

Abstract
Inflammation in the brain has been increasingly associated with the development of a number of neurological diseases. The hallmark of neuroinflammation is the activation of microglia, the resident brain immune cells. Injection of bacterial endotoxin lipopolysaccharide (LPS) into the hippocampus, cortex, or substantia nigra of adult rats produced neurodegeneration only in the substantia nigra. Although LPS appeared to impact upon mesencephalic neurons in general, an extensive loss of dopaminergic neurons was observed. Analysis of the abundance of microglia revealed that the substantia nigra had the highest density of microglia. When mixed neuron–glia cultures derived from the rat hippocampus, cortex, or mesencephalon were treated with LPS, mesencephalic cultures became sensitive to LPS at a concentration as low as 10 ng/ml and responded in a dose-dependent manner with the production of inflammatory factors and a loss of dopaminergic and other neurons. In contrast, hippocampal or cortical cultures remained insensitive to LPS treatment at concentrations as high as 10 μg/ml. Consistent with in vivo observations, mesencephalic cultures had fourfold to eightfold more microglia than cultures from other regions. The positive correlation between abundance of microglia and sensitivity to LPS-induced neurotoxicity was further supported by the observation that supplementation with enriched microglia derived from mesencephalon or cortex rendered LPS-insensitive cortical neuron–glia cultures sensitive to LPS-induced neurotoxicity. These data indicate that the region-specific differential susceptibility of neurons to LPS is attributable to differences in the number of microglia present within the system and may reflect levels of inflammation-related factors produced by these cells.
Keywords: endotoxin, lipopolysaccharide, inflammation, glia-mediated neurotoxicity, midbrain, microglia, Parkinson's disease
Under normal conditions, brain microglia are involved in immune surveillance and host defense against infectious agents. However, microglia readily become activated in response to injury or immunological challenges, as indicated by a change in morphology from a ramified resting state to an amoeboid appearance with an increase in the expression of major histocompatibility complex molecules and complement type 3 receptor (Streit et al., 1988; Kaur and Ling, 1992; Graeber et al., 1994;Kreutzberg, 1996). Microglia activation is a histopathological hallmark of several neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, multiple sclerosis, and the AIDS dementia complex (McGeer et al., 1988; Rogers et al., 1988; Matsumoto et al., 1992;Dickson et al., 1993; Raine, 1994).
Activation of microglia is believed to contribute to neurodegenerative processes through the release of proinflammatory and/or cytotoxic factors, including interleukin-1 (IL-1), tumor necrosis factor-α (TNFα), nitric oxide (NO), reactive oxygen intermediates, arachidonic acid metabolites, and quinolinic acid (Chao et al., 1992; Dickson et al., 1993; Lee et al., 1993; Brosnan et al., 1994; Matsuo et al., 1995;Minghetti and Levi, 1995; Espey et al., 1997). Production of these factors by microglia after exposure to lipopolysaccharide (LPS), the human immunodeficiency virus-1 coat protein gp120, or β-amyloid has been well documented (Boje and Arora, 1992; Chao et al., 1992; Dawson et al., 1994; Ii et al., 1996; Kong et al., 1996). Furthermore, activation of microglia and subsequent production of proinflammatory and cytotoxic factors have been attributed to increased neurotoxicity in in vitro neuron–glia cultures treated with LPS, β-amyloid, or a combination of IL-1, TNFα, and interferon-γ (Chao et al., 1992; Dawson et al., 1994; Jeohn et al., 1998), suggesting that microglia-derived factors such as NO and TNFα are important mediators of inflammation-mediated neurodegeneration.
Microglia are present in large numbers within the brain, but they are not distributed with a uniform density or morphology in all major divisions of the brain (Lawson et al., 1990). This heterogeneity may imply an unequal sensitivity to microglia-mediated neurotoxicity in different regions of the brain. In this study, we investigated whether stimulation with the bacterial endotoxin LPS could differentially effect neuronal cell death in the rat hippocampus, cortex, or mesencephalon in vivo and in mixed neuron–glia cultures derived from these regions. In addition, we examined whether the abundance, morphology, or responsiveness of microglial cells contributes to region-specific differences in LPS-induced neurotoxicity. Our data indicate that mesencephalic neurons are significantly more susceptible than hippocampal or cortical neurons to inflammation-mediated neurodegeneration, and this differential neurotoxicity may be attributable, at least in part, to differences in the abundance of microglia within a specific brain region.
MATERIALS AND METHODS
Animals. Male (225–250 gm) and timed-pregnant female rats (Fischer 344) were purchased from Charles River Laboratories (Raleigh, NC) and kept on a 12 hr light/dark cycle with ad libitum access to food and water. Male rats were acclimated to their environment for 10 d before use for experiments.
Materials. Minimal essential medium (MEM), penicillin, streptomycin, gentamicin, horse serum, and LPS (strain O111:B4) were purchased from Life Technologies (Gaithersburg, MD). [7,8-3H]dopamine (DA) (40 Ci/mmol) was obtained from Amersham Pharmacia Biotech (Arlington Heights, IL) and poly-d-lysine, biotinylated isolectin B4, and dopamine were obtained from Sigma (St. Louis, MO). Biotinylated secondary antibodies, ABC kit, and 3,3′-diaminobenzidine were from Vector Laboratories (Burlingame, CA). The polyclonal anti-tyrosine hydroxylase (TH) antiserum was a gift from Glaxo Wellcome (Research Triangle Park, NC).
LPS injection in vivo. Male rats were anesthetized with sodium pentobarbital (50 mg/kg) and positioned in a small-animal stereotaxic apparatus. Injection of LPS into specific brain regions was made using the following stereotaxic coordinates, measured from bregma (Paxinos and Watson, 1986): for the hippocampus, 3.3 mm posterior, 2.0 mm lateral, and 3.5 mm ventral; for the cortex, 1.4 mm posterior, 2.0 mm lateral, and 2.0 mm ventral; and for the substantia nigra (SN), 4.8 mm posterior, 1.7 mm lateral, and 8.2 mm ventral. LPS (5 or 10 μg in a volume of 2 μl of PBS) was injected into the right side of the hippocampus, cortex, or SN over a period of 2 min, and the injection needle was kept in place for 2 min after the injection. Control injections of PBS alone were made into the left side of the hippocampus, cortex, or SN under the conditions described above. At least six animals were used for each brain region examined.
Cell culture. Primary mixed neuron–glia cultures were prepared from the brains of embryonic day 16–17 rats. The whole brain was removed aseptically, and the blood vessels and meninges were discarded. The desired brain regions (hippocampus, cortex, or mesencephalon) were dissected, pooled, and mechanically dissociated by mild trituration in ice-cold calcium- and magnesium-free W3 buffer (in mm: 15 HEPES, pH 7.4, 145 NaCl, 5.4 KCl, 1 NaH2PO4, and 11 glucose). To each poly-d-lysine-coated well of 24-well culture plates were seeded 5 × 105cells in 0.5 ml of culture medium. The cultures were maintained at 37°C in an atmosphere of 5% CO2 and 95% air. The culture medium consisted of MEM supplemented with 10% heat-inactivated horse serum, 2 mml-glutamine, 1 mm sodium pyruvate, 100 μm nonessential amino acids, 15 mm KCl, 50 U/ml penicillin, 50 μg/ml streptomycin, and 50 μg/ml gentamicin. The cultures were replenished with fresh medium at 1 and 4 d after plating. At 7 d after plating, the cultures were treated with vehicle or LPS in a modified culture medium in which the serum concentration was reduced to 2%. When antibodies against microtubule-associated protein-2 (MAP-2) or CR3 complement receptor, a marker for rat microglia, were used to immunostain neuron–glia cultures (see below), the following composition was determined: for hippocampal cultures, 65% neurons and 5.8% microglia; for cortical cultures, 60% neurons and 2.8% microglia; and for mesencephalic cultures, 42% neurons and 20% microglia. The remaining cells were presumed to be astroglial cells.
Enriched microglia were prepared following a previously described protocol (Liu et al., 2000a). Briefly, brain tissues of cortex and mesencephalon were dissected from postnatal day 1 rat pups. The tissues were triturated, and 107 cells were seeded in a 75 cm2 culture flask. After a confluent monolayer of glial cells were obtained, microglia were shaken off and replated at 105 cells per well either alone or on top of existing neuron–glia cultures derived from the rat cortex. At 24 hr after plating the microglia-enriched population, the cells were treated with LPS or vehicle alone.
Immunocytochemistry. Cell cultures were fixed in 3.7% formaldehyde and immunostained as described previously (Liu et al., 2000a). For in vivo studies, animals were anesthetized with sodium pentobarbital and then perfused transcardially with saline, followed by 4% paraformaldehyde in PBS. Brains were removed, post-fixed overnight in 4% paraformaldehyde in PBS, and then cryoprotected in a 30% sucrose–1% paraformaldehyde solution. Coronal sections (35 μm) were cut using a microtome and immunostained as free-floating tissue sections (Liu et al., 2000b). Every sixth serial section was selected and processed for immunohistochemistry.
Neurons were identified with a monoclonal antibody to MAP-2 (Roche Molecular Biochemicals, Indianapolis, IN) and an antibody recognizing the vertebrate neuron-specific nuclear protein (NeuN) (Chemicon, Temecula, CA). Dopaminergic neurons were stained with a polyclonal anti-TH antibody (a gift from Dr. John Reinhard of Glaxo Wellcome, Research Triangle Park, NC). Microglia were visualized by staining for the CR3 complement receptor using the monoclonal antibody OX-42 (PharMingen, San Diego, CA) or with the biotinylated isolectin B4.
Briefly, cultures or brain sections were blocked for 20 min with PBS containing 0.4% Triton X-100, 1% bovine serum albumin, and 4% normal serum, followed by an overnight incubation with one of the primary antibodies (anti-MAP-2, 5 μg/ml; anti-NeuN, 1 μg/ml; anti-TH, 1:20; or OX-42, 5 μg/ml) or with the lectin (10 μg/ml). Afterward, the cells or sections were treated for 10 min with 1% H2O2, followed by incubation with appropriate biotinylated secondary antibodies and then visualized using the avidin-biotin-peroxidase complex (ABC) method with 3,3′-diaminobenzidine as the chromogen. Images of immunostained cells were recorded using a CCD camera and the Metamorph computer software (Universal Imaging Corporation, West Chester, PA).
High-affinity [3H]dopamine uptake assay. Uptake of radiolabeled dopamine was performed as described previously with modifications (Liu et al., 2000a). Cultures were washed twice with Krebs'–Ringer's buffer (119 mmNaCl, 4.7 mm KCl, 1.3 mmEDTA, 1.8 mm CaCl2, 1.2 mm MgSO4, 16 mmNaH2PO4, 16 mmNa2HPO4, and 1 mg/ml glucose), followed by incubation for 40 min at 37°C with 0.5 μCi [3H]dopamine and 1 μm unlabeled dopamine in Krebs'–Ringer's buffer. Nonspecific uptake was determined by the addition of 10 μm mazindol. Afterward, the cultures were washed three times with ice-cold buffer and then solubilized with 1 N NaOH. The lysate was mixed with scintillation fluid, and radioactivity was counted in a scintillation counter. Specific uptake was determined by subtracting the nonspecific counts from the total counts.
Nitrite and TNFα assays. The production of NO was assessed as the accumulation of nitrite in the culture supernatants using a colorimetric reaction with the Griess reagent (Green et al., 1982) as described previously (Liu et al., 2000a). The amount of TNFα in the medium was measured with a rat TNFα enzyme-linked immunosorbent assay kit (Genzyme Diagnostics, Cambridge, MA).
Statistical analysis. The data are expressed as the mean ± SEM. Statistical significance was assessed by an ANOVA, followed by Bonferroni's t test using the StatView program (Abacus Concepts, Inc., Berkeley, CA). A value ofp < 0.05 was considered statistically significant.
RESULTS
Neurotoxic effect of LPS injection in various regions of the rat brain
LPS (5 or 10 μg in a volume of 2 μl) was injected stereotaxically into the adult rat hippocampus, cortex, or SN. Seven days later, the brain was removed, and tissue sections taken through these regions were immunostained for neuronal markers to determine whether the neurons residing in distinct brain regions were differentially sensitive to LPS-induced toxicity. For all comparisons, LPS was injected into the region of interest on the right side of the brain, and as a control, the vehicle alone (PBS) was injected contralaterally. When brain sections were immunostained for the neuronal nuclear protein NeuN, no substantial differences in NeuN immunostaining were observed between LPS (10 μg) and vehicle-injected sites from either the hippocampus or cortex (Fig.1). In contrast, injection of LPS (5 μg) dramatically reduced the number of NeuN-positive neurons in the SN compared with the vehicle-injected side (Fig. 1). Moreover, serial sections of rat brain taken through the SN also demonstrated an extensive loss of dopaminergic neurons after LPS injection when compared with the vehicle-treated control side, as shown by a decrease in the number of neurons expressing TH (Fig. 1).
Fig. 1.
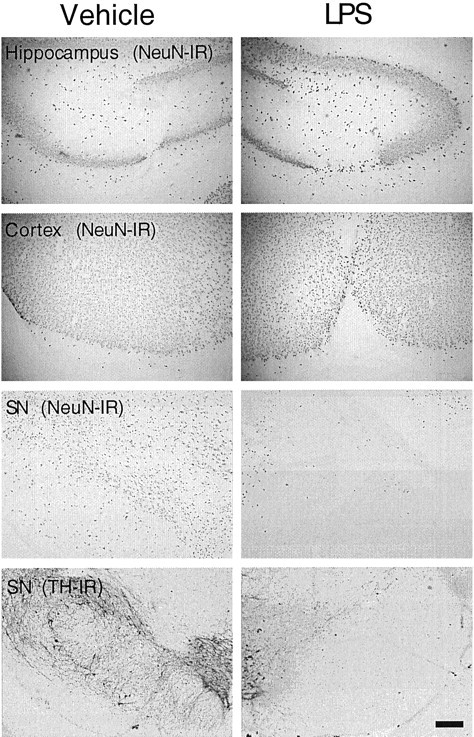
Susceptibility of neurons to LPS-induced toxicity in different rat brain regions. LPS was injected unilaterally into the adult rat hippocampus, cortex, or SN, and vehicle alone was injected contralaterally into the specific regions. The hippocampus or cortex received 10 μg of LPS, whereas the SN received 5 μg of LPS. After 7 d, the brains were removed, and tissue sections through the areas of interest were immunostained for neuronal proteins. Neurons in all regions were immunostained with an antibody against NeuN. Dopaminergic neurons in the SN were stained with an anti-TH antibody. At least six animals were used for each injection, and similar results were obtained in all animals examined. Scale bar, 250 μm.
Brain sections taken through the regions of interest were also immunostained for microglia using the monoclonal antibody OX-42. In the hippocampus, cortex, or SN of animals injected with vehicle alone, the majority of the OX-42-immunoreactive (IR) cells exhibited a resting or ramified state (Fig. 2). Only a small number of OX-42-IR cells exhibited the characteristics of activated microglia (increased cell size, staining intensity for OX-42, and shape changes), and these cells were mainly localized along the needle tract because of mechanical injury-induced activation (Fig. 2). After LPS injection, an increase in OX-42-IR was observed along the needle tracts and their termini in the hippocampus and cortex, presumably resulting from mechanical injury and high concentrations of LPS in the vicinity of the injection sites (Fig. 2). In contrast, the LPS-induced activation of microglia in the SN was not limited to the needle tracts; intensified OX-42-IR was observed in areas surrounding the injection sites (Fig. 2). Consistent with a previous report (Lawson et al., 1990), we found that the SN contains a greater number of OX-42-IR cells than the hippocampus or cortex, and these differences were even more prominent after LPS injection (Fig. 2).
Fig. 2.
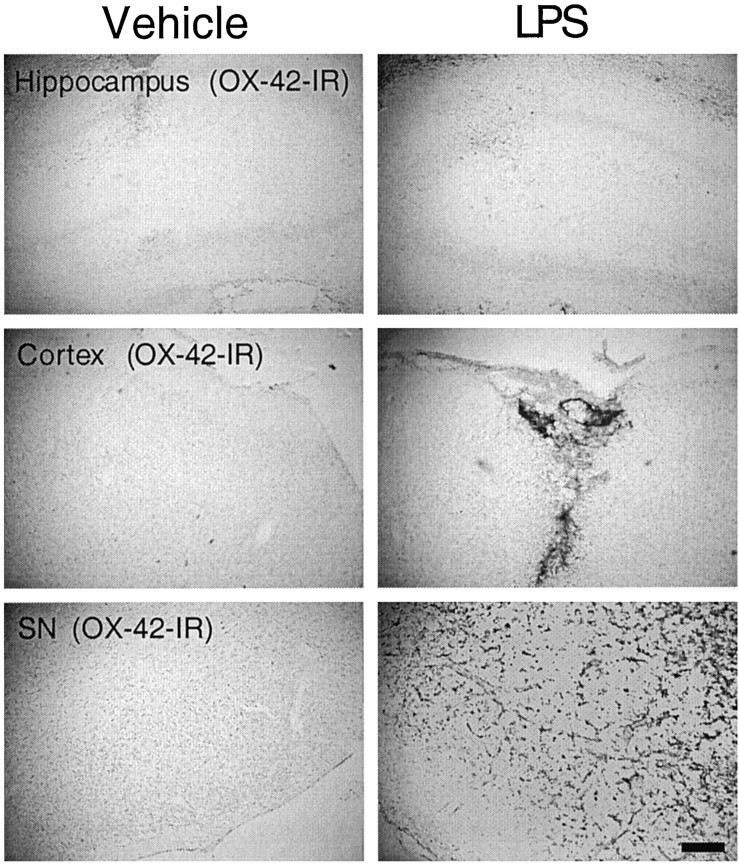
Abundance of microglia in different rat brain regions and responsiveness to LPS. LPS or vehicle was injected into the adult rat hippocampus, cortex, or SN as described in the legend to Figure 1. After 7 d, the brains were removed, and tissue sections through the areas of interest were immunostained for CR3 complement receptors using the monoclonal antibody OX-42 as a marker for microglia. At least six animals were used for each injection, and similar results were obtained in all animals examined. Scale bar, 250 μm.
Susceptibility of neurons in neuron–glia cultures from different brain regions to LPS-induced neurodegeneration
To further understand the mechanisms underlying the regional difference in the sensitivity to LPS-induced neurotoxicity observedin vivo and its relationship to activity of microglia, mixed neuron–glia cultures were established using embryonic rat brain tissues from hippocampus, cortex, or mesencephalon. Cultures were treated for 72 hr with 1 μg/ml LPS or with the vehicle alone. Neurotoxicity was assessed by morphological analysis and counting the number of neuronal cell bodies after immunostaining for MAP-2. Exposure of hippocampal or cortical neuron–glia cultures to 1 μg/ml LPS did not alter the number of MAP-2-positive cells or the morphology of these cells compared with vehicle-treated control cultures (Fig.3A). Quantification of the MAP-2-immunostained cultures did not show any significant difference between the number of MAP-2-IR cell bodies in the LPS- and vehicle-treated cultures from either the hippocampus or cortex (Fig.3B). In contrast, mesencephalic cultures treated with the same concentration of LPS (1 μg/ml) suffered dramatic degeneration of MAP-2-IR cells (Fig. 3A,B). There was a 96% decrease in the number of MAP-positive neurons in the mesencephalic cultures after stimulation with LPS (Fig. 3B). Most of the few cells remaining after treatment of mesencephalic cultures with LPS displayed shortened neurites compared with the control cultures (Fig. 3A). The loss of MAP-2-positive neurons in the mesencephalic neuron–glia cultures was dependent on the concentrations of LPS used (Fig. 3C). Significant neurotoxicity was observed with LPS concentrations as low as 3 ng/ml. At 3 or 6 ng/ml, LPS reduced the number of MAP-2-positive cells by 37 and 83%, respectively (Fig. 3C). Compared with controls, at least 95% of the MAP-2-positive neurons were destroyed with 10 ng/ml or greater (up to 1000 ng/ml) concentrations of LPS (Fig.3C). In contrast, no significant decrease in the number of MAP-2-positive neurons was observed in hippocampal or cortical neuron–glia cultures after treatment for 72 hr with LPS at concentrations ranging from 1 ng/ml to 10 μg/ml LPS (data not shown).
Fig. 3.
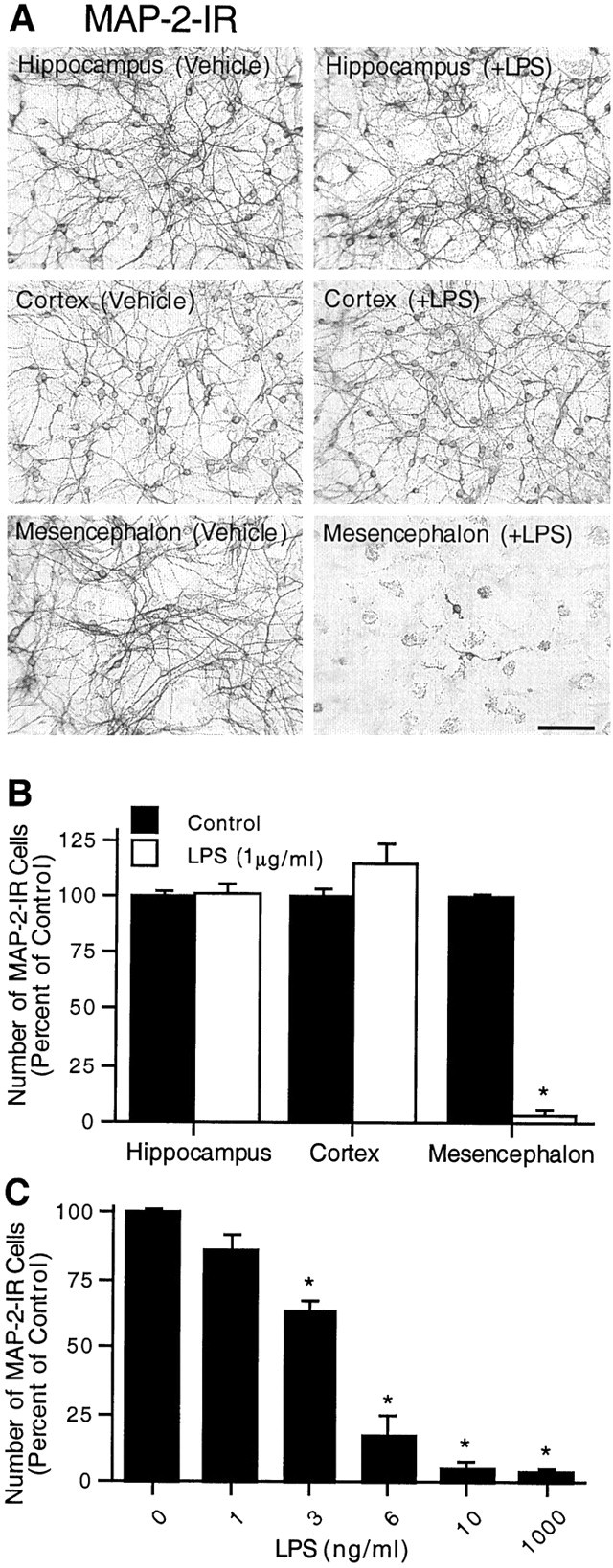
Differential neurotoxicity in mixed neuron–glia cultures from various brain regions after treatment with LPS. Mixed neuron–glia cultures were prepared from hippocampal, cortical, or mesencephalic tissues of embryonic day 16–17 rats. Cultures were treated for 72 hr with vehicle alone or 1 μg/ml LPS (A, B) or graded concentrations of LPS (C) and then immunostained with an antibody to the neuronal cytoskeletal protein MAP-2. A, Immunocytochemical analysis of MAP-2-positive neurons. Scale bar, 100 μm. B, Quantification of MAP-2-immunostained cell bodies. Significant degeneration of MAP-2-IR neurons after LPS treatment was observed in mesencephalic cultures only. For reference, the mean number of MAP-2-IR cells per well in the vehicle-treated hippocampal, cortical, and mesencephalic cultures were 6.7 × 104, 6.0 × 104, and 6.4 × 104, respectively. C, LPS-induced loss of MAP-2-positive neurons in the mesencephalic cultures was dependent on the concentrations of LPS. The data are expressed as a percent of the total number of MAP-2-IR cell bodies present in the vehicle-treated control cultures. The data represent the mean ± SEM of at least five wells taken from three independent experiments. *p < 0.0001 compared with the vehicle-treated control cultures prepared from the rat mesencephalon.
Because the loss of dopaminergic neurons in the SN is characteristic of Parkinson's disease, the effect of LPS on this population of mesencephalon-derived neurons was of particular interest. Neurotoxicity was assessed by analyzing the morphology and number of dopaminergic neurons by immunostaining for TH and also by determining the capacity of dopaminergic neurons in cultures to uptake [3H]DA. As shown in Figure4A, compared with the control cultures, treatment of mesencephalic neuron–glia cultures with 1–1000 ng/ml LPS for 72 hr resulted in a dose-dependent decrease in [3H]DA uptake. The lowest concentration of LPS required to produce a significant reduction in DA uptake was 3 ng/ml. A greater reduction in dopamine uptake capacity was observed in cultures treated with 6 ng/ml or greater concentrations of LPS (Fig.4A). Immunocytochemical staining of the cultures indicated that TH-positive neurons in the control cultures (∼0.5% of the MAP-2-positive neurons) exhibited healthy and extended neurites. After treatment for 72 hr with 1000 ng/ml LPS, a significant loss of TH-positive neurons (65% compared with control, n = 6) was detected, and the majority of the remaining dopaminergic neurons exhibited very short neurites (Fig. 4B).
Fig. 4.
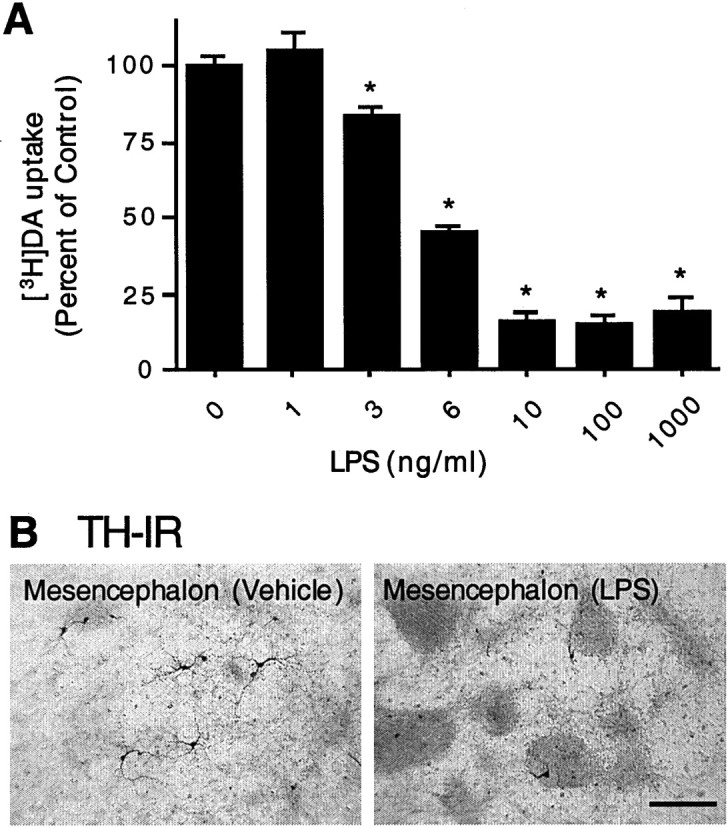
Effect of LPS on [3H]dopamine uptake and TH-IR neurons in mesencephalic neuron–glia cultures.A, Mesencephalic cultures were treated with either vehicle alone or the indicated concentrations of LPS for 72 hr, and the uptake of [3H]dopamine was assessed. The data are expressed as a percent of the dopamine uptake (minus nonspecific uptake, as determined in the presence of 10 μm mazindol) measured after treatment with the vehicle alone and represent the mean ± SEM of six wells per condition. Duplicate experiments yielded similar qualitative results. *p < 0.01 compared with the vehicle-treated control cultures. B, Immunocytochemical analysis of dopaminergic neurons. Cultures treated for 72 hr with vehicle alone or 1000 ng/ml LPS were immunostained with an anti-TH antibody. A significant loss of TH-positive neurons was induced by LPS treatment, and the remaining TH-positive neurons had significantly shorter neurites.
LPS-induced production of nitric oxide and release of TNFα in neuron–glia cultures prepared from different brain regions
Cytokines, such as TNFα, and free radicals, such as NO, are synthesized by glial cells upon stimulation with LPS and are candidates for mediating the neurotoxicity in neuron–glia cultures. To determine whether the difference in sensitivity to LPS-induced neurotoxicity could be attributable, at least in part, to differences in the generation of immune mediators, the production of NO and TNFα was examined in neuron–glia cultures after treatment with LPS. In mesencephalic neuron–glia cultures treated with vehicle alone or with 1–1000 ng/ml LPS for 24, 48, and 72 hr, a dose-dependent increase in nitrite accumulation at the three time points was observed (Fig.5A). Significant nitrite production could be seen with LPS concentrations as low as 3 ng/ml, and maximal levels of nitrite were detected at 10 ng/ml LPS or at greater concentrations (Fig. 5A). In contrast, unlike mesencephalic cultures, nitrite levels in hippocampal or cortical cultures increased only slightly in response to LPS at concentrations as high as 1000 ng/ml (Fig. 5A). For instance, the levels of nitrite in hippocampal and cortical cultures treated with 1000 ng/ml LPS for 72 hr were 5.4 and 1.7 μm, respectively (Fig.5A). In agreement with previous studies indicating that LPS-induced neurotoxicity was, at least in part, mediated via NO (Chao et al., 1992; Bronstein et al., 1995), the addition of the NO synthase inhibitorNG-nitro-l-arginine-methyl ester (1 mm) to the mesencephalic cultures inhibited the accumulation of nitrite produced after treatment for 72 hr with 1000 ng/ml LPS by 73% and reduced the loss of MAP-2-immunostained neurons by 74% (data not shown).
Fig. 5.
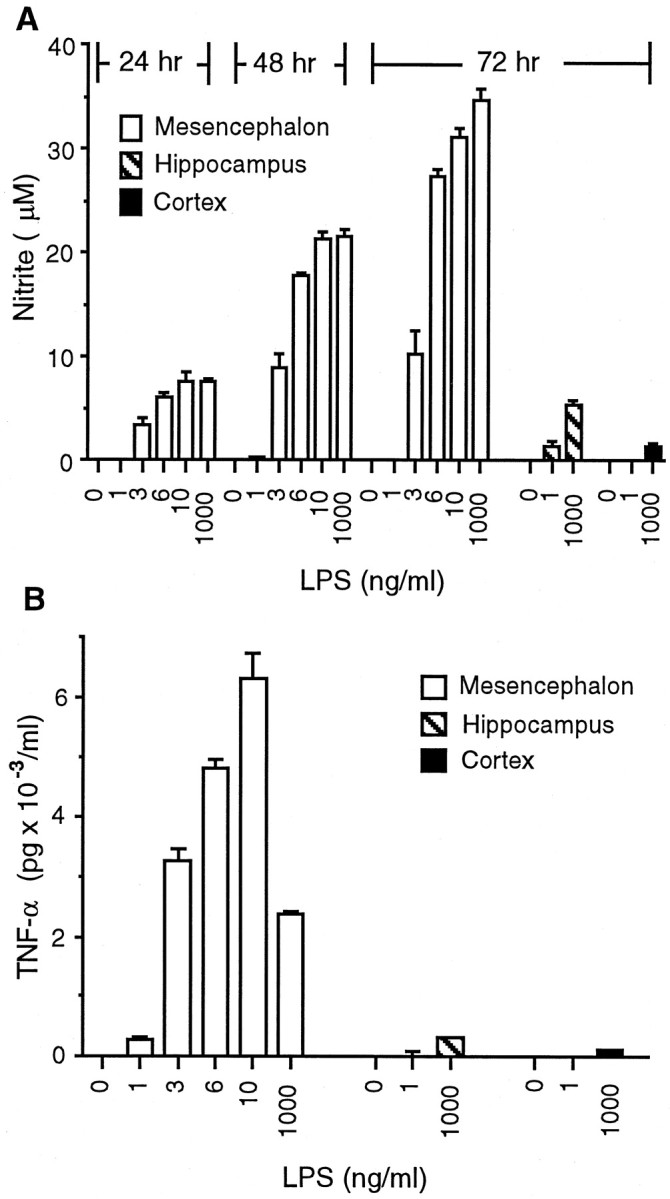
Effect of LPS on the production of NO and the release of TNFα in neuron–glia cultures derived from different brain regions. A, NO production. Mesencephalic neuron–glia cultures were treated with vehicle or the indicated concentrations of LPS, and the levels of NO production, assessed as the accumulation of nitrite, were quantified 24, 48, or 72 hr later. For comparison, the levels of NO production in hippocampal or cortical neuron–glia cultures treated with either vehicle or 1 or 1000 ng/ml LPS were measured at 72 hr. The data represent the mean ± SEM of 3–12 wells per condition. B, TNFα release. Mesencephalic neuron–glia cultures were treated with vehicle or indicated concentrations of LPS, and the levels of TNFα release were measured 6 hr later. For comparison, the levels of TNFα release in neuron–glia cultures derived from the rat hippocampus or cortex were also measured after stimulation with vehicle or 1 or 1000 ng/ml LPS. The data represent the mean ± SEM of three wells per condition. Duplicate experiments yielded similar qualitative results, although the levels of TNFα release varied between experiments.
Levels of TNFα in the culture medium were measured at 6 hr after LPS stimulation, a time at which the release of the cytokine is known to be near its maximal levels (Liu et al., 2000a). A previous report from our laboratory indicated that microglia are the major source of TNFα (Kong et al., 1996). Stimulation of mesencephalic cultures with increasing concentrations of LPS (0, 1, 3, 6, 10, or 1000 ng/ml) resulted in a dose-dependent release of TNFα into the culture medium with maximal levels of TNFα released at 10 ng/ml LPS (Fig.5B). Greater concentrations of LPS (>10 ng/ml) resulted in reduced levels of TNFα because of LPS-induced cytotoxicity of microglia (B. Liu, J. Wang and J. Hong, unpublished observations). In contrast, only low levels of TNFα (≤300 pg/ml) were released in hippocampal or cortical cultures upon stimulation with LPS at concentrations up to 1000 ng/ml (Fig.5B).
Difference in the abundance of reactive microglia among hippocampal, cortical, and mesencephalic cultures
Previous experiments have demonstrated that the LPS-induced release of TNFα and, to a degree, the production of NO are from microglial cells (Kong et al., 1996). One explanation to account for greater levels of NO formation and TNFα release in LPS-treated mesencephalic cultures compared with hippocampal or cortical cultures is that, at the time of treatment, mesencephalic cultures may have contained greater numbers of reactive microglia compared with hippocampal or cortical cultures. To examine this possibility, mixed neuron–glia cultures derived from rat mesencephalon, hippocampus, or cortex were stimulated with vehicle alone or with 10 ng/ml LPS for 72 hr, and then cells were fixed and stained with the monoclonal antibody OX-42, a marker for reactive microglia. Morphological analysis demonstrated that, under normal conditions in all three culture systems, OX-42-immunostained cells were generally small and rounded (Fig. 6A). In comparison, stimulation of neuron–glia cultures with 10 ng/ml LPS for 72 hr resulted in a dramatic change in morphology, indicative of microglia activation. For example, after endotoxin-treatment, OX-42-positive cells were larger and more intensely stained than cells in control cultures (Fig. 6A). Quantification of OX-42-IR cells demonstrated that, within each culture system, a significant increase in the number of OX-42-positive cells occurred after stimulation with 10 ng/ml LPS for 72 hr when compared with vehicle-treated cultures (Fig. 6B), indicating that all three cultures are responsive to exposure to LPS. Consistent with our data that greater levels of microglia-derived inflammatory factors are produced after LPS stimulation in mesencephalic cultures compared with hippocampal or cortical cultures, more OX-42-positive cells were present after LPS treatment of mesencephalic cultures compared with hippocampal or cortical cultures (Fig. 6B). Approximately fourfold and eightfold more OX-42-immunostained cells were present in mesencephalic cultures exposed to LPS compared with neuron–glia cultures derived from the hippocampus and cortex, respectively. Very similar numbers of immunoreactive cells and comparable morphological changes were also observed when the three culture preparations were stained with the lectin or with an antibody against inducible NO synthase (data not shown).
Fig. 6.
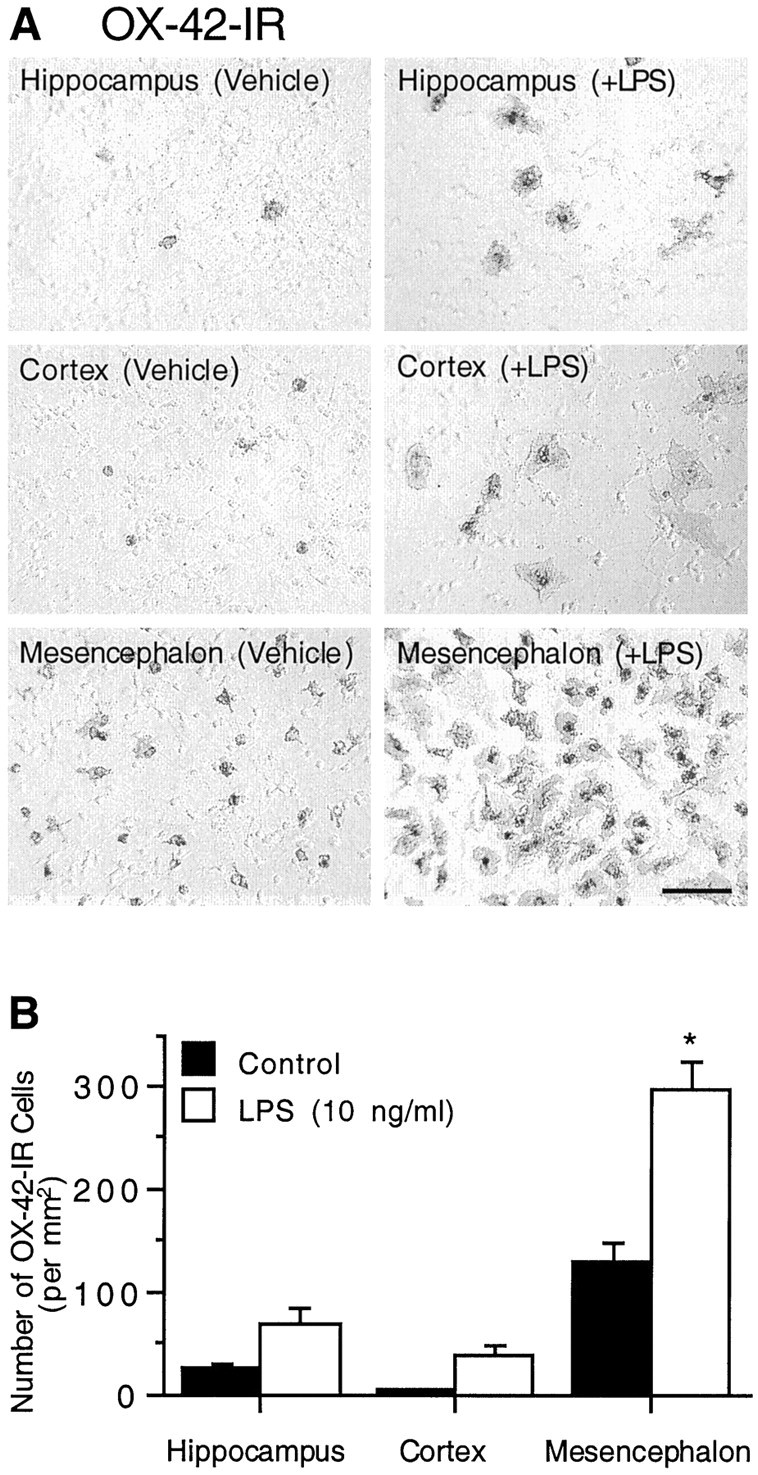
Abundance and responsiveness to LPS of cultured microglial cells derived from different brain regions. Neuron–glia cultures originated from rat hippocampus, cortex, or mesencephalon were treated with 10 ng/ml LPS for 72 hr and then immunostained with the OX-42 antibody. A, Immunocytochemical analysis of microglia. A significantly greater density of microglia was detected in both the vehicle and LPS-treated mesencephalic cultures compared with the hippocampal or cortical counterparts. Scale bar, 100 μm.B, Quantification of OX-42-immunostained cell bodies in hippocampal, cortical, or mesencephalic neuron–glia cultures. The data are expressed as the number of OX-42-IR cells per square millimeter and represent the mean ± SEM of at least three wells. *p < 0.0001 compared with the corresponding vehicle-treated control cultures, as determined by the Student'st test.
Comparison of the responsiveness of microglia derived from various brain regions to LPS
An alternative explanation to account for the differential susceptibility of mesencephalic neurons to LPS exposure might be that mesencephalon-derived microglia were more sensitive or became more reactive after LPS stimulation compared with hippocampal or cortical microglia. To examine this hypothesis, microglia from the three brain regions were plated at similar densities and then stimulated with vehicle alone or with 10 or 1000 ng/ml LPS, and levels of released TNFα were measured. Quantities of TNFα released by each of the three culture systems were normalized by immunostaining for OX-42 and counting the number of OX-42-positive cells. For normalization purposes, OX-42 immunostaining and TNFα release were measured at the same endpoint of 72 hr after endotoxin treatment, although similar trends of TNFα release were measured at 6 hr after LPS stimulation (data not shown). At 72 hr after exposure to the vehicle alone, no detectable levels of TNFα were found in the medium. Stimulation with 10 or 1000 ng/ml LPS for 72 hr resulted in approximately equivalent levels of TNFα released from the three different cultures when normalized to the number of OX-42-IR cells present in each culture system. For example, when stimulated with 10 ng/ml LPS, microglia derived from the hippocampus released 147 pg of TNFα per 1000 OX-42-IR cells. In comparison, microglia derived from the cortex or mesencephalon released 141 or 171 pg of TNFα per 1000 OX-42-IR cells, respectively. After a 72 hr stimulation with 1000 ng/ml LPS, levels of TNFα released per 1000 OX-42-IR cells were 109, 86, or 110 pg, respectively, in microglia cultures derived from the hippocampus, cortex, or mesencephalon. These results indicated an equal per unit cell capacity of microglia to produce cytokines in response to LPS, regardless of whether the cells were derived from the hippocampus, cortex, or mesencephalon. These data also suggested that the susceptibility to LPS-induced neurotoxicity in mesencephalic, but not hippocampal or cortical, neuron–glia cultures may be primarily attributable to the presence of a greater number of activated microglia.
Addition of enriched microglia rendered LPS-insensitive cortical neuron–glia cultures susceptible to LPS-induced neurotoxicity
To test directly whether LPS-induced neurotoxicity was dependent on the number of reactive microglia, equal numbers of microglia from rat cortex or mesencephalon were added to neuron–glia cultures derived from the rat cortex. The number of MAP-2-IR neurons was quantified to determine whether cortical neurons, which were insensitive to LPS-induced neurotoxicity at concentrations up to 10 μg/ml, might become sensitive to LPS-induced death in the presence of sufficient numbers of microglia. As shown in Figure7A, no significant loss of MAP-2-IR neurons was observed in cortical neuron–glia cultures treated with 10 ng/ml LPS for 72 hr, consistent with previous data from cortical or hippocampal cultures treated with 1 μg/ml LPS (Fig.3A,B). However, supplementing LPS-insensitive cortical neuron–glia cultures with additional microglia resulted in significant LPS-induced neurotoxicity (Fig.7A). For example, after a 72 hr treatment with 10 ng/ml LPS, cortical neuron–glia cultures that were supplemented with microglia (105 per well) from the cortex or mesencephalon suffered a loss of 75 or 60% of MAP-2-positive neurons, respectively (Fig. 7A). Thus, the toxicity to MAP-2-IR neurons in cortical neuron–glia cultures occurred to a similar extent, regardless of the origin of the microglia.
Fig. 7.

Addition of microglia to LPS-insensitive cortical neuron–glia cultures rendered cortical neurons susceptible to LPS-induced toxicity. Neuron–glia cultures derived from rat cortex were cultured alone or together with microglia (105) derived from either the cortex or mesencephalon and then were treated with vehicle or 10 ng/ml LPS for 72 hr. A, Quantification of the number of MAP-2-IR neurons. B, TNFα release measured at 72 hr after treatment of cultures with 10 ng/ml LPS. C, NO production at 72 hr after treatment. *p < 0.0001 compared with the corresponding vehicle-treated control cultures, as determined by the Student'st test. ND, not detected.
Consistent with the levels of neurotoxicity produced in cortical neuron–glia cultures supplemented with a microglia-enriched population, the accumulation of nitrite and levels of TNFα released into the medium at 72 hr were lowest in cortical neuron–glia cultures stimulated with vehicle alone and greatest in those cultures supplemented with a microglia-enriched population and then stimulated with LPS. As shown previously, in neuron–glia cultures derived from rat cortex, nondetectable or only low levels of TNFα release were measured in untreated or LPS-treated cultures (Fig. 7B). When 105 enriched microglia derived from the cortex or mesencephalon were added to neuron–glia cortical cultures, levels of TNFα release after exposure to the vehicle alone remained nondetectable. In comparison, levels of TNFα release in microglia-augmented cultures were increased after stimulation with 10 ng/ml LPS when compared with vehicle-treated cultures, and this increase was independent of the source of the microglia-enriched population (Fig. 7B). As with levels of TNFα release, the accumulation of nitrite was observed to be lowest in cortical neuron–glia cultures stimulated with the vehicle alone and greatest in those cultures supplemented with microglia and then stimulated with LPS (Fig. 7C). In addition, the high levels of NO production in microglia-supplemented cultures occurred independent of the origin of the additional microglia (Fig.7C). These data support the hypothesis that LPS-induced neurotoxicity is contingent on the release of proinflammatory cytokines and the generation of free radicals and that the levels of neurotoxicity may be linked to the number of activated microglia present within the system.
DISCUSSION
Using both animals and in vitro rat neuron–glia cultures, the present study demonstrates, for the first time, that neurons in various brain regions are differentially susceptible to inflammation-related degeneration. Among the three brain regions examined, neurons in the SN are most sensitive to bacterial endotoxin LPS-induced neurotoxicity, whereas neurons in hippocampus or cortex remain insensitive to treatment, even with high concentrations of LPS. The region-specific susceptibility to LPS-induced degeneration is most likely attributable to the abundance of microglia in that region.
Microglia are the primary immune cells of the brain and have been shown to be involved in the pathogenesis of numerous neurological disorders, including neurodegenerative diseases. In response to injury, infection, or inflammation, microglia readily become activated, as indicated by a change in morphology, increase in metabolic activity, and increased expression of major histocompatibility complex molecules and the complement type 3 receptor, as determined by OX-42 immunostaining (Streit et al., 1988; Kaur and Ling, 1992; Graeber et al., 1994;Kreutzberg, 1996). Activated microglia secrete a host of immunomodulatory and cytotoxic factors, among which a few are suggested to play a role in tissue repair (Streit et al., 1988) whereas others are thought to be destructive and involved in neurodegeneration through mechanisms that are not fully understood. Within the array of the microglia-derived proinflammatory and cytotoxic factors (reactive nitrogen and oxygen intermediates, arachidonic acid metabolites, proteolytic enzymes, and proinflammatory cytokines), the formation of NO is of particular interest. Under physiological conditions, NO is involved in intercellular and intracellular signaling (Garthwaite, 1991; Dawson and Snyder, 1992). However, excessive levels of NO can induce neuronal injury in vitro (Boje and Arora, 1992; Chao et al., 1992). NO can readily react with superoxide free radicals to form the highly reactive peroxynitrite species that are capable of inflicting additional neuronal damage (Beckman et al., 1990). The importance of NO in microglia-mediated neurodegeneration is further supported by observations in which the addition of NO synthase inhibitors to neuron–glia cultures inhibits the LPS-induced accumulation of nitrite and reduces neuronal cell loss (Boje and Arora, 1992; Chao et al., 1992; Bronstein et al., 1995). In addition to NO, TNFα has been suspected to be an important mediator of LPS-induced inflammation, as evidenced by the findings that levels of TNFα in the CSF of patients with bacterial meningitis correlate with concentrations of bacterial endotoxin in the brain (Arditi et al., 1990). Although TNFα alone is not capable of producing neurotoxicity in neuron–glia cultures, significant neurotoxicity can be observed when TNFα is present in combination with IL-1 and interferon-γ (Jeohn et al., 1998). Currently, it is not known whether TNFα, when present with other cytotoxic factors, exerts its effects directly on neurons in neuron–glia cultures or through an indirect mechanism, perhaps through an increased production of microglia-derived proinflammatory mediators.
The present study is the first to demonstrate that the differential susceptibility of neurons from different brain regions to LPS-induced toxicity is positively linked to the number of microglia in the various brain regions. Neurons in the SN, a region shown to contain the highest concentration of microglia in the rodent brain (Lawson et al., 1990), were sensitive to LPS-induced degeneration, whereas those in the hippocampus and cortex remained resistant to LPS insult. A similar result showing modest degeneration of dopamine-containing neurons in the SN after LPS infusion was reported by Castano et al. (1998). Thesein vivo observations corroborated data from in vitro neuron–glia cultures originating from the various brain regions. For example, in mesencephalic neuron–glia cultures, LPS dose-dependently induced microglia to become activated and to produce NO and TNFα, thereby leading to damage of both dopaminergic and other neurons in general. The sensitivity of mesencephalic neurons to LPS-induced neurodegeneration at concentrations as low as 10 ng/ml is in sharp contrast with the inability of high concentrations of LPS (10 μg/ml) to kill neurons in the hippocampal or cortical neuron–glia cultures. The insensitivity of hippocampal or cortical neurons to LPS-induced toxicity in vitro most likely resulted from low levels of NO and TNFα produced by significantly smaller numbers of microglia (fourfold to eightfold less) when compared with mesencephalic cultures. The notion that the differential sensitivity of neurons in various brain regions is dictated by the numerical difference in abundance of microglia in the region is further supported by the results that (1) microglia isolated from different brain regions are equally capable of responding to LPS stimulation and (2) the addition of microglia to otherwise LPS-insensitive cortical neuron–glia cultures rendered the neurons vulnerable to LPS-induced killing (Fig.7). These in vitro observations may help explain the sensitivity of mesencephalic neurons in vivo to LPS-induced degeneration and the insensitivity of cortical and hippocampal neurons.
It should be pointed out that, under the current experimental conditions, LPS-induced neurodegeneration both in vitro andin vivo did not appear to be selective to any specific population of neurons. However, it is noteworthy that LPS-induced neurodegeneration has been shown to be selective for a population of cholinergic forebrain neurons by several groups (Kalman et al., 1997;Willard et al., 1999). It is not known whether low doses of injected LPS and/or longer treatment times can result in a selective killing of particular neuronal populations. It has been reported that lower doses of LPS injected for longer periods of time or the long-term infusion of IL-1 resulted in the extensive, and in some cases selective, loss of neurons (Hanish et al., 1997; Hauss-Wegrzyniak et al., 2000). Nevertheless, the significant degeneration of dopaminergic neurons in the SN after LPS injection and in mesencephalic neuron–glia cultures after LPS stimulation is of particular interest because a selective and progressive loss of dopaminergic neurons in the SN is characteristic of Parkinson's disease. Although considerable effort has been made, the precise mechanisms underlying the selective degeneration of dopaminergic neurons in the SN is not fully understood. Recently, the role of inflammation in the brain has been increasingly associated with the development of Parkinson's disease. The high proportion of microglia in the SN compared with other brain regions may provide the basis by which inflammatory events may facilitate the degeneration of the nigrostriatal pathway in Parkinson's disease. Understanding the events involved in microglia-mediated cell death, therefore, is necessary for developing therapeutic interventions for such diseases.
Footnotes
We thank Dr. J. L. Maderdrut for critical reading of this manuscript.
Correspondence should be addressed to Dr. Jau-Shyong Hong, National Institute of Environmental Health Sciences/National Institutes of Health, MD: F1–01, P.O. Box 12233, Research Triangle Park, NC 27709. E-mail: hong3@niehs.nih.gov.
W.-G. Kim's present address: Korea Research Institute of Bioscience and Biotechnology, Yuson, Taejon, Korea.
REFERENCES
- 1.Arditi M, Manogue KR, Caplan M, Yogev R. Cerebrospinal fluid cachectin/tumor necrosis factor-α and platelet-activating factor concentrations and severity of bacterial meningitis in children. J Infect Dis. 1990;162:139–147. doi: 10.1093/infdis/162.1.139. [DOI] [PubMed] [Google Scholar]
- 2.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implication for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boje KM, Arora PK. Microglia-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 4.Bronstein DM, Perez-Otano I, Sun V, Mullis Sawin SB, Chan J, Wu G-C, Hudson PM, Kong L-Y, Hong J-S, McMillian MK. Glia-dependent neurotoxicity and neuroprotection in mesencephalic cultures. Brain Res. 1995;704:112–116. doi: 10.1016/0006-8993(95)01189-7. [DOI] [PubMed] [Google Scholar]
- 5.Brosnan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Reactive nitrogen intermediates in human neuropathology: an overview. Dev Neurosci. 1994;16:152–161. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- 6.Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70:1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 7.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 8.Dawson TM, Snyder SH. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. J Neurosci. 1992;14:5147–5159. doi: 10.1523/JNEUROSCI.14-09-05147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson VL, Brahmbhatt HP, Mong JA, Dawson TM. Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology. 1994;33:1425–1430. doi: 10.1016/0028-3908(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 10.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer's disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 11.Espey MG, Chernyshev ON, Reinhard JF, Jr, Namboodiri MA, Colton CA. Activated human microglia produce the excitotoxin quinolinic acid. NeuroReport. 1997;8:431–434. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- 12.Garthwaite J. Glutamate, nitric oxide, and cell-cell signaling in the nervous system. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- 13.Graeber MB, Bise K, Mehraein P. CR3/43, a marker for activated human microglia: application to diagnostic neuropathology. Neuropathol Appl Neurobiol. 1994;20:406–408. doi: 10.1111/j.1365-2990.1994.tb00987.x. [DOI] [PubMed] [Google Scholar]
- 14.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15 N] nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 15.Hanish UK, Neuhaus J, Rowe W, Van Rossum D, Moller T, Kettenmann H, Quirion R. Neurotoxic consequences of central long-term administration of interleukin-2 in rats. Neuroscience. 1997;79:799–818. doi: 10.1016/s0306-4522(97)00040-7. [DOI] [PubMed] [Google Scholar]
- 16.Hauss-Wegrzyniak B, Vannucchi MG, Wenk GL. Behavioral and ultrastructural changes induced by chronic neuroinflammation in young rats. Brain Res. 2000;859:157–166. doi: 10.1016/s0006-8993(00)01999-5. [DOI] [PubMed] [Google Scholar]
- 17.Ii M, Sunamoto M, Ohnishi K, Ichimori Y. β-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- 18.Jeohn G-H, Kong L-Y, Wilson B, Hudson P, Hong J-S. Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol. 1998;85:1–10. doi: 10.1016/s0165-5728(97)00204-x. [DOI] [PubMed] [Google Scholar]
- 19.Kalman J, Engelhardt JI, Le WD, Xie W, Kovacs I, Kasa P, Appel SH. Experimental immune-mediated damage of septal cholinergic neurons. J Neuroimmunol. 1997;77:63–74. doi: 10.1016/s0165-5728(97)00062-3. [DOI] [PubMed] [Google Scholar]
- 20.Kaur C, Ling EA. Activation and re-expression of surface antigen in microglia following an epidural application of kainic acid in the rat brain. J Anat. 1992;180:333–342. [PMC free article] [PubMed] [Google Scholar]
- 21.Kong L-Y, McMillian MK, Maronport R, Hong J-S. Protein tyrosine kinase inhibitors suppress the production of nitric oxide in mixed glia, microglia-enriched or astrocyte-enriched cultures. Brain Res. 1996;729:102–109. [PubMed] [Google Scholar]
- 22.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 23.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 24.Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1β. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- 25.Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000a;293:607–617. [PubMed] [Google Scholar]
- 26.Liu B, Jiang JW, Wilson BC, Du L, Yang SN, Wang JY, Wu GC, Chao XD, Hong JS (2000b) Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J Pharmacol Exp Ther, in press. [PubMed]
- 27.Matsumoto Y, Ohmori K, Fujiwara M. Microglial and astroglial reactions to inflammatory lesions of experimental autoimmune encephalomyelitis in the rat central nervous system. J Neuroimmunol. 1992;37:23–33. doi: 10.1016/0165-5728(92)90152-b. [DOI] [PubMed] [Google Scholar]
- 28.Matsuo M, Hamasaki Y, Fujiyama F, Miyazaki S. Eicosanoids are produced by microglia, not by astrocytes, in rat glial cell cultures. Brain Res. 1995;685:201–204. doi: 10.1016/0006-8993(95)00490-h. [DOI] [PubMed] [Google Scholar]
- 29.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 30.Minghetti L, Levi G. Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J Neurochem. 1995;65:2690–2698. doi: 10.1046/j.1471-4159.1995.65062690.x. [DOI] [PubMed] [Google Scholar]
- 31.Paxinos G, Watson C. The rat brain in stereotaxic coordinates Ed 2. Academic; Orlando, FL: 1986. [DOI] [PubMed] [Google Scholar]
- 32.Raine CS. Multiple sclerosis: immune system molecule expression in the central nervous system. J Neuropathol Exp Neurol. 1994;53:328–337. doi: 10.1097/00005072-199407000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. 1988;9:339–349. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 34.Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1:301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- 35.Willard LB, Hauss-Wegrzyniak G, Wenk GL. Pathological and biochemical consequences of acute and chronic neuroinflammation within the basal forebrain cholinergic system of rats. Neuroscience. 1999;88:193–200. doi: 10.1016/s0306-4522(98)00216-4. [DOI] [PubMed] [Google Scholar]