV injection of either vehicle (A, C) or BDNF (B, D) (n = 3/group). Brain sections from the hippocampus (A, B) and cortex (C, D) were labeled with anti-neuronal nucleus antibody NeuN (red) and anti-phospho-ERK1/2 antibody specific to the phosphorylated form of ERK1/2 (green). Arrows inB and D indicate yellowcells in which NeuN and p-ERK1/2 are colocalized. Scale bar, 20 μm.
BDNF neuroprotection appears to be mediated via the ERK pathway
Our previous studies have demonstrated that BDNF is strongly neuroprotective against H-I-induced injury to the developing rat brain (Cheng et al., 1997; Han et al., 2000). Thus, we were interested in determining which BDNF-stimulated intracellular signaling pathway was responsible for BDNF's neuroprotective effects. We first sought to determine whether we could inhibit the increase in p-ERK1/2 after BDNF stimulation. PD98059 and U0216 are selective inhibitors of ERK1/2 kinase (MAP kinase kinase, MEK1, and MEK2) (Dudley et al., 1995; Favata et al., 1998). We pursued our in vivo studies with U0216 because it is 100× more potent than PD98059 and does not have problems with solubility that we have found to occur at concentrations of PD98059 >50 μm in aqueous solutions. We found that a single ICV injection of as little as 1 nmol of U0216 was able to inhibit markedly the increase in p-ERK1/2 resulting from a single ICV injection of BDNF (Fig. 3). In contrast, U0126 did not block the ability of BDNF to increase p-AKT (Fig. 3). We then asked whether U0126 would block BDNF's neuroprotective effects against H-I.
Fig. 3.

U0126 blocks BDNF-mediated ERK1/2 phosphorylationin vivo. P7 rats were ICV injected with vehicle (1 μl of DMSO in 4 μl of PBS) or U0126 (0.2 or 1 nmol/animal) 20 min before receiving an ICV injection of vehicle or BDNF (5 μg/animal) as indicated. Cortical tissues prepared 1 hr after the injection were lysed, and proteins (50 μg/lane) were separated by SDS-PAGE and subjected to immunoblotting with an anti-phospho-ERK1/2 antibody specific to phosphorylated ERK1/2. Blots were stripped and subsequently reprobed with anti-ERK, anti-phospho-AKT, and anti-AKT. Data shown are representative of six independent experiments.
We used a well characterized model of neonatal H-I in which P7 rats undergo unilateral (left) carotid ligation followed by exposure to 2.5 hr of hypoxia (8% oxygen) as described previously (Rice et al., 1981;Cheng et al., 1997, 1998). This treatment results in robust brain injury to the cortex, hippocampus, striatum, and thalamus ipsilateral but not contralateral to the side of the carotid ligation. A component of the neuronal death in this model is caspase dependent (Cheng et al., 1998), and BDNF is particularly effective at blocking caspase-3-like activation (Han et al., 2000). This ability correlates well with BDNF's overall neuroprotective effects (Han et al., 2000). P7 rats were treated with either VEH, U0126, BDNF, or both U0126 and BDNF just before left carotid ligation and hypoxia. In addition, one group of animals received U0126 alone and was not subjected to H-I. As shown previously (Cheng et al., 1998; Han et al., 2000), there was a large increase in caspase-3-like activity observed ipsilateral but not contralateral to the carotid ligation followed by hypoxia in P7 pups treated with VEH (Fig. 4). Administration of U0126 followed by H-I did not result in significantly more caspase-3-like activity than did VEH treatment. This suggests that endogenous ERK activity is not contributing to H-I-induced injury in this neonatal model. In agreement with previous experiments (Han et al., 2000), ICV BDNF treatment virtually abolished the increase in caspase-3-like activity induced by H-I (Fig. 4). Interestingly U0126 significantly attenuated the ability of BDNF to block caspase-3-like activity in both the hippocampus and cortex (Fig. 4). In addition to blocking BDNF's ability to inhibit caspase-3 activation, U0126 also significantly inhibited BDNF's protection against tissue loss (Fig. 5). These results suggest that BDNF's neuroprotective effects against H-I on developing hippocampal and cortical neurons in vivo appear to be mediated predominantly via the ERK pathway.
Fig. 4.
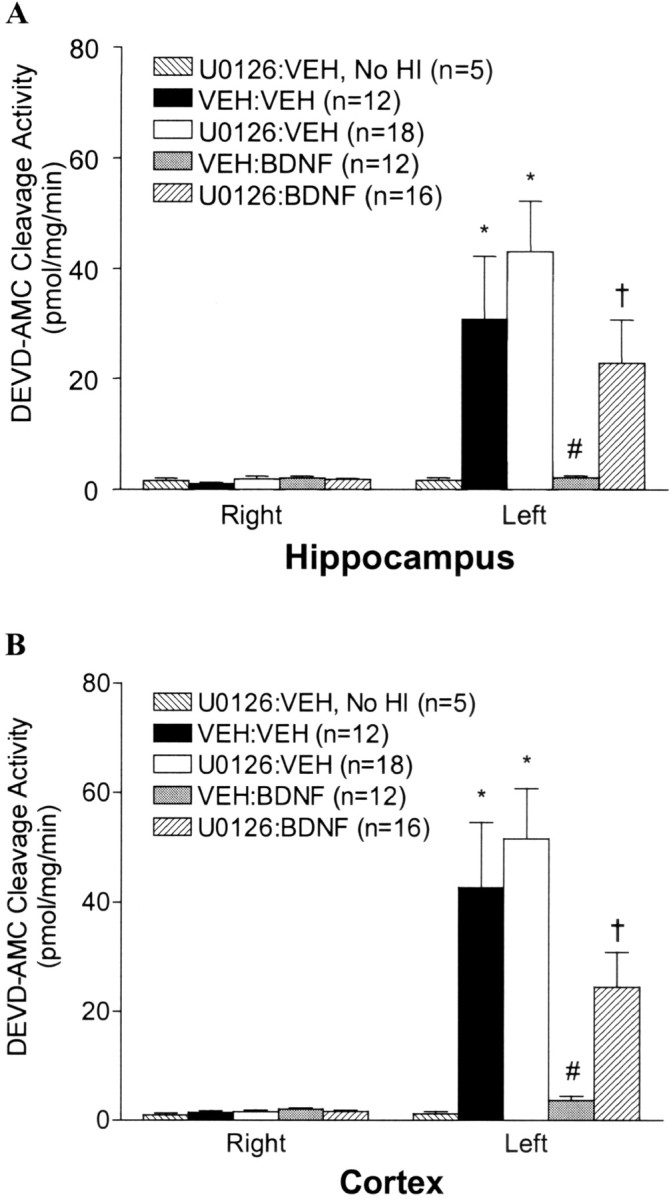
Inhibition of the ERK1/2 pathway blocks BDNF-mediated inhibition of caspase-3-like activity. P7 rats received ICV injections of vehicle or U0126 (1 nmol/animal) followed 15 min later by an ICV injection of vehicle or BDNF (5 μg/animal) just before the carotid ligation and exposure to 2.5 hr of hypoxia. Twenty-four hours after H-I, brain tissues from the hippocampus (A) and cortex (B) contralateral (right) and ipsilateral (left) to the ligation were dissected and subjected to DEVD-AMC cleavage assay as described in Materials and Methods. Note that there is no activation of caspase-3-like activity in the group treated with U0126 without subsequent H-I (No HI). Data represent the mean ± SEM; *p < 0.05 compared with the contralateral (right) hemisphere, #p < 0.05 comparing ipsilateral hemispheres of VEH:BDNF with that of the VEH:VEH group, and †p < 0.05 comparing ipsilateral hemispheres from theU0126:BDNF group with that of theVEH:BDNF group. Data were analyzed by ANOVA and Dunn's multiple comparison method.
Fig. 5.

Inhibition of the ERK1/2 pathway blocks BDNF-mediated neuroprotection against neonatal H-I. P7 rats were treated with VEH:VEH, VEH:BDNF, orU0126:BDNF before the carotid ligation and exposure to hypoxia for 2.5 hr. One week later, animals were killed, and brain sections were stained with cresyl violet. A, Examples of the degree of injury of representative animals from each treatment group are shown. Note the marked unilateral hemispheric tissue loss in the cortex and hippocampus in VEH:VEH andU0126:BDNF animals, whereas there is little to no tissue loss in the VEH:BDNF animal. B,BDNF-mediated neuroprotection was significantly inhibited by U0126. Regional area loss from the striatum, hippocampus, and cortex of each group was assessed as described in Materials and Methods. Data represent the mean ± SEM; *p < 0.05 comparing VEH:BDNF with either theVEH:VEH or the U0126:BDNF group. Data were analyzed by ANOVA and Dunn's multiple comparison method.
BDNF activation of the PI3-kinase pathway does not appear to mediate BDNF's protective effects
To address further the possible role of the PI3-kinase pathway in BDNF's neuroprotective effects, we assessed whether we could block the ability of BDNF to activate the PI3-kinase pathway without inhibiting its ability to activate the ERK pathway. We found that the BDNF-mediated increase in p-AKT was blocked by as little as 0.02 and 0.1 nmol of the PI3-kinase inhibitor wortmannin given ICV just before BDNF (Fig. 6A). Importantly, 0.1 nmol of wortmannin was able to block BDNF-stimulated increases in p-AKT over the entire 12 hr period during which p-AKT levels are increased by BDNF (Fig. 6B). This dose of wortmannin did not block the ability of BDNF to increase p-ERK1/2 (Fig.6). We then asked whether wortmannin would block BDNF's neuroprotective effects. P7 rats were treated with either VEH, wortmannin, BDNF, or both wortmannin and BDNF before carotid ligation and hypoxia. In addition, one group of animals received wortmannin alone and was not subjected to H-I. BDNF treatment again prevented the increase in caspase-3-like activity induced by H-I (Fig.7). In contrast to the effects of U0126, wortmannin had no significant effect on the ability of BDNF to block caspase-3-like activity in both the hippocampus and cortex (Fig. 7). ICV wortmannin given just before H-I or in the absence of H-I had no additional effect on caspase-3 activity. In addition to results obtained assessing caspase-3-like activity, we also found that wortmannin had no significant effect on the ability of BDNF to protect against tissue loss observed 1 week after H-I (Fig. 7C). These data suggest that BDNF activation of PI3-kinase is not the major pathway by which BDNF protects the neonatal rat brain against H-I injury.
Fig. 6.

BDNF-mediated AKT but not ERK1/2 phosphorylation is blocked by wortmannin in vivo. P7 rats received ICV injections of vehicle (1 μl of DMSO in 4 μl of PBS) or wortmannin (Wort; 0.1 or 0.02 nmol/animal in A; 0.1 nmol/animal in B) followed 15 min later by an ICV injection of vehicle or BDNF (5 μg/animal) as indicated. Cortical tissues prepared 1 hr after the injection in A or at later time points in B were lysed, and proteins (50 μg/lane) were separated by SDS-PAGE and subjected to immunoblotting with an anti-phospho-AKT antibody specific to phosphorylated AKT. Blots were stripped and subsequently reprobed with anti-phospho-ERK1/2, anti-ERK, and anti-AKT. Data shown are representative of four independent experiments.
Fig. 7.
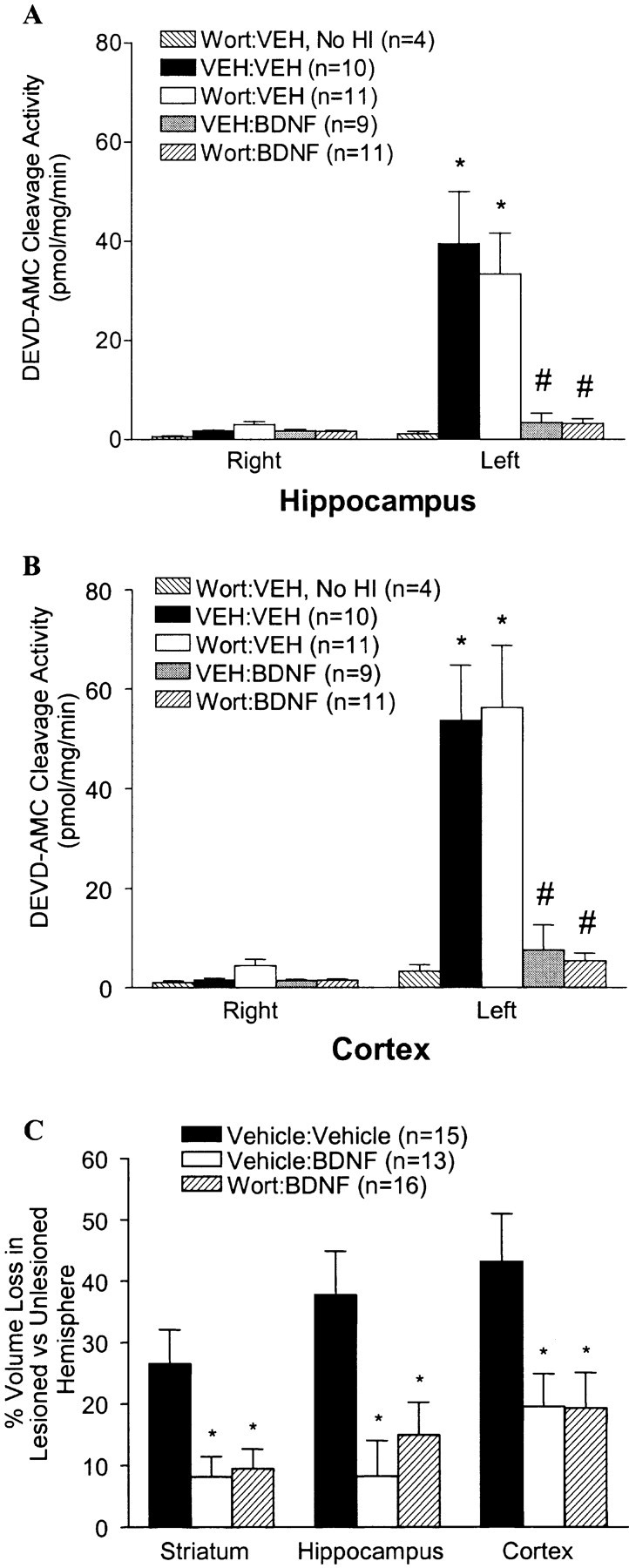
Inhibition of the AKT pathway does not block BDNF-mediated neuroprotection against neonatal H-I. A, B, P7 rats received ICV injections of VEH or wortmannin (0.1 nmol/animal) followed 15 min later by ICV injections of VEH or BDNF (5 μg/animal) before the carotid ligation and exposure to hypoxia for 2.5 hr. Twenty-four hours after H-I, brain tissues from the hippocampus (A) and cortex (B) contralateral (right) and ipsilateral (left) to the ligation were dissected and subjected to DEVD-AMC cleavage assay as described in Materials and Methods. Note that there is no activation of caspase-3-like activity in the group treated with wortmannin without subsequent H-I. Data represent the mean ± SEM; *p < 0.05 comparing VEH:VEH and Wort:VEHipsilateral with contralateral hemispheres; #p < 0.05 comparing ipsilateral hemispheres of VEH:BDNF andWort:BDNF with ipsilateral hemispheres of eitherVEH:VEH or Wort:VEH groups. Data were analyzed by ANOVA and Dunn's multiple comparison. C, P7 rats were treated with Vehicle:Vehicle,Vehicle:BDNF, or Wort:BDNF before the carotid ligation and exposure to hypoxia for 2.5 hr. One week later, animals were killed, and brain sections were stained with cresyl violet. The regional area loss from the striatum, hippocampus, and cortex of each group was assessed as described in Materials and Methods. Data represent the mean ± SEM; *p < 0.05 comparing Vehicle:Vehicle with either theVehicle:BDNF or the Wort:BDNF group. Data were analyzed by ANOVA and Dunn's multiple comparison method.
DISCUSSION
Since the discovery of potent survival-promoting effects of neurotrophic factors, there has been promise that they could be used in the treatment of diseases of the PNS and CNS. One step toward testing this possibility is to elucidate which factors can protect against death and dysfunction of particular neuronal populations in specific disease states. An important corollary is to understand which cellular signaling cascades activated by neurotrophic factors are responsible for their protective effects in models of relevant diseases of the nervous system. Using inhibitors of specific intracellular signaling pathways, we found that BDNF's neuroprotective effects in a rat model of neonatal H-I brain injury appear to be mediated by stimulation of the ERK pathway. These findings may have important implications in terms of understanding the cellular signaling responsible for BDNF actions in CNS neurons in vivo as well as for developing treatments to protect the neonatal brain against H-I damage.
H-I injury to the prenatal and perinatal brain is a major contributor to morbidity and mortality to infants and children (Vanucci, 1990;Volpe, 1995), often leading to mental retardation, seizures, and motor impairment (cerebral palsy). In the well studied Levine model, H-I treatment is given to the P7 rat brain at a developmental age believed to be approximately equivalent to 32–36 weeks of human gestation (Johnston, 1983; Vanucci, 1990). The injury resulting is similar to that seen in the term human infant that has been exposed to an H-I insult. We have found recently that BDNF markedly protects against the H-I injury in this model, a portion of which has features of apoptotic-like death (Ferrer et al., 1994; Mehmet et al., 1994; Hill et al., 1995; Sidhu et al., 1997; Silverstein et al., 1997; Hasegawa et al., 1998; Pulera et al., 1998) and is caspase dependent (Cheng et al., 1998; Han et al., 2000). Our results in the present study supporting the involvement of the ERK pathway in this protection may have important clinical implications. Theoretically, a protein such as BDNF could be administered to a neonate at very high risk for H-I brain injury. Because BDNF does not cross the blood—brain barrier (BBB) to an appreciable extent, it would have to be given ICV. In contrast, it is possible that neuronal stimulation of the ERK pathway by agents permeable to the BBB will have protective effects similar to that of BDNF. In light of this, it is interesting to note that stimulation of neurotransmitter receptors such as muscarinic acetylcholine receptors results in prolonged activation of ERK1/2 both in vitro andin vivo (Rosenblum et al., 2000). A variety of agents such as ligands for specific neurotransmitter receptors (Ferraguti et al., 1999; Hayashi et al., 1999; Mukherjee et al., 1999; Yan et al., 1999), nitric oxide (Yun et al., 1998), and estrogen (Singh et al., 1999) can activate the neuronal ERK pathway in vitro. A search for agents that can activate the ERK pathway or its downstream mediators in neurons in vivo seems warranted to attempt to develop better therapies against neonatal H-I brain injury.
Several in vitro studies have demonstrated that trophic factor stimulation of either PI3-kinase or ERK can be the dominant survival-promoting pathway depending on factors such as neuronal cell type as well as mode of cellular stress. A recent study by Hetman et al. (1999) emphasizes that the stressful stimulus can determine which pathway is dominant even within the same cell type. It was found that, in response to serum deprivation, BDNF mediated survival of cultured cortical neurons via the PI3-kinase pathway. In contrast, when the same cells were treated with campothecin, a DNA synthesis inhibitor, BDNF mediated protection via the ERK pathway. Our study emphasizes the importance of the ERK pathway to BDNF-mediated protection not only of cortical but also of hippocampal neurons in vivo. Furthermore, it demonstrates that intracellular signaling pathways that mediate neuroprotection in vivo cannot be predicted a priori and need to be determined as a function of both development as well as the injurious stimulus.
Our studies address the role of the ERK pathway during development after a specific injurious stimulus in vivo. The role of this pathway in the setting of different CNS diseases and in the adult brain is not clear. The ability of different neurotrophic factors to act on neurons is often developmentally regulated (Knusel et al., 1994;Cheng et al., 1997; Hata et al., 2000), and the impact of developmental maturation on effects of intracellular signaling has not been determined. Also, the mechanism and time course of neuronal death in response to different injuries can be age dependent (Easton et al., 1997) and potentially influence the response to intracellular signaling. Finally, the role of endogenous alterations in ERK signaling after brain injury such as H-I may be quite distinct from that caused by trophic factor stimulation. For example, a recent study using an adult focal H-I model suggests that the ERK pathway may contribute to damage resulting from focal cerebral ischemia (Alessandro et al., 1999). In our study, we found that administration of the ERK pathway inhibitor U0126 alone before neonatal H-I did not lessen brain damage. This suggests that after an H-I stimulus to the neonatal brain, endogenous ERK signaling does not play a major role in the ensuing injury. We have, however, observed that ERK activation does occur in the P7 rat brain after H-I. In contrast to the effects of BDNF, this activation occurs in astrocytes but not in neurons (B. H. Han and D. M. Holtzman, unpublished observations). In future studies, it will be important to determine the cell type-specific role of ERK activation in vivo both in the normal brain and under different disease-related conditions.
The intracellular mechanisms downstream of neuronal ERK activation that are required for protection against injury in vivo will be important to elucidate. The caspase-dependent component of cell death after neonatal H-I does not peak until 12–24 hr after injury. This suggests that there is a prolonged therapeutic window in which effects of ERK activation on both transcription-dependent and -independent mechanisms could influence survival. Recent findings in cultured cerebellar granule neurons suggest that BDNF-activated ERK signaling promotes survival via a dual mechanism by phosphorylating and inhibiting the antiapoptotic protein BAD and by inducing expression of prosurvival genes via the transcription factor CRE-binding protein (CREB) (Bonni et al., 1999). Interestingly, both of these mechanisms appear to be mediated by a member(s) of the pp90 ribosomal S6 kinase family (Rsks). Members of the Rsk family of protein kinases are activated by ERK signaling (Blenis, 1993). It will be important to determine whether effects of ERK activation on the phosphorylation of BAD and CREB via Rsks are relevant to protective effects of neurotrophins in vivo both in neonatal H-I as well as in other in vivo models of brain injury.
Footnotes
This work was supported by National Institutes of Health Grant NS 35902 to D.M.H. We thank Maia Parsadanian for her technical support; Eugene Johnson, Mohanish Deshmukh, Laura Dugan, Jeff Gidday, and Donna Ferriero for helpful comments; and Amgen, Inc. for recombinant BDNF.
Correspondence should be addressed to Dr. David M. Holtzman, Washington University School of Medicine, Department of Neurology, 660 South Euclid Avenue, Box 8111, St. Louis, MO 63110. E-mail:holtzman@neuro.wustl.edu.
REFERENCES
- 1.Alessandro A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blenis J. Signal transduction via the MAP kinases: proceed at your own Rsk. Proc Natl Acad Sci USA. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1365. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 4.Chao MV. Neurotrophin receptors: a window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- 5.Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J Neurosci. 1997;17:7288–7296. doi: 10.1523/JNEUROSCI.17-19-07288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM. Marked age-dependent neuroprotection by BDNF against neonatal hypoxic-ischemic brain injury. Ann Neurol. 1997;41:521–529. doi: 10.1002/ana.410410416. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Jr, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and AKT protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolcet X, Egea J, Soler R, Martin-Zanca D, Comella JX. Activation of the phosphatidylinositol 3-kinase, but not extracellular-regulated kinases, is necessary to mediate neurotrophic factor-induced motoneuron survival. J Neurochem. 1999;73:521–531. doi: 10.1046/j.1471-4159.1999.0730521.x. [DOI] [PubMed] [Google Scholar]
- 11.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 12.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from bax deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- 15.Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci. 1997;17:7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyck DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 17.Ferraguti F, Baldani-Guerra B, Corsi M, Nakanishi S, Corti C. Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur J Neurosci. 1999;11:2073–2082. doi: 10.1046/j.1460-9568.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- 18.Ferrer I, Tortosa A, Macaya A, Sierra A, Moreno D, Munell F, Bianco R, Squier W. Evidence of nuclear DNA fragmentation following hypoxia-ischemia in the infant rat brain, and transient forebrain ischemia in the adult gerbil. Brain Pathol. 1994;4:115–122. doi: 10.1111/j.1750-3639.1994.tb00821.x. [DOI] [PubMed] [Google Scholar]
- 19.Gidday JM, Fitzgibbons JC, Shah AR, Park TS. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett. 1994;168:221–224. doi: 10.1016/0304-3940(94)90455-3. [DOI] [PubMed] [Google Scholar]
- 20.Han BH, D'Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A, Deshmukh M, Holtzman DM. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000;7:38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa K, Litt L, Espanol MT, Sharp FR, Chan PH. Expression of c-fos and hsp70 mRNA in neonatal rat cerebrocortical slices during NMDA-induced necrosis and apoptosis. Brain Res. 1998;785:262–278. doi: 10.1016/s0006-8993(97)01410-8. [DOI] [PubMed] [Google Scholar]
- 22. Hata Y, Ohshima M, Ichisaka S, Wakita M, Fukuda M, Tsumoto T. Brain-derived neurotrophic factor expands ocular dominance columns in visual cortex in monocularly deprived and nondeprived kittens but does not in adult cats. J Neurosci 20 2000. RC57 (1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi T, Umemori H, Mishina M, Yamamoto T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature. 1999;397:72–76. doi: 10.1038/16269. [DOI] [PubMed] [Google Scholar]
- 24.Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-related kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 25.Hill IE, MacManus JP, Rasquinha I, Tuor UI. DNA fragmentation indicative of apoptosis following unilateral cerebral hypoxia-ischemia in the neonatal rat. Brain Res. 1995;676:398–403. doi: 10.1016/0006-8993(95)00145-g. [DOI] [PubMed] [Google Scholar]
- 26.Johnston MV. Neurotransmitter alterations in a model of perinatal hypoxic-ischemic brain injury. Ann Neurol. 1983;13:511–518. doi: 10.1002/ana.410130507. [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of the C-Myc-induced apoptosis by ras signaling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 28.Klein R, Parada LF, Coulier F, Barbacid M. trk B, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J. 1989;8:3701–3709. doi: 10.1002/j.1460-2075.1989.tb08545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- 30.Knusel B, Rabin S, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1–17. [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehmet H, Yue X, Squier MV, Lorek A, Cady E, Penrice J, Sarraf C, Wylezinska M, Kirkbride V, Cooper C, Brown GC, Wyatt JS, Reynolds EOR, Edwards AD. Increased apoptosis in the cingulate sulcus of newborn piglets following transient hypoxia-ischaemia is related to the degree of high energy phosphate depletion during the insult. Neurosci Lett. 1994;181:121–125. doi: 10.1016/0304-3940(94)90574-6. [DOI] [PubMed] [Google Scholar]
- 34.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit trkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 36.Mukherjee PK, DeCoster MA, Campbell FZ, Davis RJ, Bazan NG. Glutamate receptor signaling interplay modulates stress-sensitive mitogen-activated protein kinases and neuronal cell death. J Biol Chem. 1999;274:6493–6498. doi: 10.1074/jbc.274.10.6493. [DOI] [PubMed] [Google Scholar]
- 37.Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 39.Pulera MR, Adams LM, Liu HT, Santos DG, Nishimura RN, Yang FS, Cole GM, Wasterlain CG. Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke. 1998;29:2622–2629. doi: 10.1161/01.str.29.12.2622. [DOI] [PubMed] [Google Scholar]
- 40.Qiu YH, Zhao X, Hayes RL, Perez-Polo JR, Pike BR, Huang L, Clifton GL, Yang K. Activation of phosphatidylinositol 3-kinase by brain-derived neurotrophic factor gene transfection in septo-hippocampal cultures. J Neurosci Res. 1998;52:192–200. doi: 10.1002/(SICI)1097-4547(19980415)52:2<192::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 41.Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 42.Rosenblum K, Futter M, Jones M, Hulme EC, Bliss TVP. ERKI/II regulation by the muscarinic acetylcholine receptors in neurons. J Neurosci. 2000;20:977–985. doi: 10.1523/JNEUROSCI.20-03-00977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sidhu S, Tuor UI, Del Bigio MR. Nuclear condensation and fragmentation following cerebral hypoxia-ischemia occur more frequently in immature than older rats. Neurosci Lett. 1997;223:129–132. doi: 10.1016/s0304-3940(97)13426-7. [DOI] [PubMed] [Google Scholar]
- 44.Silverstein FS, Barks JD, Hagan P, Liu XH, Ivacko J, Szaflarski J. Cytokines and perinatal brain injury. Neurochem Int. 1997;30:375–383. doi: 10.1016/s0197-0186(96)00072-1. [DOI] [PubMed] [Google Scholar]
- 45.Singh M, Setalo G, Jr, Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19:1179–1188. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Snider WD. Functions of the neurotrophins during development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 47.Taylor DL, Edwards AD, Mehmet H. Oxidative metabolism, apoptosis and perinatal brain injury. Brain Pathol. 1999;9:93–117. doi: 10.1111/j.1750-3639.1999.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanucci RC. Experimental biology of cerebral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res. 1990;27:317–326. doi: 10.1203/00006450-199004000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Vemuri GS, McMorris FA. Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development. 1996;122:2529–2537. doi: 10.1242/dev.122.8.2529. [DOI] [PubMed] [Google Scholar]
- 50.Volpe JJ. Neurology of the newborn. Saunders; Philadelphia: 1995. [Google Scholar]
- 51.Xia X, Dickens M, Raingeaud J, Davis RJ, Greenberg M. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 52.Yan Z, Feng J, Fienberg AA, Greengard P. D2 dopamine receptors induced mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc Natl Acad Sci USA. 1999;96:11607–11612. doi: 10.1073/pnas.96.20.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao R, Cooper GM. Requirement for phophatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 54.Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-d-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci USA. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]