

Abstract
Cholinergic markers in the middle layers of rat auditory cortex are transiently upregulated during the second postnatal week, at which time α7 nicotinic acetylcholine receptors (nAChRs) selectively regulate NMDA receptor (NMDAR)-mediated EPSPs. To investigate the developmental role of this regulation, we determined whether manipulating nAChR function at specific times during the first 4 weeks after birth could alter subsequent neuronal function. Rat pups were injected twice daily with nicotine (1 or 2 mg/kg) or saline during approximately the first, second, or fourth postnatal week (i.e., before, during, or after the peak upregulation of nAChRs). Glutamate EPSPs and intrinsic membrane properties were measured during whole-cell recordings from visually identified pyramidal neurons in layers II–IV of brain slices prepared at least 15 hr after the last injection. Chronic nicotine exposure (CNE) had little effect on intrinsic membrane properties and during week 1 or 4 did not affect synaptic function. However, CNE during week 2 resulted in EPSPs with long durations, multiple peaks, and enhanced NMDAR components. These changes remained significant even 10 d after CNE. Rapid application of nicotine, which in control neurons selectively enhances NMDAR EPSPs during week 2, produced only weak effects after CNE. Receptor binding studies showed that CNE-induced EPSP alterations occurred in the absence of altered α7 nAChR numbers or agonist binding affinity. Thus, altered stimulation of nAChRs by CNE during week 2, but not before or after, disrupts the development of glutamate synapses in rat auditory cortex.
Keywords: acetylcholine, auditory, cortex, development, EPSP, glutamate, nicotine, NMDA, intrinsic properties, synaptic
Nicotine, the neuroactive ingredient in tobacco, is a highly addictive substance that has multiple effects on the nervous system (for review, see Benowitz, 1996; Dani and Heinemann, 1996; Role and Berg, 1996). The effects of nicotine extend to nonsmokers by way of secondhand smoke exposure, and both infants and fetuses are particularly vulnerable. Children born to mothers who smoked during pregnancy are often born prematurely and exhibit decreased body weight and size, increased body tremor, and impaired respiratory function (for review, see Nash, 1988). Perinatal nicotine exposure may also produce sensory deficits. For example, infants exposed to nicotine either in utero or after birth show decreased ability to habituate to sounds and to orient toward an auditory stimulus (Saxton, 1978; Picone et al., 1982) and continue to show auditory and auditory-related cognitive impairments as they mature (Sexton et al., 1990; McCartney et al., 1994; Fried et al., 1997). Thus, early exposure to nicotine can adversely affect nervous system development, including that of the auditory system.
During the second postnatal week in rats, primary sensory cortex displays a dramatic increase in expression of the cholinergic hydrolytic enzyme, acetylcholinesterase (AChE) (Kristt, 1979; Prusky et al., 1988; Robertson et al., 1991), and α7 nicotinic acetylcholine receptors (nAChRs) (Fuchs, 1989; Broide et al., 1995, 1996) in the middle cortical layers. This period corresponds to the third trimester of human development, when thalamic fibers innervate auditory neocortex and columns of AChE appear transiently in its middle layers (Krmpotic-Nemanic et al., 1980, 1983). Recently, we demonstrated that during the period of enhanced cholinergic expression in rat auditory cortex, rapid application of nicotine to pyramidal neurons selectively enhances the NMDAR component of glutamate EPSPs (Aramakis and Metherate, 1998). This effect is most prominent during postnatal days (P) 8–12 (>80% of neurons affected) and virtually disappears after P19 (<6% of neurons). These findings suggest that nAChR regulation of NMDAR activity during the second postnatal week in the rat may be important for auditory cortex development.
The goal of this study was to determine whether manipulation of nAChRs via chronic nicotine exposure (CNE) could affect the functional development of auditory cortex. We report that CNE during the second postnatal week, but not during the first or fourth weeks, disrupts synaptic development by selectively altering NMDAR-mediated synaptic transmission. Thus, postnatal week 2 appears to be a sensitive period for the disruptive actions of nicotine that may lead to long-term impairment of auditory cortex function.
MATERIALS AND METHODS
CNE treatment. Pups from timed-pregnant Sprague Dawley rats (Charles River, Wilmington, MA) were maintained on a 12 hr light/dark cycle. The date of birth was recorded as P0. Litters ranged in size from 10 to 16 pups, which were randomly assigned to saline or CNE treatment groups. Nicotine hydrogen tartrate was injected subcutaneously at a dose of either 1 or 2 mg/kg (0.35 or 0.7 mg/kg free base) in a 2 ml/kg volume using a 50 or 100 μl Hamilton syringe. Saline-treated littermates received an equivalent volume of 0.9% sterile saline. Injections were made twice daily, at 8:00–9:00 A.M. and 4:00–5:00 P.M. After injections, pups were placed individually in an open container to monitor behavior for 2–15 min and then returned to their home cage and litter. Nicotine- and saline-treated pups gained weight at the same rate (p > 0.10).
Four CNE schedules (CNE groups I–IV) were used. These protocols were designed to expose pups at different developmental times relative to the α7 nAChR upregulation in cortical layer IV, which begins shortly after birth, peaks at ∼P10, and ends after P15 (Broide et al., 1996). CNE group I pups were injected with nicotine (2 mg/kg) from P1 through P8–9 (treatments were maintained up to the day before electrophysiological recordings; slices were prepared the morning after the last injection). CNE group II rats were injected with either 1 or 2 mg/kg nicotine from P8 through P11–14, and slices were again prepared the next morning. CNE group III pups received 2 mg/kg nicotine from P8 through P16 and were allowed to survive an additional 7–10 d, and slices were made on P23 or P26. CNE group IV received 2 mg/kg nicotine from P20 through P23–25, and slices were prepared the morning after the last injection.
The majority of experiments used a nicotine dose of 2 mg/kg (0.7 mg/kg free base, CNE groups I–IV). This dose is commonly used in CNE studies (Abdulla et al., 1996; Rowell and Li, 1997) to approximate blood levels of nicotine in smokers (Isaac and Rand, 1972; Murrin et al., 1987;Henningfield et al., 1993). Some animals in CNE group II received a lower dose of 1 mg/kg nicotine (0.35 mg/kg free base).
Slice preparation and maintenance. Male and female rats (8–26 d old) were decapitated, and their brains were rapidly removed and placed in cold ACSF containing (in mm): NaCl 125.0, KCl 2.5, NaHCO3 25.0, KH2PO4 1.25, MgSO4 1.2, CaCl2 2.0, dextrose 10.0, bubbled with 95% O2, 5% CO2, pH 7.4 (osmolarity 298–302 mOsm/kg). Brains were blocked, and coronal sections (300–400 μm) containing auditory cortex (Paxinos and Watson, 1986; Sally and Kelly, 1988) were made using a vibroslicer (WPI, Sarasota, FL). The location of auditory cortex was determined based on landmarks (e.g., the dorsoventral extent of the hippocampus and distance dorsal to the rhinal fissure) and comparison with previous results using AChE histochemistry, which delineates primary sensory cortex in juvenile rats (Robertson et al., 1991; Aramakis and Metherate, 1998). Layers II–IV were identified within the recording chamber by micrometer measurements of distance from the pia (2.5 μm resolution) and scaled to published values (Roger and Arnault, 1989). Slices were placed in a holding chamber containing oxygenated ACSF at room temperature. For recordings, a single slice was placed in a submersion recording chamber on the fixed stage of an upright microscope (Zeiss, Axioskop FS) and maintained at 34°C with an ACSF flow rate of 3.5–4.0 ml/min.
Electrophysiological recording and stimulation. Neurons were visualized with infrared differential interference contrast (IR-DIC) optics with a video camera (Hamamatsu Photonics, Tokyo, Japan) and monitor (Stuart et al., 1993). Whole-cell recordings were made from the somas of layer II–IV neurons with clear ascending apical dendrites (i.e., presumed pyramidal neurons). Patch pipettes were made from filamented glass capillary tubes (1.5 mm outer diameter, A-M Systems, Seattle, WA), with tip diameters of ∼2.5 μm and DC resistances of 5–8 MΩ using a horizontal micropipette puller (P-97, Sutter Instrument, Novato, CA). The pipettes were filled with (in mm): 125.0 K-MeS03, 0.05 CaCl2, 0.5 NaCl, 2 Mg-ATP, 0.5 Na-GTP, 1.6 EGTA, and 10 HEPES. The pH was adjusted to 7.3 with KOH (1m), and final osmolarity was 270–280 mOsm/kg.
ACSF-filled micropipettes with tip diameters of ∼5 μm were used for electrical stimulation of afferent fibers. The electrode was placed a distance of 50–125 μm (mean 74 ± 2 μm, n = 106) lateral to the recording site, i.e., within the same cortical layer. Stimulation consisted of monophasic constant current pulses (200 μsec, 5–100 μA).
Recordings were obtained using an intracellular amplifier (Axoclamp 2B,Axon Instruments, Foster City, CA). Seal resistance was 5–12 GΩ. Series resistance ranged from 3 to 25 MΩ, but most often was <12 MΩ, and was compensated using the bridge balance. Membrane potential (Vm) was not corrected for liquid junction potential estimated to be approximately −13 mV (Metherate and Aramakis, 1999). Vm was monitored on an oscilloscope, digitized at 5.5 kHz, and stored on computer. Software (Axodata, Axograph, Axon Instruments) controlled data acquisition and analysis.
Pharmacological agents. Pharmacological agents included (±)-2-amino-5-phosphonovaleric acid (APV; RBI, Natick, MA) and (−)-nicotine hydrogen tartrate (nicotine; Sigma, St. Louis, MO). APV was prepared as a concentrated stock solution in purified H2O and diluted to its final concentration with ACSF. Nicotine for subcutaneous injection was prepared weekly as a 0.5 or 1 mg/ml solution in 0.9% sterile saline, adjusted to pH 7.0–7.5 (1m NaOH), and refrigerated. For rapid application to neurons in brain slices, nicotine was dissolved in ACSF (25 μm) and applied by pressure ejection from a pipette with a 5 μm tip, using a picospritzer (General Valve, Fairfield, NJ). The nicotine-filled pipette was placed within 30 μm of the neuron's apical dendrite.
Electrophysiology analysis. For each cell, afferent stimulus intensities ranged from below the threshold for eliciting an EPSP to above the threshold for eliciting an action potential. For all ages and treatments, EPSP measurements were obtained using stimulus intensities that were just subthreshold for generating spikes (“maximum subthreshold EPSPs”). EPSP amplitude was measured at the absolute peak, and latency to peak was measured from stimulus onset. EPSP duration was measured at one-third, one-half, and two-thirds peak amplitude. Measurements were made from averages of two to six responses taken at 10 sec intervals; however, traces used in Figures are single responses, not averages, except as noted in Figure 7. Mean data are presented ± 1 SE. Because synaptic and membrane properties develop rapidly during early postnatal life (Metherate and Aramakis, 1999; present study), a Factorial ANOVA was used for age-matched comparisons between treatment groups. For CNE groups I and II, data were grouped by individual days, whereas for CNE groups III and IV data were pooled within two age ranges, P22–24 and P25–26, because of the small number of cells at each age and the relatively slow rate of change in neuronal properties by P22. Except where noted, additional within-cell and between-group comparisons used ttests for paired or unpaired samples, respectively. Differences were considered significant at p < 0.05.
Fig. 7.

Effect on EPSPs of acute, rapid nicotine application to control and CNE group II neurons. All data are from P13 neurons. A, In a control neuron, pressure-ejected nicotine enhanced the late portion of the EPSP;Vm −70 mV. In a CNE group II neuron, nicotine increased the EPSP magnitude only slightly;Vm −62 mV. Each trace is an average of five responses at 10 sec interstimulus intervals.B, Rapid application of nicotine (25 μm, 10–40 msec, 20 psi) significantly enhanced EPSPs in control neurons (n = 17; paired t test,p < 0.02), but not CNE group II neurons (n = 8; p > 0.2).C, EPSP enhancement for the 25 neurons whose responses contribute to the histograms in B is shown for neurons that had either an increase (●) or no effect (○) in response to rapid nicotine application. For control neurons, nicotine increased EPSP areas 34–289% (mean 114%). In contrast, increases for CNE group II neurons were only 30–34% (mean 32%). Dashed linesindicate statistical significance at the 0.05 level (see Materials and Methods). Arrows indicate measurements for responses shown in A.
Effects of rapid nicotine application to neurons in vitrowere determined as described previously (Aramakis and Metherate, 1998). Synaptic potentials were elicited by a single stimulus pulse every 10 sec. After several control responses were obtained, pressure-ejected nicotine was paired with the afferent stimulus for several trials, with each pressure pulse beginning 15 msec before the afferent stimulus. For each condition, three to five traces were averaged, and EPSP area was measured. To establish a threshold value for a statistically significant effect, a pre-drug mean EPSP area and SD were calculated for each of five cells. The SD was then expressed as a percentage of the mean, and this value was averaged across cells. The threshold for a significant change in area was established at twice the average SD (95% confidence interval). This threshold value was 28.8 and 27.4% for control and CNE neurons, respectively (data from five neurons for each group). Thus, changes in EPSP area of ∼30% or greater were considered significant. To ensure that pressure-ejected nicotine was applied to control and CNE neurons under similar conditions, only neurons from P13 animals were compared, and the nicotine concentration in the pressure pipette was kept constant at 25 μm.
In situ hybridization and receptor binding. After CNE or saline treatments, rats were decapitated, and their brains were rapidly removed, frozen, and processed for α7 mRNA levels, α-bungarotoxin (α-BTX) binding sites, and nicotine inhibition of α-BTX binding, as detailed previously (Broide et al., 1996; Ospina et al., 1998). Briefly, 20 μm tissue sections were cryostat cut and mounted onto either gelatin-coated slides (for [125I] α-BTX binding), or slides with an additional coating of poly-l-lysine (for in situ hybridization) and kept at −20°C. Sections for [125I] α-BTX binding were stored desiccated at 4°C for 2 hr and then at −20°C until use. Sections for in situ hybridization were post-fixed with 4% paraformaldehyde in 0.1 m PBS, pH 7.4, for 1 hr at 22°C, then washed in PBS and air-dried.
A 2.1 kb cDNA encoding the entire rat α7 subunit cloned into pBluescript II SK+ was obtained from Dr. Jim Boulter (University of California Los Angeles), and35S-labeled uridine triphosphate was used in synthesizing cRNA riboprobes. These probes were further subjected to alkaline hydrolysis using the method of Cox et al. (1984). Tissue sections were incubated for 18 hr at 60°C with a hybridization solution (50% formamide, 10% dextran sulfate, 0.02% Ficoll, 0.02% polyvinyl pyrolidone, 0.02% bovine serum albumin, 500 μg/ml tRNA, 10 mm dithiothreitol, 0.3 m NaCl, 10 mm Tris, pH 8, and 1 mm EDTA, pH 8) containing labeled cRNA probes in the antisense orientation. Sections were then incubated with RNase A (20 μg/ml) for 30 min at 37°C, followed by washes of decreasing salinity (2–0.5× SSC buffer) at 22°C, and a 30 min wash in 0.1× SSC at 70°C. Tissue sections were dehydrated, dried, and apposed to β-max film for 3 d at 4°C.
Tissue sections for [125I] α-BTX binding were processed using a modification of the method described byFuchs (1989). Sections were preincubated in a solution containing 120 mm NaCl and 50 mm Tris-HCl, pH 7.4, for 15 min at 22°C. Slides were then incubated at 22°C for 2 hr in the same buffer containing 5 nm[125I] α-BTX (specific activity >200 Ci/mmol) in the absence or presence of α-cobratoxin (1 mm). For competition experiments, tissues were incubated as above, or with 4 or 40 μm nicotine. After incubation, slides were washed two times for 10 min each in Tris buffer at 4°C followed by two dips in distilled water, then air-dried. Labeled sections were exposed to radiation-sensitive Hyperfilm, along with14C standards, for 48 hr at room temperature. After film development, tissue sections were stained with cresyl violet for anatomical analysis.
Autoradiograms were quantified with a computer-based image analysis system (MCID, Imaging Research, St. Catherine, Ontario, Canada) using calibrated standards of reference. A calibration curve of optical density against radioligand concentration (disintegrations per minute per milligrams of tissue for in situ hybridization and femtomols per milligrams of tissue for binding films) was constructed using [14C] brain paste standards of known radioactivity. For binding, the curve was calibrated for reading [125I] emissions, as described by Miller and Zahniser (1987). Optical densities in the discrete regions of the autoradiogram were measured, and values of radioactivity were calculated by interpolation from the calibration curve. The measurements for specific hybridization were calculated by subtracting values of radioactivity in sections hybridized with sense probe from those hybridized with antisense. The measurements for specific α-BTX binding were obtained by subtracting values of nonspecific binding in the presence of cobratoxin from the values of total binding. Nissl-stained slides were used to verify the localization of α7 mRNA and binding sites in the brain. Cortical layers were identified by examination of the pattern and distribution of Nissl-stained cells. A transparency with the outline of each cortical layer was constructed from the Nissl-stained section and used as a template to guide the analysis of the respective autoradiogram.
RESULTS
Effects of CNE on electrophysiological measures of auditory cortex development were determined using slices from nicotine-injected, saline-injected, and untreated rat pups. Data were obtained from 190 pyramidal neurons in layers II–IV of slices from 68 pups ranging in age from P8 to P26. Of these, 45 neurons were from animals that had been injected with nicotine, and 145 neurons were from saline-injected or untreated pups. Synaptic and intrinsic properties from saline-injected pups did not differ from age-matched untreated pups (p > 0.10); these data were therefore combined and are referred to as controls. An additional 16 animals injected with nicotine or saline were used for receptor binding and in situ hybridization studies.
Postnatal development of EPSPs in auditory cortex
To illustrate synaptic changes during normal postnatal development, data on maximal subthreshold EPSPs were grouped into four age ranges: P8–12, P13–16, P20–22, and P24–26. At the youngest ages, EPSPs were of relatively small amplitude, long duration (measured at one-third, one-half, and two-thirds peak amplitude), and long latency to peak, and required higher stimulus intensities to be elicited (Fig. 1, Table1). EPSPs from the oldest age range had the largest amplitude, shortest duration, and shortest latency to peak, and required the lowest stimulus intensities. A continuum in these parameters was observed over intermediate ages. [Distances from the stimulating electrode to the recorded neuron averaged 72 ± 2 μm and did not differ between age groups (p > 0.05)]. These data on postnatal development of synaptic properties served as age-matched controls for the effects of CNE.
Fig. 1.

Normal development of EPSPs in pyramidal neurons of layers II–IV of auditory cortex. A,Maximal subthreshold EPSPs recorded in neurons from control P8–26 rats. B, Data on maximal subthreshold EPSPs grouped into four age ranges (values in Table 1). Bi, Latency to peak decreased over development. Bii, EPSP amplitude increased with age. Biii, EPSP duration, measured at1/3, 1/2, and 2/3 peak (PK) (A, arrows), decreased with age.
Table 1.
EPSPs in layers II–IV pyramidal neurons of rat auditory cortex during normal development and after CNE1-a
| Age/condition | Stimulus intensity (μA) | Latency to peak (msec) | Peak (mV) | EPSP width @ one-third peak | EPSP width @ one-half peak | EPSP width @ two-thirds peak | n |
|---|---|---|---|---|---|---|---|
| Development | |||||||
| P8–12 | 26.5 ± 7.0 | 16.8 ± 4.7 | 12.1 ± 1.3 | 107.8 ± 12.3 | 73.0 ± 8.5 | 50.6 ± 6.6 | 12 |
| P13–16 | 22.7 ± 3.6 | 9.2 ± 0.7 | 15.3 ± 1.2 | 66.5 ± 7.7 | 48.3 ± 6.9 | 28.4 ± 1.9 | 17 |
| P20–22 | 15.5 ± 5.0 | 5.7 ± 0.4 | 17.7 ± 1.0 | 34.5 ± 1.8 | 24.5 ± 1.4 | 16.4 ± 1.1 | 13 |
| P24–26 | 12.4 ± 3.5 | 4.9 ± 0.5 | 23.0 ± 2.5 | 27.1 ± 2.4 | 18.3 ± 2.2 | 12.8 ± 1.6 | 5 |
| Control | 38.8 ± 12.4 | 20.9 ± 8.7 | 11.0 ± 1.7 | 102.9 ± 16.7 | 72.7 ± 14.6 | 52.4 ± 12.1 | 6 |
| CNE group I | 50.0 ± 15.8 | 20.0 ± 5.5 | 7.9 ± 2.2 | 115.5 ± 14.7 | 80.6 ± 10.2 | 53.6 ± 6.7 | 5 |
| Control | 20.7 ± 2.9 | 10.2 ± 1.2 | 14.9 ± 1.1 | 76.3 ± 8.6 | 53.4 ± 6.2 | 32.8 ± 2.9 | 22 |
| CNE group II | |||||||
| 1 mg/kg | 19.0 ± 2.6 | 14.5 ± 2.9 | 13.5 ± 1.3 | 112.3 ± 11.81-b | 82.9 ± 10.41-b | 61.9 ± 8.91-b | 13 |
| 2 mg/kg | 17.5 ± 1.4 | 17.5 ± 4.1 | 12.9 ± 1.2 | 118.5 ± 14.91-b | 82.4 ± 11.71-b | 60.8 ± 10.01-b | 9 |
| Control | 10.1 ± 2.5 | 4.7 ± 0.3 | 21.2 ± 2.0 | 27.3 ± 1.9 | 18.5 ± 1.5 | 12.7 ± 1.1 | 8 |
| CNE group III | 9.5 ± 1.8 | 14.8 ± 4.6 | 16.3 ± 2.0 | 46.6 ± 5.91-b | 36.6 ± 5.51-b | 28.8 ± 5.11-b | 7 |
| Control | 10.1 ± 2.5 | 4.7 ± 0.3 | 21.2 ± 2.0 | 27.3 ± 1.9 | 18.5 ± 1.5 | 12.7 ± 1.1 | 8 |
| CNE group IV | 12.1 ± 2.6 | 5.6 ± 1.2 | 20.3 ± 1.6 | 33.7 ± 2.2 | 24.3 ± 2.4 | 17.1 ± 2.5 | 9 |
Measurements from maximal subthreshold EPSPs.
Significantly different from control; ANOVA, p < 0.05.
Effects of CNE on synaptic development
As described above (Materials and Methods), four groups of rat pups were exposed chronically to nicotine during different phases of the postnatal nAChR upregulation (CNE groups I–IV). A qualitative assessment of the behavioral effects of nicotine treatments indicated that within ∼30 sec of the subcutaneous nicotine injection (1 or 2 mg/kg; 0.35 or 0.7 mg/kg free base), young animals (<P10) displayed increased resting tremor and performed “swimming” movements. In older pups, nicotine produced increased locomotion, often resulting in circling movements. Such behavior persisted for up to 15 min, after which time nicotine-injected animals could not be distinguished behaviorally from controls.
CNE group I: CNE from P1 to P8–9 does not affect synaptic development
Neurons from pups injected with 2 mg/kg nicotine from P1 to P8–9 had maximal subthreshold EPSPs similar to those from age-matched controls (Fig. 2, Table 1). Nicotine-treated animals had similar EPSP peak amplitudes, peak latencies, and durations as control EPSPs and were elicited by stimuli of similar intensity (Table 1) [distances from the stimulating electrode to the recorded neuron did not differ between any CNE group (CNE I–IV) and its control group; p > 0.05]. Thus CNE for 8–9 d, including the entire first postnatal week, did not affect the measurements of synaptic development used in this study.
Fig. 2.
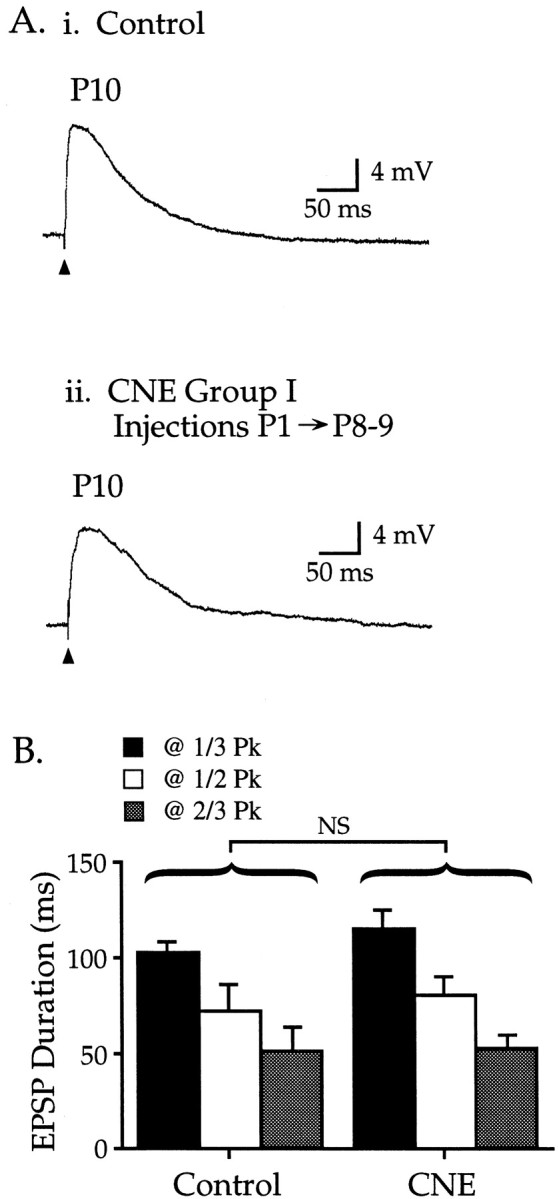
CNE group I: nicotine treatment for approximately the first postnatal week does not affect glutamate EPSPs.Ai, Representative EPSP in a control P10 neuron;Vm −66 mV. Aii, Nicotine injections from P1–9 resulted in an EPSP that was similar to controls; P10 neuron, Vm −61 mV. B, CNE group I EPSP durations (measured at 1/3,1/2, and 2/3 Pk) did not differ from age-matched controls (Table 1; p > 0.10).
CNE group II: CNE from P8 to P11–14 disrupts synaptic development
Nicotine injections (1 or 2 mg/kg) for 4–7 d from P8 through P11–14 dramatically altered EPSPs in two ways (Fig.3, Table 1). First, EPSPs from nicotine-treated animals had longer durations, measured at one-third, one-half, or two-thirds peak amplitude (Fig.3A,C). Second, multiple, discrete fluctuations resembling miniature EPSPs appeared prominently on the descending phase of EPSPs (Fig. 3Aii, asterisks). These miniature events occurred spontaneously, but their frequency increased markedly for tens of milliseconds after an afferent stimulus (Fig. 3Bii shows total number of events for 22 cells; comparison of events 50 msec before and after the afferent stimulus demonstrates a statistically significant increase; paired ttest, p < 0.001). Similar miniature events occurred at all stimulus intensities (data not shown). Few if any miniature events occurred in control cells (Fig. 3Bi shows total number of events in 22 control cells; small individual events can be seen in traces from P8 and P20 cells in Fig. 1A), nor did control cells show an increase in miniature frequency after an afferent stimulus (p > 0.3). Nicotine doses of 1 and 2 mg/kg were both effective in altering EPSP characteristics (Fig.3A,C) (comparison of EPSP durations shown in Fig. 3C; p > 0.1). Nicotine-treated EPSPs also tended to have smaller amplitudes and longer peak latencies than controls, but these differences were not significant (Table 1). Together, these data indicate that chronic exposure to nicotine for 4–7 d during the second postnatal week dramatically altered EPSP development in auditory cortex.
Fig. 3.

CNE group II: nicotine treatment during postnatal week 2 alters EPSPs. Ai, Representative EPSP from a control P13 neuron; Vm −73 mV.Aii, Nicotine injections (1 or 2 mg/kg) from P8 through P11–14 produced EPSPs that were larger in duration and magnitude and had multiple peaks on their descending phase (asterisks). Top trace,Vm −66 mV; middle trace,Vm −66 mV; bottom trace,Vm −77 mV. B, The time of occurrence of miniature fluctuations is plotted for 50 msec before and 260 msec after the stimulus (10 msec bins; comparisons using Fisher's PLSD). Bi, In 22 control neurons (P9–15), the number of events occurring before or 60 msec after the stimulus did not differ (p > 0.10). Bii, For 22 CNE group II neurons (1 and 2 mg/kg doses combined), the number of events within 60 msec after the stimulus was greater than during either the prestimulus period or the same 60 msec poststimulus period in control neurons (p > 0.10). C, EPSP durations recorded from CNE group II neurons (measured at , ½, and peak; histogram shading as in Fig. 2) were longer than age-matched controls (Table 1; p < 0.01). The increase in duration was similar for the two nicotine doses (p > 0.10).
CNE group III: long-term effects of CNE
To determine whether the effects of CNE during the second postnatal week were long lasting, rat pups were injected with 2 mg/kg nicotine for 9 d from P8 to P16 and then allowed to survive for an additional 7–10 d before electrophysiological experiments were performed. EPSPs from P23–26 pups in CNE group III were significantly longer than age-matched controls (Fig. 4, Table 1) (p < 0.01), but not as long as CNE group II EPSPs (comparison with CNE group II pups treated with 2 mg/kg,p < 0.05). EPSPs in CNE group III also displayed a small but significant increase in presumed miniature events within the first 20 msec after the stimulus (p < 0.05) (e.g., Fig. 6Bi). Group III EPSPs tended to have smaller amplitudes and longer peak latencies than controls, but these differences were not significant (Table 1). These data indicate that nicotine exposure during the second postnatal week can produce effects that last well into the fourth week. However, the normal developmental trends appear to resume after cessation of CNE.
Fig. 4.
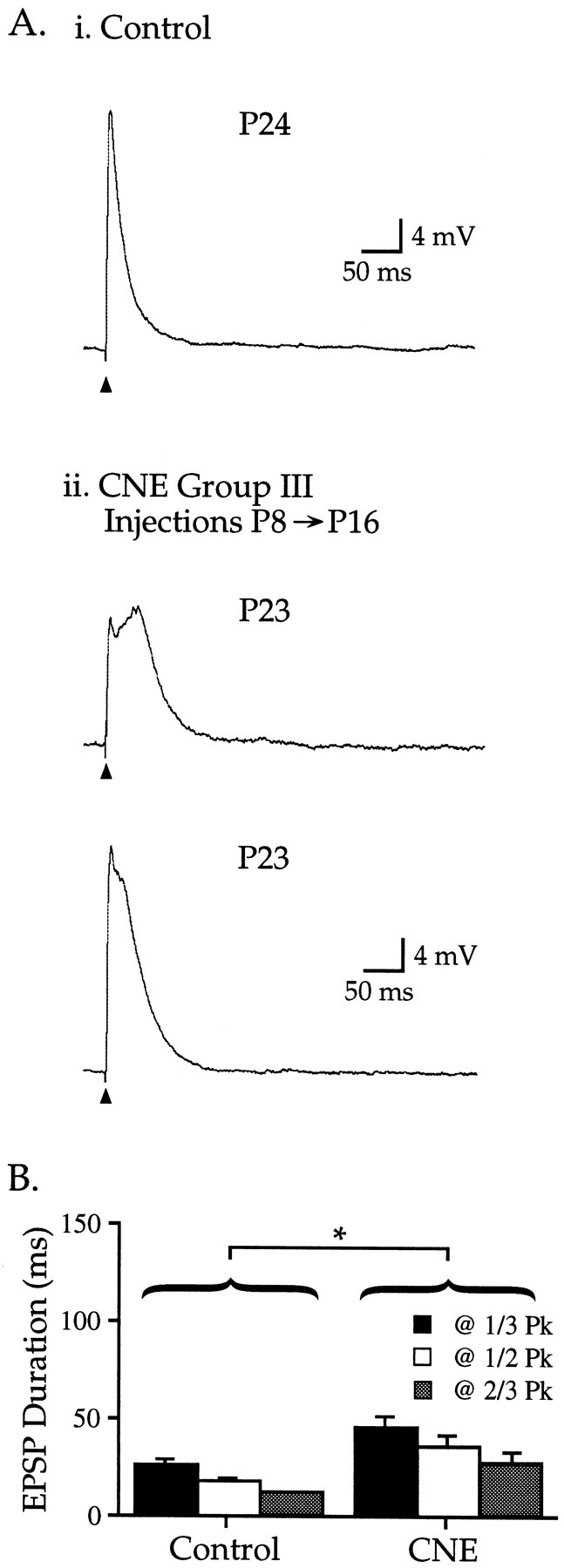
CNE group III: EPSP alterations produced by CNE treatment during week 2 last for at least 1 week. Ai, Representative EPSP from a control P24 neuron;Vm −80 mV. Aii, EPSPs recorded 7 d after nicotine injections from P8–16. Top trace, Vm −68 mV; bottom trace, Vm −79 mV. B, CNE group III EPSP durations (measured at1/3, 1/2, and 2/3 Pk) were longer than age-matched controls (Table 1; p < 0.01).
Fig. 6.
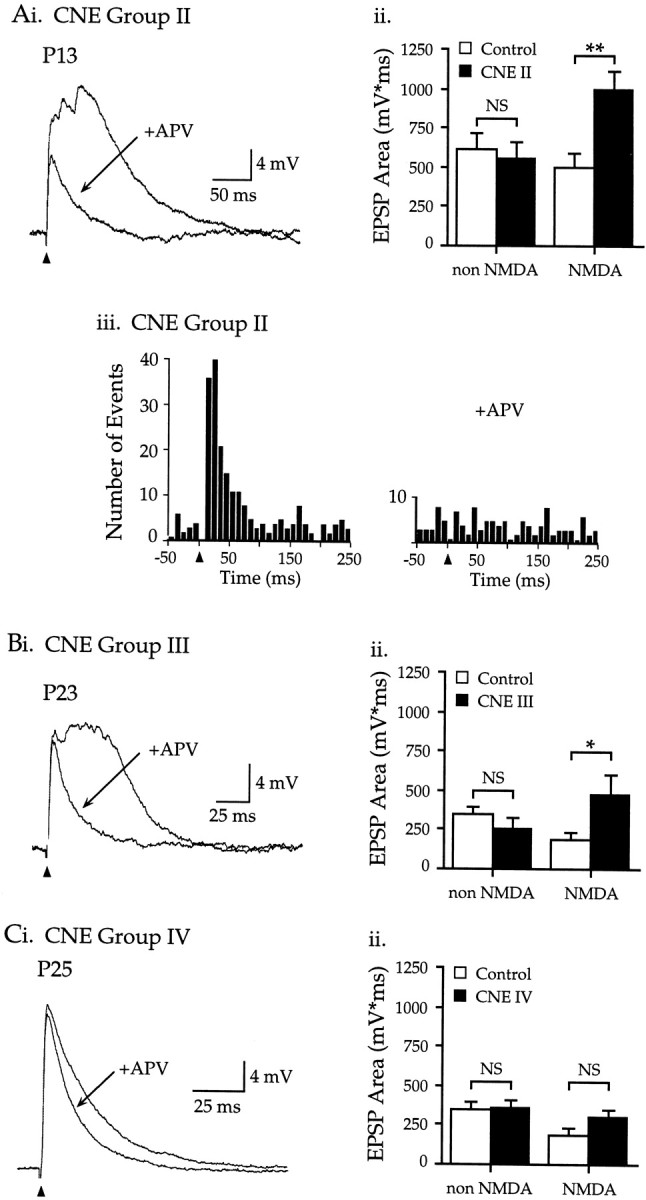
NMDARs mediate the enhanced EPSPs and stimulus-evoked miniature events produced by CNE during week 2.Ai, CNE group II: APV (50 μm) reduced the long-duration and multiple-peaked EPSP in a P13 neuron;Vm −70 mV. Aii, The area of the non-NMDAR EPSP (the EPSP in APV) in CNE group II neurons did not differ from control (control, 619 ± 104 mV · msec,n = 7; CNE II, 557 ± 99 mV · msec,n = 15; p > 0.10). In contrast, the area of the NMDAR EPSP (pre-APV area minus area in APV) was twice as large in CNE group II neurons as in control neurons (control, 493 ± 102 mV · msec; CNE II, 994 ± 117 mV · msec; p < 0.01). Aiii, APV reduced the stimulus-evoked increase in miniature events (p < 0.01, n = 11 neurons; for each neuron, miniature events were counted in three consecutive EPSPs, 10 sec interstimulus interval). Bi, CNE group III: APV decreased EPSP area in a P23 neuron;Vm −68 mV. Bii, The non-NMDAR EPSP area was similar in control and CNE group III neurons (control, 344 ± 44 mV · msec, n = 7; CNE III, 258 ± 69 mV · msec, n = 5;p > 0.10). However, the NMDAR EPSP area was more than twice as large in CNE group III neurons as in control neurons (control, 180 ± 48 mV · msec; CNE III, 476 ± 131 mV · msec; p < 0.05). Ci, CNE group IV: APV produced a small decrease in the late EPSP; P25 neuron,Vm −79 mV. Cii, The non-NMDAR EPSP area was similar in control and CNE group IV neurons (control, 344 ± 44 mV · msec, n = 7; CNE IV, 360 ± 49 mV · msec, n = 9;p > 0.10). Similarly, the NMDAR EPSP area did not differ between control and CNE group IV neurons (control, 180 ± 48 mV · msec; CNE IV, 306 ± 40 mV · msec;p > 0.10).
CNE group IV: CNE from P20 to P23–25 does not affect synaptic development
A final group of animals was injected with 2 mg/kg nicotine for 4–6 d from P20 to P23–25, and slices were made on the morning after the last injection. In seven of nine neurons, EPSPs were indistinguishable from those of age-matched controls (Fig.5Ai, Aii, compare P26 neurons). In two neurons, maximal subthreshold EPSPs had longer durations that qualitatively resembled EPSPs in CNE group III (P24 neuron in Fig. 5Aii), although unlike group III, lower intensity stimuli elicited EPSPs resembling controls (Fig.5Aii, see smaller EPSP for P24 neuron). Nevertheless, group data for all nine neurons in CNE group IV showed that EPSP duration, amplitude, latency to peak, and miniature fluctuations did not differ from control values (Fig. 5B, Table 1) (p > 0.10; data for miniature fluctuations not shown). Thus, nicotine exposure during the fourth postnatal week did not significantly affect the measurements of synaptic development used in this study.
Fig. 5.
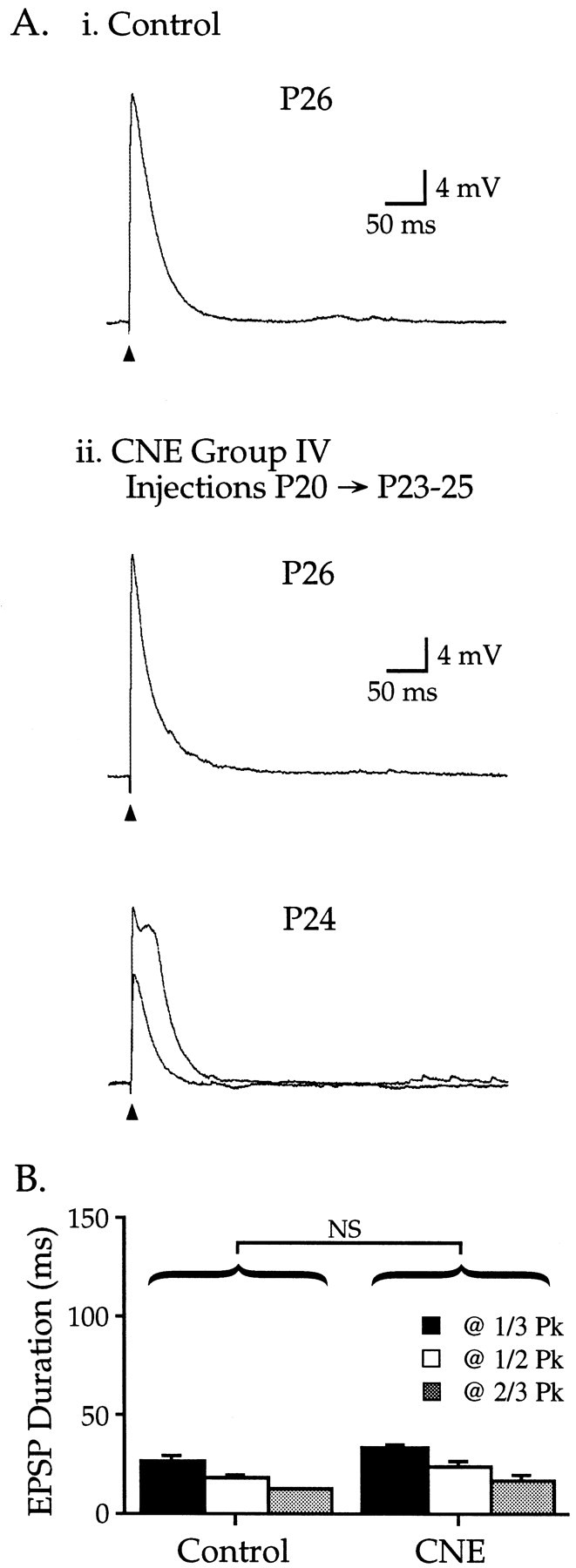
CNE group IV: nicotine treatment during postnatal week 4 does not affect EPSPs. Ai, Representative EPSP from a control P26 neuron; Vm −78 mV.Aii, CNE from P20 through P23–25 resulted in EPSPs that in general were similar to age-matched controls. Top trace, Vm −79 mV. However, in two of nine neurons, maximal subthreshold EPSPs had enhanced durations, whereas lower intensity stimuli produced control-like durations (bottom traces, Vm −75 mV).B, CNE group IV EPSP durations did not differ from age-matched controls (Table 1; p > 0.10).
Effects of CNE on NMDAR EPSPs
Because our previous study provided evidence that nAChRs selectively regulate EPSPs mediated by NMDARs (Aramakis and Metherate, 1998), we might expect that manipulations of nAChR function, such as the CNE treatment used here, would selectively affect NMDAR EPSPs. We therefore compared the contribution of NMDARs to EPSPs in neurons from nicotine-exposed and control pups.
CNE group II
As described above (Fig. 3), nicotine treatment from P8 to P11–14 produced long-duration EPSPs with multiple miniature fluctuations. In neurons from pups treated with 2 mg/kg nicotine, bath application of the NMDAR antagonist APV (50 μm) greatly decreased the magnitude and duration of EPSPs (Fig.6A) (n= 5; note that treatment with 1 mg/kg nicotine produced similar, statistically significant effects in 10 additional neurons, but to facilitate comparisons across CNE groups these data are not shown). Data from APV application allowed comparison of the NMDAR component of the EPSP (obtained by subtracting the EPSP area in APV from the pre-APV area) and the non-NMDAR component (the area in APV) in control and nicotine-treated neurons. As shown in Figure 6Aii, the NMDAR EPSP in CNE group II neurons clearly was enhanced relative to control neurons (102% increase, p < 0.01), whereas the non-NMDAR EPSP was unchanged (p > 0.10). Importantly, the prominent stimulus-evoked increase in miniature fluctuations seen in CNE group II neurons did not occur in the presence of APV (Fig. 6Aiii). Thus, both the CNE-induced miniature fluctuations and increased EPSP magnitude depended on NMDARs.
CNE group III
EPSPs recorded 7–10 d after nicotine exposure from P8 to P16 also contained enhanced NMDAR components. APV significantly reduced EPSPs in CNE group III neurons (Fig. 6Bi) (n = 5), and explicit comparison of the non-NMDAR and NMDAR components of the EPSP area (Fig. 6Bii) demonstrated that CNE more than doubled the NMDAR component (164% increase, p < 0.05) but did not change the non-NMDAR component (p > 0.10). These findings are consistent with the data from CNE group II and indicate that CNE during the second postnatal week selectively enhanced the NMDAR component of the EPSP for at least 10 d.
CNE group IV
APV also reduced the area of EPSPs in neurons from pups exposed to nicotine from P20 to P23–25 (Fig. 6C). However, non-NMDAR and NMDAR components of the EPSP were similar in control and nicotine-treated neurons (Fig. 6Cii) (p > 0.05; note that although group differences were not significant, the two CNE group IV neurons mentioned above with prominent, wide EPSPs also had prominent APV-sensitive components). On average, therefore, nicotine exposure in the fourth postnatal week did not affect the NMDAR component of the EPSP.
Effects of acute (pressure-ejected) nicotine on EPSPs after CNE
Previously we had demonstrated that rapid application of nicotine directly to neurons selectively enhances NMDAR-mediated synaptic transmission in neurons from untreated animals (Aramakis and Metherate, 1998). Because CNE group II neurons in the present study had enhanced EPSPs (Fig. 3) caused by selective enhancement of NMDAR components (Fig. 6), we sought to determine whether these EPSPs could be enhanced further by acute nicotine application (pressure-pulse application from a nicotine-containing micropipette placed adjacent to the apical dendrite of the neuron). Only P13 neurons were used, to examine untreated neurons close to their peak nicotine sensitivity (at ∼P10) (Aramakis and Metherate, 1998), as well as CNE neurons that had been treated through the peak period (i.e., from P8 to P12). Acute nicotine was applied to 17 untreated neurons and 8 CNE group II neurons (Fig.7). On average, acute nicotine significantly enhanced the EPSP area in untreated neurons (Fig.7B) (from 940 ± 128 to 1323 ± 199 mV · msec; paired t test, p < 0.02) but did not affect CNE neurons (1171 ± 170 to 1259 ± 176 mV · msec;p > 0.2). Data from individual neurons (Fig.7C) show that acute nicotine significantly enhanced 9 of 17 (53%) untreated neurons by an average of 114% and 2 of 8 (25%) CNE neurons by an average of 32% (significant enhancement for individual neurons indicates an increase in EPSP area greater than ∼30%; see Materials and Methods for details). Although comparisons between untreated and CNE groups did not reveal statistically significant differences, these data raise the possibility that CNE-induced increases in NMDAR EPSPs (Figs. 3, 6) may limit further enhancement by acute nicotine application (Fig. 7). Future experiments should resolve this issue.
Effects of CNE on the development of intrinsic membrane properties
Intrinsic membrane properties of layer II–IV pyramidal neurons change dramatically during early postnatal life (Metherate and Aramakis, 1999). Data from control neurons indicate that from P8 to P26 resting potential becomes more negative, input resistance and time constant decrease, spike amplitude increases, spike width decreases, and spike threshold shows no change (Table2). These data document the normal development of intrinsic membrane properties, against which the effects of CNE can be compared.
Table 2.
Intrinsic membrane properties of layers II–IV pyramidal neurons in rat auditory cortex during normal development and after CNE
| Age/condition | RMP (mV) | Ri (MΩ) | τ (msec) | Spike threshold (mV) | Spike height (mV) | Spike width (msec) | n |
|---|---|---|---|---|---|---|---|
| Development | |||||||
| P8-12 | −64.3 ± 0.7 | 282.2 ± 10.6 | 22.9 ± 0.7 | −38.7 ± 0.5 | 65.6 ± 1.0 | 1.2 ± .03 | 53 |
| P13-16 | −68.4 ± 0.6 | 195.6 ± 7.7 | 18.1 ± 0.4 | −41.1 ± 0.4 | 72.8 ± 0.8 | 1.1 ± 0.3 | 70 |
| P20-22 | −72.9 ± 1.6 | 72.5 ± 7.2 | 11.9 ± 0.6 | −40.9 ± 0.6 | 74.3 ± 1.1 | 0.74 ± .03 | 15 |
| P24-26 | −76.8 ± 1.5 | 49.8 ± 6.1 | 8.2 ± 0.5 | −38.9 ± 1.8 | 76.4 ± 2.6 | 0.68 ± .03 | 7 |
| Control | −64.8 ± 1.1 | 305.2 ± 19.6 | 21.6 ± 1.0 | −38.2 ± 0.8 | 63.8 ± 1.5 | 1.1 ± .04 | 16 |
| CNE Group I | −60.8 ± 1.2 | 300.0 ± 24.2 | 23.3 ± 2.3 | −33.1 ± 2.42-a | 50.8 ± 3.42-a | 1.5 ± .122-a | 5 |
| Control | −67.5 ± 0.6 | 220.5 ± 7.5 | 19.6 ± 0.5 | −40.6 ± 0.4 | 71.3 ± 0.9 | 1.1 ± .02 | 71 |
| CNE Group II | −64.3 ± 1.3 | 200.4 ± 11.0 | 20.2 ± 0.9 | −40.0 ± 0.5 | 67.3 ± 1.2 | 0.95 ± 0.022-a | 22 |
| Control | −75.1 ± 1.9 | 59.0 ± 10.4 | 8.3 ± 0.3 | −39.7 ± 1.3 | 75.4 ± 2.2 | 0.66 ± 0.03 | 10 |
| CNE Group III | −75.7 ± 2.1 | 60.9 ± 9.0 | 8.6 ± 0.8 | −42.0 ± 1.7 | 75.8 ± 2.1 | 0.75 ± 0.06 | 7 |
| Control | −75.1 ± 1.9 | 59.0 ± 10.4 | 8.3 ± 0.3 | −39.7 ± 1.3 | 75.4 ± 2.2 | 0.66 ± 0.03 | 10 |
| CNE Group IV | −76.7 ± 1.2 | 53.3 ± 4.3 | 8.3 ± 0.3 | −43.0 ± 1.4 | 79.3 ± 2.4 | 0.72 ± 0.03 | 9 |
Significantly different from control; ANOVA, p < 0.05.
CNE during the first postnatal week altered some intrinsic properties, whereas nicotine exposure during the second and fourth weeks had little effect. CNE group I neurons had lower spike thresholds, smaller spike amplitudes, and longer spike widths than age-matched controls, whereas resting potential, input resistance, and time constant were not affected (Table 2). Intrinsic properties for CNE group II neurons were not affected by nicotine exposure except for a small reduction in spike width, and CNE groups III and IV showed no effects on any measured parameter. Thus, of the membrane properties measured, CNE during the first postnatal week altered those related to spike generation but not passive membrane characteristics. CNE in subsequent weeks exerted little effect on developing membrane properties.
Effects of CNE on α7 nAChRs
To determine whether nicotine-induced changes in synaptic development resulted from changes in α7 nAChRs, we determined levels of α7 mRNA and [125I] α-BTX binding (which reflects levels of α7 nAChR protein) (Seguela et al., 1993;Drisdel and Green, 2000) in nicotine- and saline-injected littermates. As with CNE groups II and III, nicotine or saline injections were performed from P8 to P12, and brains were removed on either P13 or P23. Table 3 shows that nicotine exposure produced a trend toward increased α7 mRNA levels and α-BTX binding at P13 and P23; however, only the increase in α-BTX binding at P23 reached statistical significance (repeated measures ANOVA,p < 0.001). Thus, the CNE-induced alteration of glutamate EPSPs during postnatal week 2 took place without significant changes in α7 nAChR levels.
Table 3.
Density of α7 mRNA and [125I]α-BTX binding sites in auditory cortex of rats treated from P8-12 with saline or nicotine (CNE)
| α7 mRNA (cpm/mg tissue) | [125I]α-BTX (fmol/mg tissue) | |||
|---|---|---|---|---|
| Saline | CNE | Saline | CNE | |
| P13 | ||||
| Layer II/III | 1292 ± 94 | 1681 ± 116 | 3.2 ± 0.1 | 3.3 ± 0.3 |
| Layer IV | 1529 ± 213 | 1950 ± 149 | 4.1 ± 0.1 | 4.4 ± 0.2 |
| Layer V/VI | 1611 ± 188 | 1915 ± 230 | 5.0 ± 0.1 | 5.3 ± 0.2 |
| P23 | ||||
| Layer II/III | 1134 ± 120 | 1212 ± 28 | 2.1 ± 0.1 | 2.4 ± 0.33-a |
| Layer IV | 1267 ± 130 | 1336 ± 59 | 2.5 ± 0.1 | 2.9 ± 0.23-a |
| Layer V/VI | 1291 ± 117 | 1361 ± 53 | 3.8 ± 0.1 | 4.3 ± 0.23-a |
Data are mean ± SEM for four animals in each group. Saline- versus nicotine-treated groups compared with repeated measures ANOVA.
p < 0.001.
Despite the apparent lack of change in total α7 nAChRs in week 2 (P13 data), it is possible that CNE altered their functional state. We therefore determined the agonist binding affinity of α7 nAChRs in nicotine- and saline-injected pups. Nicotine inhibition of α-BTX binding was no different in the two groups (Table4), either 1 d after P8–12 injections (at P13) or 10 d later (P23), although agonist binding affinity increased over this time (compare P13 and P23 data in Table4). Thus CNE did not alter the agonist binding affinity of α7 nAChR sites, at least not beyond the CNE period itself.
Table 4.
Nicotine inhibition of α-BTX binding in auditory cortex of rats treated from P8-12 with saline or nicotine (CNE)
| 4 μm nicotine | 40 μm nicotine | |||
|---|---|---|---|---|
| Saline | CNE | Saline | CNE | |
| P13 | 8.0 ± 2.0 | 11.5 ± 1.8 | 56.9 ± 8.6 | 53.9 ± 3.7 |
| P23 | 25.9 ± 2.9 | 25.9 ± 1.5 | 66.6 ± 2.6 | 71.3 ± 1.7 |
Data are percentage inhibition (mean ± SEM) for four animals in each group. Data across layers did not differ significantly and are averaged. No significant differences between CNE and saline groups.
DISCUSSION
To investigate whether nAChR function plays a role in auditory cortex development, we exposed rats chronically to nicotine during postnatal weeks 1, 2, or 4, and subsequently examined physiological and anatomical development. CNE during week 1 or 4 had little measurable effect (CNE groups I or IV). However, exposure during week 2 (CNE group II) produced long-duration, multi-peaked EPSPs, without changing the levels of α7 nAChRs. The physiological effects were seen even 10 d after the nicotine treatment (CNE group III), at which time an ∼15% increase in α7 nAChR levels was observed. Pharmacological manipulations indicated that CNE selectively enhanced the NMDAR component of EPSPs. CNE-enhanced EPSPs were only weakly increased further by acute nicotine application, in contrast to the robust enhancement observed in control neurons. These data identify postnatal week 2 as a period during which CNE can alter synaptic development in rat auditory cortex.
Previously, we have shown in normal animals that rapid activation of α7 nAChRs during postnatal week 2 selectively enhances NMDAR EPSPs (Aramakis and Metherate, 1998). The results implied that presynaptic α7 nAChRs regulated glutamate release at synapses that had only NMDARs postsynaptically, i.e., pure-NMDAR (“silent”) synapses. Our present results showing that CNE selectively affects NMDAR EPSPs is consistent with this hypothesis (see below). Although CNE will affect any nAChR, our previous result serves to focus our interpretation and the subsequent discussion on α7 nAChRs.
nAChR function during and after CNE
Nicotinic AChRs are ionotropic receptors that gate cation currents, and nAChRs containing α7 subunits are noteworthy for their high Ca2+ permeability and rapid desensitization (Seguela et al., 1993; Zhang et al., 1994; Castro and Albuquerque, 1995; for review, see Role and Berg, 1996). Presynaptic and postsynaptic α7 nAChRs are found throughout the brain where they can enhance neurotransmitter release and mediate fast synaptic transmission, respectively (for review, see Role and Berg, 1996;Wonnacott, 1997; Jones et al., 1999). During development, α7 nAChRs function in neurite retraction and synapse formation (Pugh and Berg, 1994; Zheng et al., 1994) and regulate glutamate release in sensory cortex (Gil et al., 1997; Aramakis and Metherate, 1998). Other nAChRs may mediate synaptic transmission in developing sensory cortex (Roerig et al., 1997). Thus CNE can impact cortical development directly via nAChRs.
The CNE protocols used in the present study mimic nicotine blood levels during smoking, which may reach ∼0.6 μm and remain above baseline levels for hours (Isaac and Rand, 1972; Henningfield et al., 1993; for review, see Benowitz, 1996; Dani and Heinemann, 1996). Exposure to 0.3–0.5 μm nicotine for several minutes will prevent regulation of glutamate EPSPs by α7 nAChRs in auditory cortex (Aramakis and Metherate, 1998), presumably by way of receptor desensitization. Thus, our original intent for the present study was to maintain nAChRs in a desensitized state using the CNE protocol. However, it is not clear what level of desensitization actually occurs and whether it is maintained for the full period between nicotine injections. Low doses of nicotine may not significantly desensitize α7 nAChRs (Fenster et al., 1997), and although chronic exposure can induce prolonged (hours or days) desensitization of nAChRs expressed in oocytes and cell lines (Lukas, 1991; Peng et al., 1994), α7 nAChRs in cultured rat cortical cells can recover within minutes (Kawai and Berg, 1999). Further studies will be needed to characterize nAChR function during the CNE protocol.
Understanding changes in nAChR function after CNE is complicated by developmental regulation of nAChRs. In mouse, CNE from P10 to P16 decreases the total number of nicotine binding sites in P17 mice; this effect is associated with decreased low-affinity and increased high-affinity nicotine binding sites (Nordberg et al., 1991). In rat, CNE from P8 to P16 increases [3H]nicotine binding in adult cortex, with a shift in agonist binding from a low- to high-affinity state (Miao et al., 1998). However, mRNA levels for several nAChR subunits, including α7, remain unchanged (Miao et al., 1998). These studies conclude that CNE in young rodents can produce long-lasting changes in the numbers and agonist binding affinity of nAChRs, and that postnatal week 2 is a critical period for CNE effects.
In the present study on rat auditory cortex, CNE from P8 to P12 did not significantly alter α7 nAChR levels ([125I] α-BTX binding sites) at P13, but it increased levels ∼15% after 10 d. CNE also did not affect agonist binding affinity of α7 nAChRs, although affinity increased during development. Although we focused on α7 nAChRs in rat auditory cortex because of our earlier study (Aramakis and Metherate, 1998), effects of CNE on other nAChR subtypes and in other cortical areas may differ (previous paragraph), given the differential distribution of nAChRs among cortical areas and layers (Fuchs, 1989;Naeff et al., 1992; Broide et al., 1996). However, these data indicate that the striking changes in glutamate-mediated EPSPs produced 1 d after CNE are not associated with changes in either the numbers or agonist binding affinity of α7 nAChRs. Altered EPSPs more likely result from altered nAChR stimulation during the CNE protocol than from changes in receptor function per se.
Postnatal week 2 is a “critical period” in rat auditory cortex development
Several postnatal events are particularly relevant to the present discussion. First, the dramatic upregulation of AChE and α7 nAChRs in the middle layers of rat auditory cortex begins early in week 1, peaks during week 2, and then declines to low (adult) levels by the end of week 3 (Fuchs, 1989; Robertson et al., 1991; Broide et al., 1995). Second, the composition of NMDARs and their contribution to glutamate EPSPs change markedly over the first postnatal weeks, in part driven by sensory experience (Agmon and O'Dowd, 1992; Burgard and Hablitz, 1993;Monyer et al., 1994; Quinlan et al., 1999; for review, see Fox et al., 1999). Some early glutamate synapses are pure NMDAR synapses that are converted, possibly in an experience-dependent process, to mixed AMPA/NMDAR synapses (Liao et al., 1995; Wu et al., 1996; Isaac et al., 1997). A large body of evidence indicates that cholinergic and glutamatergic functions regulate cortical development and may contribute to postnatal critical periods (Bear and Singer, 1986;Hohmann et al., 1991; Robertson et al., 1998; for review, see Hohmann and Berger-Sweeney, 1998; Fox et al., 1999; Yuste and Sur, 1999). For the auditory system, early week 2 is further distinguished by the onset of hearing and the subsequent, rapid maturation of the cortical auditory evoked potential (Jewett and Romano, 1972; Iwasa and Potsic, 1982). Thus, α7 nAChR regulation of NMDAR synapses occurs during a period of intense synaptogenesis and neural circuit formation.
The present study indicates that the second postnatal week may be critical for auditory cortex development, in that CNE produces a dramatic and long-lasting alteration of glutamatergic synaptic transmission. The degree to which this alteration affects acoustic information processing, and hence the designation of week 2 as a critical or sensitive period, remains to be determined. Several mechanisms may underlie the CNE effects. Desensitization of α7 nAChRs would prevent regulation of NMDAR EPSPs (Aramakis and Metherate, 1998), which in turn could prevent or delay the maturation of NMDAR synapses. A delay would produce EPSPs with larger NMDAR components than in age-matched controls, as seen in the present study, because NMDAR components normally decrease with age (Agmon and O'Dowd, 1992; Burgard and Hablitz, 1993). Our results suggest a delay in development rather than a permanent halt, because the magnitudes of NMDAR EPSPs resume their decline after cessation of CNE.
An additional finding that is not consistent with a simple delay of maturation, however, is the CNE-induced appearance of stimulus-evoked miniature events. These events, which are not prominent in normal development, imply a presynaptic effect of CNE to prolong glutamate release (note also the lack of CNE effect on postsynaptic membrane properties). Nicotine-induced Ca2+ fluxes in presynaptic terminals (McGehee et al., 1995; Gray et al., 1996) may enhance the slow (asynchronous) component of evoked glutamate release (Goda and Stevens, 1994), leading to longer EPSPs and discrete miniature events. Evidence consistent with this suggestion has been obtained in hippocampal neurons, where repeated stimulation of presynaptic α7 nAChRs can produce a long-lasting increase in the frequency of miniature postsynaptic events (Radcliffe and Dani, 1998). Obviously, this scenario is inconsistent with the notion of maintained nAChR desensitization during CNE. An alternative explanation is that nAChRs recover between nicotine injections, such that repeated injections lead to repeated, potent stimulation of α7 nAChRs and long-lasting enhancement of glutamate release. This could also explain the finding that, after CNE, the effect of acute nicotine (i.e., rapid application directly to neurons) appeared to be reduced. That is, glutamate release already enhanced by CNE might not be enhanced further by acute nicotine application. Future studies must distinguish among these alternatives.
Implications for human auditory development
If α7 nAChR-mediated regulation of NMDARs contributes to the maturation of cortical circuits, then CNE could disrupt subsequent auditory function. Supportive evidence exists in studies of human development where the analogous developmental period, including transient cholinergic upregulation in auditory cortex, occurs during the third trimester (Krmpotic-Nemanic et al., 1980, 1983). Children exposed prenatally to nicotine via secondhand smoke show decreased auditory responsiveness (habituation or orientation) and auditory-related cognitive deficits (see introductory remarks). Future studies should determine the developmental role of nAChRs in shaping auditory function and the effects of perinatal CNE on auditory dysfunction.
Footnotes
This work was supported by the National Institute on Deafness and Other Communication Disorders (DC02967), the University of California Tobacco-Related Disease Research Program (8RT-0059), and the University of California Irvine Council on Research, Computing, and Library Resources. We thank Dr. Heather Oliff for participating in initial experiments, and Yiling Chen and Michelle Cheng for technical assistance.
Correspondence should be addressed to Dr. Raju Metherate, Department of Neurobiology and Behavior, University of California, Irvine, 2205 Biological Sciences II, Irvine, CA 92697-4550. E-mail:rmethera@uci.edu.
REFERENCES
- 1.Abdulla FA, Bradbury E, Calaminici MR, Lippiello PM, Wonnacott S, Gray JA, Sinden JD. Relationship between up-regulation of nicotine binding sites in rat brain and delayed cognitive enhancement observed after chronic or acute nicotinic receptor stimulation. Psychopharmacology (Berl) 1996;124:323–331. doi: 10.1007/BF02247437. [DOI] [PubMed] [Google Scholar]
- 2.Agmon A, O'Dowd DK. NMDA receptor-mediated currents are prominent in the thalamocortical synaptic response before maturation of inhibition. J Neurophysiol. 1992;68:345–349. doi: 10.1152/jn.1992.68.1.345. [DOI] [PubMed] [Google Scholar]
- 3.Aramakis VB, Metherate R. Nicotine selectively enhances NMDA receptor-mediated synaptic transmission during postnatal development in sensory neocortex. J Neurosci. 1998;18:8485–8495. doi: 10.1523/JNEUROSCI.18-20-08485.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- 5.Benowitz NL. Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- 6.Broide RS, O'Connor LT, Smith MA, Smith JAM, Leslie FM. Developmental expression of α7 neuronal nicotinic receptor messenger RNA in rat sensory cortex and thalamus. Neuroscience. 1995;67:83–94. doi: 10.1016/0306-4522(94)00623-d. [DOI] [PubMed] [Google Scholar]
- 7.Broide RS, Robertson RT, Leslie FM. Regulation of alpha-7 nicotinic acetylcholine receptors in the developing rat somatosensory cortex by thalamocortical afferents. J Neurosci. 1996;16:2956–2971. doi: 10.1523/JNEUROSCI.16-09-02956.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgard EC, Hablitz JJ. Developmental changes in NMDA and non-NMDA receptor-mediated synaptic potentials in rat neocortex. J Neurophysiol. 1993;69:230–240. doi: 10.1152/jn.1993.69.1.230. [DOI] [PubMed] [Google Scholar]
- 9.Castro NG, Albuquerque EX. The α-bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J. 1995;68:516–524. doi: 10.1016/S0006-3495(95)80213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox KH, DeLeon DV, Angerer LM, Angerer RC. Detection of mrnas in sea urchin embryos by in situ hybridization using asymmetric RNA probes. Dev Biol. 1984;101:485–502. doi: 10.1016/0012-1606(84)90162-3. [DOI] [PubMed] [Google Scholar]
- 11.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 12.Drisdel RC, Green WN. Neuronal α-bungarotoxin receptors are α7 subunit homomers. J Neurosci. 2000;20:133–139. doi: 10.1523/JNEUROSCI.20-01-00133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox K, Henley J, Isaac J. Experience-dependent development of NMDA receptor transmission. Nat Neurosci. 1999;2:297–299. doi: 10.1038/7203. [DOI] [PubMed] [Google Scholar]
- 15.Fried PA, Watkinson B, Siegel LS. Reading and language in 9- to 12-year olds prenatally exposed to cigarettes and marijuana. Neurotoxicol Teratol. 1997;19:171–183. doi: 10.1016/s0892-0362(97)00015-9. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs JL. [125I]α-Bungarotoxin binding marks primary sensory areas of developing rat neocortex. Brain Res. 1989;501:223–234. doi: 10.1016/0006-8993(89)90640-9. [DOI] [PubMed] [Google Scholar]
- 17.Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 18.Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci USA. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 20.Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- 21.Hohmann CF, Berger-Sweeney J. Cholinergic regulation of cortical development and plasticity. New twists to an old story. Perspect Dev Neurobiol. 1998;5:401–425. [PubMed] [Google Scholar]
- 22.Hohmann CF, Kwiterovich KK, Oster-Granite ML, Coyle JT. Newborn basal forebrain lesions disrupt cortical cytodifferentiation as visualized by rapid golgi staining. Cereb Cortex. 1991;1:143–157. doi: 10.1093/cercor/1.2.143. [DOI] [PubMed] [Google Scholar]
- 23.Isaac JTR, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 24.Isaac PF, Rand MJ. Cigarette smoking and plasma levels of nicotine. Nature. 1972;236:308–310. doi: 10.1038/236308a0. [DOI] [PubMed] [Google Scholar]
- 25.Iwasa H, Potsic WP. Maturational change of early, middle, and late components of the auditory evoked responses in rats. Otolaryngol Head Neck Surg. 1982;90:95–102. doi: 10.1177/019459988209000117. [DOI] [PubMed] [Google Scholar]
- 26.Jewett DL, Romano MN. Neonatal development of auditory system potentials averaged from the scalp of rat and cat. Brain Res. 1972;36:101–115. doi: 10.1016/0006-8993(72)90769-x. [DOI] [PubMed] [Google Scholar]
- 27.Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- 28.Kawai H, Berg DK. Long-term nicotinic effects on acetylcholine receptors containing alpha-7 subunits. Abstr Tobacco-Related Disease Research Program Annual Meeting C06. 1999.
- 29.Kristt DA. Development of neocortical circuitry: histochemical localization of acetylcholinesterase in relation to the cell layers of rat somatosensory cortex. J Comp Neurol. 1979;186:1–15. doi: 10.1002/cne.901860102. [DOI] [PubMed] [Google Scholar]
- 30.Krmpotic-Nemanic J, Kostovic I, Kelovic Z, Nemanic D. Development of acetylcholinesterase (AChE) staining in human fetal auditory cortex. Acta Otolaryngol. 1980;89:388–392. doi: 10.3109/00016488009127153. [DOI] [PubMed] [Google Scholar]
- 31.Krmpotic-Nemanic J, Kostovic I, Kelovic Z, Nemanic D, Mrzljak L. Development of the human fetal auditory cortex: growth of afferent fibers. Acta Anat. 1983;116:69–73. [PubMed] [Google Scholar]
- 32.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 33.Lukas RJ. Effects of chronic nicotinic ligand exposure on functional activity of nicotinic acetylcholine receptors expressed by cells of the PC12 rat pheochromocytoma or the TE671/RD human clonal line. J Neurochem. 1991;56:1134–1145. doi: 10.1111/j.1471-4159.1991.tb11403.x. [DOI] [PubMed] [Google Scholar]
- 34.McCartney JS, Fried PA, Watkinson B. Central auditory processing in school-age children prenatally exposed to cigarette smoke. Neurotoxicol Teratol. 1994;16:269–276. doi: 10.1016/0892-0362(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 35.McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 36.Metherate R, Aramakis VB. Intrinsic electrophysiology of neurons in thalamorecipient layers of developing rat auditory cortex. Dev Brain Res. 1999;115:131–144. doi: 10.1016/s0165-3806(99)00058-9. [DOI] [PubMed] [Google Scholar]
- 37.Miao H, Liu C, Bishop K, Gong ZH, Nordberg A, Zhang X. Nicotine exposure during a critical period of development leads to persistent changes in nicotinic acetylcholine receptors of adult rat brain. J Neurochem. 1998;70:752–762. doi: 10.1046/j.1471-4159.1998.70020752.x. [DOI] [PubMed] [Google Scholar]
- 38.Miller JA, Zahniser NR. The use of 14C-labeled tissue paste standards for the calibration of 125I-labeled ligands in quantitative autoradiography. Neurosci Lett. 1987;81:345–350. doi: 10.1016/0304-3940(87)90408-3. [DOI] [PubMed] [Google Scholar]
- 39.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 40.Murrin LC, Ferrer JR, Zeng WY, Haley NJ. Nicotine administration to rats: methodological considerations. Life Sci. 1987;40:1699–1708. doi: 10.1016/0024-3205(87)90020-8. [DOI] [PubMed] [Google Scholar]
- 41.Naeff B, Schlumpf M, Lichtensteiger W. Pre- and postnatal development of high-affinity [3H]nicotine binding sites in rat brain regions: an autoradiographic study. Dev Brain Res. 1992;68:163–174. doi: 10.1016/0165-3806(92)90058-5. [DOI] [PubMed] [Google Scholar]
- 42.Nash JE. Embryopathic risks of cigarette smoking. Exp Pathol. 1988;33:65–73. doi: 10.1016/s0232-1513(88)80127-0. [DOI] [PubMed] [Google Scholar]
- 43.Nordberg A, Zhang XA, Fredriksson A, Eriksson P. Neonatal nicotine exposure induces permanent changes in brain nicotinic receptors and behaviour in adult mice. Dev Brain Res. 1991;63:201–207. doi: 10.1016/0165-3806(91)90079-x. [DOI] [PubMed] [Google Scholar]
- 44.Ospina JA, Broide RS, Acevedo D, Robertson RT, Leslie FM. Calcium regulation of agonist binding to α7-type nicotinic acetylcholine receptors in adult and fetal rat hippocampus. J Neurochem. 1998;70:1061–1068. doi: 10.1046/j.1471-4159.1998.70031061.x. [DOI] [PubMed] [Google Scholar]
- 45.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 46.Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- 47.Picone TA, Allen LH, Olsen PN, Ferris ME. Pregnancy outcome in North American women. II. Effects of diet, cigarette smoking, stress, and weight gain on placentas, and on neonatal physical and behavioral characteristics. Am J Clin Nutr. 1982;36:1214–1224. doi: 10.1093/ajcn/36.6.1214. [DOI] [PubMed] [Google Scholar]
- 48.Prusky GT, Arbuckle JM, Cynader MS. Transient concordant distributions of nicotinic receptors and acetylcholinesterase activity in infant rat visual cortex. Brain Res. 1988;467:154–159. doi: 10.1016/0165-3806(88)90078-8. [DOI] [PubMed] [Google Scholar]
- 49.Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind α-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci. 1994;14:889–896. doi: 10.1523/JNEUROSCI.14-02-00889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- 51.Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci. 1998;18:7075–7083. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robertson RT, Mostamand F, Kageyama GH, Gallardo KA, Yu J. Primary auditory cortex in the rat: transient expression of acetylcholinesterase in developing geniculocortical projections. Dev Brain Res. 1991;58:81–95. doi: 10.1016/0165-3806(91)90240-j. [DOI] [PubMed] [Google Scholar]
- 53.Robertson RT, Gallardo KA, Claytor KJ, Ha DH, Ku K-H, Yu BP, Lauterborn JC, Wiley RG, Yu J, Gall CM, Leslie FM. Neonatal treatment with 192 IgG-saporin produces long-term forebrain cholinergic deficits and reduces dendritic branching and spine density of neocortical pyramidal neurons. Cereb Cortex. 1998;8:142–155. doi: 10.1093/cercor/8.2.142. [DOI] [PubMed] [Google Scholar]
- 54.Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17:8353–8362. doi: 10.1523/JNEUROSCI.17-21-08353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roger M, Arnault P. Anatomical study of the connections of the primary auditory area in the rat. J Comp Neurol. 1989;287:339–356. doi: 10.1002/cne.902870306. [DOI] [PubMed] [Google Scholar]
- 56.Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 57.Rowell PP, Li M. Dose–response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J Neurochem. 1997;68:1982–1989. doi: 10.1046/j.1471-4159.1997.68051982.x. [DOI] [PubMed] [Google Scholar]
- 58.Sally SL, Kelly JB. Organization of auditory cortex in the albino rat: sound frequency. J Neurophysiol. 1988;59:1627–1638. doi: 10.1152/jn.1988.59.5.1627. [DOI] [PubMed] [Google Scholar]
- 59.Saxton D. The behavior of infants whose mothers smoke in pregnancy. Early Hum Dev. 1978;2:363–369. doi: 10.1016/0378-3782(78)90063-4. [DOI] [PubMed] [Google Scholar]
- 60.Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sexton M, Fox NL, Hebel JR. Prenatal exposure to tobacco: II. Effects on cognitive functioning at age three. Int J Epidemiol. 1990;19:72–77. doi: 10.1093/ije/19.1.72. [DOI] [PubMed] [Google Scholar]
- 62.Stuart GJ, Dodt H-U, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 63.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 64.Wu G-Y, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 65.Yuste R, Sur M. Development and plasticity of the cerebral cortex: from molecules to maps. J Neurobiol. 1999;41:1–6. doi: 10.1002/(sici)1097-4695(199910)41:1<1::aid-neu1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Z, Vijayaraghavan S, Berg DK. Neuronal acetylcholine receptors that bind alpha-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 67.Zheng JQ, Felder M, Connor JA, Poo M-M. Turning of nerve growth cones induced by neurotransmitters. Nature. 1994;368:140–144. doi: 10.1038/368140a0. [DOI] [PubMed] [Google Scholar]