

Abstract
Previous studies suggest that the vesicular cysteine-string protein (CSP) may modulate presynaptic Ca2+ channel activity in fast neurotransmitter release. To test this hypothesis, we analyzed the dynamics of presynaptic Ca2+ ion influx with the Ca2+ indicator fluo-4 AM at csp mutant neuromuscular junctions of Drosophila. From 24 to 30°C, stimulus-evoked, relative presynaptic Ca2+ signals were increasingly larger in csp mutant boutons than in controls. Above 30°C, Ca2+ signals declined and were similar to controls at 34°C. A prolonged decay of Ca2+ signals in mutant boutons at high temperatures indicated abnormally slow Ca2+ clearance. Cytosolic Ca2+ at rest was determined with the ratiometric Ca2+ indicator fura-2 AM and was similar in mutant and control boutons at 24°C but higher in mutant boutons at 34°C. Despite larger Ca2+ signals in mutant boutons, evoked neurotransmitter release was always reduced in cspmutants and exhibited pronounced facilitation. Thus, a lack of Ca2+ entry cannot explain the reduction of neurotransmitter release in csp mutants. At all temperatures tested, raising extracellular Ca2+ increased transmitter release elicited by single stimuli in cspmutants. Collectively, these data suggest multiple functions for CSP at synaptic terminals. Increased Ca2+ signals coupled with reduced release suggest a direct function of CSP in exocytosis downstream from Ca2+ entry. Because the reduction of evoked release in csp mutants is counteracted by increased Ca2+ levels, we suggest that CSP primarily increases the Ca2+ sensitivity of the exocytotic machinery.
Keywords: cysteine-string protein, Ca2+ channel, syntaxin, neurotransmitter release, exocytosis, synaptic transmission, Ca2+ clearance, Drosophila, neuromuscular junction, Ca2+measurement
Cysteine-string protein (CSP), originally identified as a synapse-associated antigen in the nervous system of Drosophila (Zinsmaier et al., 1990), is associated with secretory vesicles and is conserved from invertebrates to man (for review, see Umbach et al., 1995; Buchner and Gundersen, 1997). Genetic studies in Drosophila have shown that CSP is critical for viability and regulated neurotransmitter release. In particular, a complete gene deletion reduces evoked neurotransmitter release by 50% at 22°C and abolishes evoked but not spontaneous release above 29°C (Umbach et al., 1994; Zinsmaier et al., 1994). A similar reduction of evoked neurotransmitter release occurs with injection of CSP antibodies into frog motor neurons (Poage et al., 1999).
Coexpression of Torpedo CSP RNA in frog oocytes altered the activity of ectopically expressed N-type Ca2+channels (Gundersen and Umbach, 1992). Previous studies of cytosolic Ca2+ in csp mutant Drosophilanerve terminals indicated severely attenuated Ca2+signals at temperatures above 29°C shortly after repetitive stimulation (Umbach et al., 1998). These and other studies suggested that CSP may modulate presynaptic Ca2+ channel activity and potentially link synaptic vesicles to Ca2+ channels (Mastrogiacomo et al., 1994; Leveque et al., 1998).
Regulation of Ca2+ channels, however, is unlikely to be the only function of CSP. An increase or decrease of CSP levels in PC12 and insulin-secreting cells severely reduced exocytosis without affecting transmembrane Ca2+ fluxes, suggesting that CSP may mediate a direct step of exocytosis (Brown et al., 1998;Chamberlain and Burgoyne, 1998; Zhang et al., 1998, 1999). Similarly, overexpression of CSP in chromaffin cells inhibited the extent of exocytosis and slowed the kinetics of individual release events, indicating a key role of CSP close to fusion pore opening during Ca2+-regulated exocytosis (Graham and Burgoyne, 2000). In agreement with these studies, neurosecretory neuromuscular terminals of csp mutant Drosophila exhibited normal presynaptic Ca2+ currents at restrictive temperatures (Morales et al., 1999).
The ability of CSP to interact with syntaxin (Nie et al., 1999; Wu et al., 1999) and synaptobrevin/vesicle-associated membrane protein (Leveque et al., 1998) is compatible with a role in regulating Ca2+ channel activity or SNARE [solubleN-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor] complex function in vesicle fusion. Thus, two alternative interpretations of CSP function exist. First, CSP may have different functions in “fast” versus “slow” secretory systems. Second, CSP may mediate a direct step of fast neurotransmitter exocytosis independent of Ca2+ channel activation. To establish which possibility is more likely, we assayed neurotransmitter release and Ca2+ entry in presynaptic terminals ofcsp mutants under similar conditions to correlate deficits of evoked release with Ca2+ entry. In contrast to a previous study (Umbach et al., 1998), we found that Ca2+ entry during nerve stimulation is not abolished at high temperatures in csp mutant Drosophilaneuromuscular junctions, although neurotransmitter release is severely reduced. Consequently, the reduction of neurotransmitter release cannot be caused solely by a reduction of Ca2+ entry. Our observations support the conclusion that CSP mediates multiple roles in neurotransmitter release, including a late step in exocytosis and the stabilization of Ca2+ entry and extrusion.
MATERIALS AND METHODS
Fly stocks
Flies were raised on standard molasses food at 25°C. The homozygous semi-lethal cspU1allele is a molecular null mutation deleting the entire cspgene and was generated by heat-shock-enhanced recombination between two P elements flanking the csp gene (Eberle et al., 1998). ThecspX1 allele was obtained by a P element jump-out mutagenesis and deletes the csp promoter, including the first exon containing the translational start site (Zinsmaier et al., 1994). The allele is phenotypically indistinguishable from molecular null mutations (Zinsmaier et al., 1994; Eberle et al., 1998). Both mutations were kept genetically balanced with a TM6Balancer chromosome in a white genetic background. The dominant Tubby mutation of TM6 was used to identify genotypes; csp homozygous larvae were of non-Tubby phenotype. Genotypes were as follows:w1118 (control),w1118; cspU1/TM6 Tb Sb and w1118;cspX1/TM6 Tb Sb.
Electrophysiology
Both voltage-clamp and current-clamp recordings were made at the indicated temperatures from muscle 6 in the anterior ventral abdomen (primarily abdominal segment A3) of control and mutant climbing third instar larvae raised at 18°C. Dissections and recordings were made in HL3 medium (Stewart et al., 1994). The composition (in mm) was: NaCl 70, KCl 5, MgCl2 20, CaCl2 1 (unless otherwise indicated), NaHCO3 10, Trehalose 5, HEPES 5, and sucrose 115. Larvae were pinned down at the head and tail to Sylgard (Dow Corning, Midland, MI) covering the bottom of a 35 × 10 mm Petri dish, cut longitudinally, and pinned out. For recordings, the dissection saline was replaced with fresh recording saline and continuously superfused over the preparation using a gravity feed system coupled to a vacuum outlet. Intracellular electrodes were pulled (Sutter P-87; Sutter Instruments, Novato, CA) from 1.5 mm outer diameter glass capillaries (World Precision Instruments, Sarasota, FL) containing an internal filament.
For voltage recordings, the intracellular electrode was filled with 3m KCl and had a resistance of 20–40 MΩ. Voltage signals were amplified with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA), filtered at 1 kHz, and digitized at 5 kHz directly to disk with a DigiData 1200 interface and pClamp 6.0 software (Axon Instruments). To evoke an excitatory junction potential (EJP), we stimulated the segmental nerve (0.1 msec pulse duration) at 2.5–3 times the stimulus amplitude required for a threshold response with a fire-polished glass suction electrode (10 μm diameter tip opening) filled with extracellular saline. The whole-cell EJPs were the combined responses of the two axons innervating muscles 6 and 7 (Kurdyak et al., 1994).
Current recordings were made using the two-electrode voltage-clamp (TEVC) configuration to clamp the muscle at −80 mV. The voltage-sensing electrode had a resistance of 18–30 MΩ, and the current-passing electrode had a resistance of 6–12 MΩ. Both electrodes were filled with 3 m KCl. The current-passing electrode was placed in the middle of the muscle fiber, and the voltage-sensing electrode was placed ∼50–100 μm away from it. Both electrode assemblies were electrically shielded to expose only the tips of the microelectrodes and mounted such that the angle between the two electrodes was at least 60°C. Current signals were amplified with an Oocyte Clamp OC-725C (Warner Instruments, Hamden, CT), filtered at 1 kHz, and digitized at 20 kHz directly to disk with a DigiData 1200 interface and pClamp 6.0 software (Axon Instruments). Voltage deviations during the current responses were <5 mV. The segmental nerve was stimulated with a suction electrode as described above; stimulating pulses 5 V in amplitude and 0.1 msec in duration were applied. A saturating response was always confirmed with a higher stimulus (10 V). Current responses were analyzed with pClamp 6.0 software (Axon Instruments). Voltage response trains were analyzed using the Mini Analysis Program (Synaptosoft Inc., Leonia, NJ). Plots were made using Origin 4.0 (Microcal Software Inc., Northampton, MA).
Calcium imaging procedures
Loading of nerve terminal boutons with the calcium indicator fluo-4 AM for Ca2+ imaging was performed using a modified version of the protocol described by Karunanithi et al (1997).
Stock solutions. A 100 mm stock solution of the zinc chelator N,N,N′,N′-tetrakis (2-pyridylmethyl) ethlenediamine (TPEN) (Molecular Probes, Eugene, OR) was made by adding 4.24 mg of TPEN to 0.1 ml of 100% ethanol. A stock solution of pluronic acid (a permeability enhancer) was made by adding 50 mg of pluronic acid to 250 μl of DMSO. Stock solution (1 mm) of the Ca2+ indicator fluo-4 AM (Molecular Probes) was made by adding 45.6 μl of DMSO to a 50 μg vial of fluo-4 AM.
Loading solution. TPEN stock solution (5 μl) was added to 25 ml of HL3, and then 5 μl of the HL3/TPEN solution was added to 0.988 ml of Schneider's medium (Life Technologies, Gaithersburg, MD) giving a final TPEN concentration of 20 μm. The TPEN-containing solution was placed in an Eppendorf tube and then vortexed for 1–2 sec. Pluronic acid stock solution (2 μl) was added to the same Eppendorf tube, giving a final concentration of 0.04% (w/v) pluronic acid, and vortexed for 1–2 sec. Finally, 12 μl of fluo-4 AM stock solution was added for a final concentration of 12 μm and vortexed for 30 sec. The final loading solution contained 0.8% (v/v) DMSO and 0.02% ethanol in addition to TPEN, pluronic acid, and fluo-4 AM.
Experimental solution. The solution used for imaging was prepared in the same manner as the loading solution; however, HL3 was used instead of Schneider's medium, and 5 μl (5 μm) rather than 12 μl of fluo-4 AM was used to give a final concentration of 12 μm fluo-4 AM.
Procedure for loading the calcium indicator. The larval neuromuscular preparation, dissected and secured in a standard dissection dish, was incubated in the loading solution in the dark for 40 min at 15°C. After loading, the incubation solution was replaced with the experimental solution.
Calcium crimson AM and fura-2 AM. For the experiments using calcium crimson AM and fura-2 AM, nerve terminals were loaded as described above for fluo-4 AM. However, for the calcium crimson experiments, we used 2 mm[Ca2+]e in the experimental and loading solutions. For each indicator, a final concentration of 5 μm was used for loading.
Ratiometric imaging procedures. A ratiometric calcium indicator, fura-2 AM (Molecular Probes), was used to determine the absolute [Ca2+]i levels in boutons at rest. After loading the indicator as indicated above, [Ca2+]i was determined ratiometrically from a standard fluorescence microscope system (Nikon Optiphot-2; Nikon, Tokyo, Japan) fitted with 350 and 385 nm excitation filters (Omega Filters) mounted on a filter wheel (Empix). Nerve terminals were visualized using a 40× Olympus Optical (Tokyo, Japan) water immersion objective, and frames were captured using an intensified CCD camera (IC-100; Photon Technologies International, Monmouth, NJ) at two frames/sec, using Axon Imaging Workbench 2.1 (Axon Instruments). Images were acquired in pairs by briefly exposing the preparation (133 msec/image) to alternating excitation wavelengths of 350 and 385 nm. The 350 nm images were divided by their corresponding 385 nm images (pixel by pixel), thus creating a series of ratio images. Background fluorescence was determined from a region of muscle near the nerve terminals, imaged before ratios were calculated. Background values were subtracted from the nerve terminal values. For determination of resting [Ca2+]i, individual terminals were selected, and the mean ratios for each selected terminal were determined. Ratios were then averaged over a 5 sec acquisition period. Resting [Ca2+]i ratios for each nerve terminal were converted into absolute calcium concentrations using Equation 5 of Grynkiewicz et al. (1985). The calibrations used for determining [Ca2+]i from fura-2 measurements followed those described by Delaney et al. (1989), Ravin et al. (1997), and Msghina et al. (1999).
Stimulation and imaging. Indicator-loaded preparations in dissection dishes were secured with dental wax to a ceramic Peltier thermoregulator (Melcor Corp., Trenton, NJ), which was mounted on a 0.25-inch-thick aluminium plate, secured in turn to the stage of an upright microscope. A copper–constantan thermocouple (0.2 mm diameter, BAT-12; Sensortek, Clifton, NJ), coated with epoxy at the tip, was placed 2 mm from the preparation in the dissection dish. The Peltier thermoregulator was connected to a 12 V power supply with a rheostatic control and a built-in voltmeter and ammeter. The Peltier device provided good control of the temperature (±0.1°C) in the preparation dish when the immersion objective of the microscope was placed over the preparation. The experiments were conducted over a range of temperatures (19–35°C).
Stimulus trains 5 sec in duration were delivered at 3–4 min intervals. Ca2+ signals were recorded from motor nerve terminals innervating muscle 6 or 7. Imaging was performed on an upright microscope (Nikon Optiphot-2) with a Bio-Rad (Hercules, CA) MRC 600 confocal laser scanner and a 40× (0.55 nA) Nikon water immersion objective. The 488 nm excitation line of the argon laser was attenuated to 1% of its maximum power.
For the confocal microscopy experiments done with fluo-4 AM and calcium crimson AM, the following set up was used. Emission was monitored through a low-pass optical filter with a cutoff at 515 nm. The pinhole of the photomultiplier tube was fully open to allow maximum sensitivity. Selected boutons were imaged consecutively (30 times each time trial) before, during, and after trains at the different frequencies of stimulation. Images of 127 × 170 pixels were accumulated at 630 msec intervals. The fluorescence response, normalized to resting fluorescence (ΔF/Frest), was expressed as the change in fluorescence (Fstim −Frest) divided by resting fluorescence (Frest): ΔF/Frest (%) = 100 × (Fstim −Frest)/Frest. Time constants of the Ca2+ signal decay were obtained using nonlinear regression with a double variable exponential decay (y = ae−bx +ce−dx), fitted to the average signal after stimulation at each frequency.
Comparisons between terminals of mutant and control flies at different temperatures were made using two-way ANOVAs with a Student–Newman–Keuls repeated measures test and the Student'st test. p < 0.05 was deemed significant.
RESULTS
Ca2+ signals persist in csp mutants at temperatures above 30°C
In previous reports on Drosophila csp mutants, both evoked neurotransmitter release elicited by single stimuli (Umbach et al., 1994; Zinsmaier et al., 1994; Umbach and Gundersen, 1997), and presynaptic cytosolic Ca2+ levels in synaptic boutons measured after repetitive stimulation were abolished at restrictive temperatures above 30°C (Umbach et al., 1998). These results had been consistent with the hypothesis that CSP may regulate presynaptic Ca2+ channel activity through an association between synaptic vesicles and Ca2+channels (Mastrogiacomo et al., 1994). On the other hand, calcium currents in neurosecretory boutons (type III ending on larval muscle 12) were not altered in csp mutants (Morales et al., 1999). To resolve this apparent discrepancy and to better correlate the putative defect of evoked Ca2+ transmembrane fluxes with neurotransmitter release, we conducted additional experiments. Our goal was to verify whether calcium entry in type I glutamatergic boutons is fully blocked at nonpermissive temperatures (above 30°C). We used the calcium indicators fluo-4 AM (found in preliminary trials to enter terminals with minimal background signal in the muscle fibers), calcium crimson AM (used by Umbach et al., 1998), and fura-2 AM to measure [Ca2+]i at rest. Fluo-4 AM was used in the majority of the experiments because it yielded a brighter signal than calcium crimson AM.
Surprisingly, both control and csp null mutant boutons loaded with fluo-4 AM showed robust increases in presynaptic Ca2+ signals when stimulated at temperatures above 30°C (Fig. 1A), even after prolonged thermal equilibration. Mutant boutons exhibited a much slower time course of decay for the Ca2+ signal, especially at the higher temperatures (Fig. 1A). Sometimes boutons of csp mutants failed to regain their resting fluorescence values, unlike control boutons. For example, when the original “standard” Drosophila solution (Jan and Jan, 1976) was used rather than a more haemolymph-like HL3 solution (Stewart et al., 1994), boutons often retained high fluorescence values after stimulation (Fig. 1B). This suggests thatcsp mutant boutons are physiologically less able to cope with Ca2+ loads than control boutons and more easily compromised by adverse conditions.
Fig. 1.
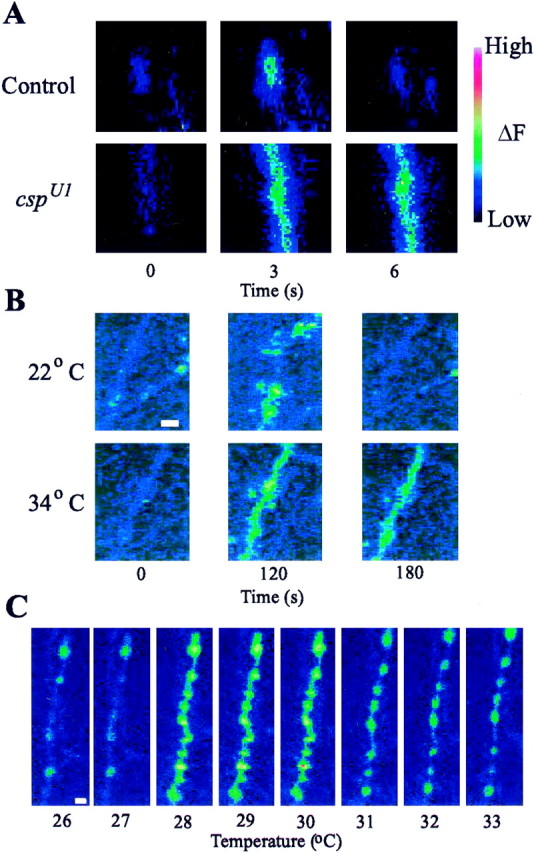
Ca2+ imaging of cspmutant and control neuromuscular junctions with fluo-4 AM.A, At 34°C, control andcspU1 null mutant terminals were stimulated at 40 Hz for 5 sec in HL3 solution. Both terminals exhibit a large Ca2+ signal; the csp mutant terminal recovers more slowly than the control terminal. B, Images of boutons in cspU1 mutants stimulated for 2 min (0–120 sec) at 10 Hz in standard solution at 22 and 34°C. Signals are observed at both temperatures, but recovery was incomplete at 34°C. C, Spontaneous Ca2+ flash phenomenon observed incspU1 boutons in HL3 solution containing 3 mm external Ca2+ when the temperature is increased at a rate of 1°/min. At 28°C, thecspU1 boutons exhibited a large spontaneous Ca2+ signal; high Ca2+ persisted in the terminals at higher temperatures. Scale bars, 2 μm.
Because the results obtained with fluo-4 AM differed from those reported by Umbach et al. (1998) who used the calcium indicator calcium crimson AM, we next determined whether the contradictory results could be attributed to different calcium indicators (fluo-4 AM versus calcium crimson AM) or different solutions (standard versus HL3 solution) used in the two studies. Although the standard solution appears to compromise the isolated preparations more rapidly than HL3 solution, we observed similar fluo-4 signals at room and elevated temperatures in either solution (data not shown). Calcium crimson AM with 2 mm external Ca2+([Ca2+]e) in standard (used byUmbach et al., 1998) or in HL3 solution also showed robust Ca2+ signals in boutons of csp mutants at all temperatures (Fig. 2). Thus, stimulus-evoked Ca2+ signals were observed upon nerve stimulation at temperatures above 30°C with two different calcium indicators and in two different experimental solutions.
Fig. 2.

Ca2+ imaging of cspmutant neuromuscular junctions with calcium crimson AM in 2 mm extracellular calcium. A, Images ofcspU1 mutant boutons stimulated for 2 min at 10 Hz in standard solution at 21 and 34°C. Signals are observed in the same preparation at both temperatures. Note higher resting fluorescence at 34°C. B, Mean peak Ca2+signals at the two temperatures 21 and 34°C after 2 min of stimulation at 10 Hz. A larger signal is observed at 34°C, as shown with the Ca2+ indicator fluo-4 AM.
Although Ca2+ entry persisted at nonpermissive temperatures, csp mutant boutons were more readily incapacitated. The occasional occurrence of spontaneous Ca2+ “flashes” at high external Ca2+ (>2 mm) suggested greater susceptibility of mutant boutons to increased temperature (Fig.1C). In the example shown, the temperature was slowly raised. At 28°C, a sudden large increase in Ca2+occurred, affecting all boutons in one branch of the nerve terminal. Once the spontaneous flash had occurred, elevated Ca2+ persisted in the boutons, and they became unresponsive to stimulation of their parent motor axon. Sometimes, an apparently irreversible change (manifested as persistent elevation in [Ca2+]i) occurred spontaneously in csp mutant boutons in HL3 solution, provided that 3 or 4 mm Ca2+ was present; however, such events were not observed in control boutons and rarely in mutant boutons in solutions containing 1 mm[Ca2+]e. The observations indicate that csp mutant boutons are adversely affected by high temperature and sometimes irreversibly altered because unregulated Ca2+ accumulation. Nevertheless, changes in Ca2+ can almost always be detected in mutant boutons at high temperatures in HL3 solution containing 1 mm Ca2+, which preserves responsiveness of the boutons to stimulation of the motor axon.
Enhanced facilitation of neurotransmitter release incsp mutants
Having established that nerve-evoked Ca2+signals can be detected in both control and csp null mutant boutons over a wide range of temperatures, we investigated the dynamic features of neurotransmitter release during repetitive stimulation to study the relationship between depolarization-dependent Ca2+ entry and neurotransmitter release incsp mutants. Neurotransmitter release was assayed at larval neuromuscular junctions of muscle 6 by TEVC recordings of nerve-evoked EJCs in [Ca2+]e ranging from 1 to 8 mm at 23°C. In 1 mm[Ca2+]e, 30 Hz stimulation rapidly depressed control EJC amplitudes (77 ± 3%, mean ± SEM) but facilitated EJCs of cspX1mutants by 258 ± 42% (Fig.3A). Similar results were obtained with other csp null alleles, such ascspU1 (data not shown). Facilitation of EJCs at mutant NMJs decreased progressively as [Ca2+]e was increased (Fig.3B–D). Specifically, mutant preparations showed much less facilitation in 4 (159 ± 20%), 6 (130 ± 18%), and 8 (116 ± 18%) mm than at 1 mm[Ca2+]e; in fact, facilitation was negligible in [Ca2+]e of 8 mm (Fig. 3D). Control responses, however, showed a further relative depression of neurotransmitter release in elevated [Ca2+]e; EJCs declined to 53% of the initial amplitude during repetitive stimulation in 8 mm [Ca2+]e.
Fig. 3.

Facilitation of transmission at cspmutant neuromuscular junctions. Postsynaptic responses at 23°C incsp null mutants show facilitation of evoked neurotransmitter release under conditions in which control responses show a depression. EJCs at Drosophila larval NMJs were recorded under two-electrode voltage clamp at different [Ca2+]e while stimulating the motor nerve at 30 Hz. Symbols represent mean EJC amplitudes normalized to the first response. Error bars indicate SEM.A, In 1 mm, pronounced facilitation occurs in the cspX1 null mutant. Normalized mutant amplitudes measured for pulses 2–10 are significantly different from control (p < 0.02, Student'st test). B, At 4 mmCa2+, facilitation of mutant responses is reduced compared with the results obtained in 1 mmCa2+, and all responses are significantly different from control (p < 0.02, Student's ttest). C, Facilitation of csp mutant responses is further reduced in 6 mmCa2+ (p < 0.02, Student'st test). D, In 8 mmCa2+, facilitation of csp mutant responses becomes negligible, because none of the subsequent mutant responses (2–10) is significantly different from controls (p > 0.06, Student's t test).E, Absolute EJC amplitudes in 1 mmCa2+ normalized for muscle size by dividing the current amplitude by the cell capacitance. All mutant EJC amplitudes in 1 mm Ca2+ are significantly different from control (p < 0.04, Student's ttest). F, Absolute EJC amplitudes in 8 mmCa2+. None of the mutant EJC amplitudes are significantly different from control (p > 0.1, Student's t test).
A comparison of absolute EJC amplitudes of csp and control NMJs showed that, despite greater facilitation (Fig 3A), neurotransmitter release in csp mutants was nevertheless significantly reduced compared with controls after repetitive stimulation in 1 mm[Ca2+]e (Fig. 3E). During the 30 Hz stimulus, the initial EJC amplitude in csp mutants in 1 mm [Ca2+]e was reduced by 84 ± 2% compared with controls, whereas the amplitude of the 10th EJC was reduced by 44 ± 4% (Fig. 3E). The convergence of absolute EJC amplitudes from control and cspmutants was attributable to depression of control and facilitation of mutant responses. Consistently, in 8 mm[Ca2+]e, this convergence led to similar absolute EJC amplitudes in mutants and controls (p > 0.1, Student's t test) (Fig.3F), because facilitation of mutant responses was negligible, whereas control responses were further depressed (Fig.3D).
The “rescue” of the mutant defect in neurotransmitter release was not dependent on the combination of high [Ca2+]e and high-frequency stimulation. High [Ca2+]e was sufficient to rescue csp mutant responses elicited by single stimuli at 0.2 Hz (Fig. 4). In controls, EJC amplitudes did not significantly increase beyond 4 mm [Ca2+]e. Mutant EJC amplitudes, however, increased gradually with increasing [Ca2+]e and were significantly different from controls up to 6 mm[Ca2+]e (p < 0.007, Student's t test). At 8 mm[Ca2+]e, csp mutant EJC amplitudes became statistically similar to controls (p > 0.1, Student's t test).
Fig. 4.

Ca2+ dependence of evoked release elicited by single stimuli at 23°C in csp mutants. High [Ca2+]e restores the loss of evoked release at cspX1 mutant NMJs.A, Typical two-electrode voltage-clamp recordings of EJCs elicited at 0.2 Hz from larval NMJs in 1 and 8 mm[Ca2+]e. Each EJC represents an average of 20 trials. B, Recorded EJC amplitudes at the indicated [Ca2+]e were normalized for muscle size by dividing the current amplitude by the cell capacitance. Twenty trials were averaged for n larvae. Eachpoint represents the mean EJC amplitude of at least four larvae. Error bars indicate SEM. In 1, 4, and 6 mm[Ca2+]e,cspX1 amplitudes are significantly different from control (p < 0.007, Student'st test). In 8 mm[Ca2+]e,cspX1 mutant amplitudes are no longer significantly different from control (p = 0.15, Student's t test). Mean EJC amplitudes were 15.6 ± 2.1 nA/nF (mean ± SEM) for control and 3.4 ± 0.7 nA/nF forcspX1 mutants in 1 mm[Ca2+]e, and 49.1 ± 8.8 nA/nF for control and 31.1 ± 6.4 nA/nF forcspX1 mutants in 8 mm[Ca2+]e.
By Ca2+ imaging, we established that depolarization-dependent Ca2+ entry into boutons of both csp mutants and controls gradually increased with higher [Ca2+]e (Fig.5). Thus, the facilitated EJC amplitudes were correlated with more Ca2+ entry at higher [Ca2+]e, as would be expected from entry of Ca2+ through voltage-activated Ca2+ channels. However, in the present experiments, we did not simultaneously measure Ca2+ signals and transmitter release from the same bouton.
Fig. 5.

Ca2+ signals in boutons at different external Ca2+ levels at 21°C. As [Ca2+]e was increased, a larger Ca2+ signal was observed for both controls andcspU1 mutants. A,B, Ca2+ changes in type Ib boutons of controls (A, n = 12) andcspU1 mutants (B,n = 5) with increased [Ca2+]e: 0.5, 1, and 3 mm.Symbols indicate mean ± SEM values at each time point. Preparations were stimulated in HL3 solution at 5 Hz for 5 sec (solid horizontal bar). Large error bars shown at 3 mm [Ca2+]e during stimulation resulted from contraction of the muscle. C, Peak values for intracellular Ca2+ signal with different extracellular Ca2+ concentrations.
Rescue of neurotransmitter release in csp mutants at temperatures above 30°C
Previous reports have described a temperature-sensitive block of neurotransmitter release elicited by single stimuli at temperatures above 29°C in csp mutants (Umbach et al., 1994; Zinsmaier et al., 1994; Umbach and Gundersen, 1997). Because cspmutants showed a pronounced facilitation of neurotransmitter release at permissive temperatures, we speculated that facilitation may persist at restrictive temperatures, leading to a rescue of neurotransmitter release during repetitive stimulation. Neurotransmitter release was assayed at larval neuromuscular junctions by current-clamp recordings of nerve-evoked EJPs. Consistent with previous reports (Umbach et al., 1994; Umbach and Gundersen, 1997), we observed that transmitter release evoked by single stimuli is virtually abolished at 30°C. At a low frequency of stimulation (0.2 Hz), the temperature-sensitive block of neurotransmitter release was confirmed, and no evoked EJPs were recorded after heat equilibration in HL3 solution containing 1 mm Ca2+, although spontaneous miniature EJPs were frequent (Fig.6A). However, evoked release could be partially restored by high-frequency stimulation or by higher [Ca2+]e. Stimulation at higher frequency (10 Hz in Fig. 6B) produced well defined but variable EJPs. Interestingly, after 3–4 failures, EJP amplitudes appeared suddenly, did not show a further, gradual increase in amplitude, and never reached control amplitudes. The partially rescued EJP responses were highly variable in amplitude, as indicated by occasional failures to produce an EJP. The temperature-sensitive block of neurotransmission was also counteracted by raising the [Ca2+]e to 4 mm without increasing the stimulation frequency (Fig. 6C). Both procedures were found to increase intraterminal Ca2+levels in the present study; this appears to partially circumvent the loss of normal CSP function.
Fig. 6.

Rescue of the temperature-sensitive block of evoked release at 30°C in csp mutants. When single stimuli (0.2 Hz) failed to evoke neurotransmission in csp null mutants at 30°C, it was possible to partially rescue the temperature-sensitive defect with high-frequency stimulation or high [Ca2+]e. Traces represent typical recordings under current clamp at 30°C. A, Evoked EJPs were blocked in cspU1 null mutants with 0.2 Hz stimulation and 1 mm[Ca2+]e. B, After neurotransmission was blocked, the preparation was stimulated at 10 Hz. The mean amplitude of the first 40 rescued responses was 4.6 ± 1.7 mV (n = 6, ±SEM). C, After neurotransmission at 0.2 Hz was blocked in 1 mm[Ca2+]e, the bath solution was switched to 4 mm[Ca2+]e. Subsequently, stimulation at 0.2 Hz evoked EJPs; the mean amplitude of 12 responses was 19.1 ± 6.1 mV (n = 4).
Temperature-dependence of evoked neurotransmitter release and Ca2+ entry
To correlate defects of cytosolic Ca2+ entry and neurotransmitter release in csp mutants over the same temperature range, we used the same stimulation to measure both Ca2+ entry and neurotransmitter release. In contrast with a previous report showing that Ca2+ signals do not increase in csp mutant boutons after 10 Hz stimulation for 2 min at restrictive temperatures (Umbach et al., 1998), we found a temperature-dependent increase in cytosolic Ca2+ incsp mutant terminals during stimulation at 5 Hz for 5 sec (Fig. 7). In controls, the relative change of the Ca2+ signal was not greatly influenced by temperature and was very consistent, with small variations for each trial (Fig. 7A, C). Typically, Ca2+ signals increased to a maximum value (fluorescence increased by ∼15.4 ± 4.3%) in 1–2 sec, after which a plateau was sustained during the train, with a decline to resting level in ∼1 sec at the end of the train. These results are similar to those reported previously with fluo-3 in HL3 solution (Karunanithi et al., 1997). In contrast, Ca2+signals in csp mutants showed a much greater relative increase, much larger variation, and a much slower recovery after the train (Fig. 7B, C). Significantly, prominent evoked Ca2+ signals peaked in csp mutant boutons at temperatures of 30°C; fluorescence values increased by 43.9 ± 4.3% at 24°C and 77.6 ± 14.3% at 30°C. However, the amplitude of the normalized Ca2+ signal was reduced when the temperature was elevated above 30°C, and values of relative fluorescence much closer to controls were recorded at 34°C (Fig. 7B, C). In contrast with the pattern of increased Ca2+ levels in csp mutants, EJP amplitudes elicited by the same repetitive stimulation (5 Hz for a period of 5 sec) were reduced at permissive temperature by 66% and gradually declined from a mean value of 10.8 ± 0.4 mV (mean ± SEM) at 24°C to 4.8 ± 2.8 mV at 32°C (Fig. 7D). For control preparations, mean EJP values were 31.4 ± 3.9 mV at 24°C and 17.5 ± 3.1 mV at 32°C.
Fig. 7.

Temperature dependence of evoked release and cytosolic Ca2+ levels in csp mutants and controls. A–C, Comparison of Ca2+signals in type Ib boutons of cspU1mutants and controls at different temperatures. Preparations were stimulated in HL3 solution containing 1 mmCa2+ at 5 Hz for 5 sec (solid horizontal bar). Symbols indicate mean ±SEM values for each time.A, Ca2+ signals in control boutons at temperatures between 24 and 34°C, showing a small increase with little change at higher temperatures. B, Ca2+ signals in cspU1boutons at temperatures between 24 and 34°C. Larger Ca2+ signals and increased variability compared with controls are apparent at all temperatures. C, Maximum relative Ca2+ changes in Ib boutons.cspU1 mutants show a significantly larger change in Ca2+ than controls at all temperatures except 34°C. D, Temperature dependence of mean EJP responses evoked by repetitive stimulation. EJP recordings were made after 30 min at each temperature in 1 mmCa2+ HL3 solution. For each specimen, the mean of 25 responses was obtained; the mean values for all specimens were then averaged for each point on the graph.
Together, these experiments show that the relative increase of intraterminal Ca2+ is generally larger incsp mutants than in controls, that recovery (clearance) of intraterminal Ca2+ at high temperatures is much slower, and that the reduction of neurotransmitter release cannot be entirely explained by a loss of Ca2+ entry into the nerve terminal.
Comparison of resting [Ca2+]i incsp and control boutons
Relative changes in fluorescence were often larger incsp mutant boutons than in controls, even at nonpermissive temperatures, whereas transmitter release was markedly reduced under the same conditions. This result could suggest that a primary defect of Ca2+-triggered exocytosis may be partially compensated by increased Ca2+ entry incsp mutants. Alternatively, it could be that resting [Ca2+]i is actually lower incsp mutants than in controls. If this were the case, the observed relative changes in fluorescence would not necessarily indicate a larger Ca2+ accumulation incsp mutant boutons, because peak fluorescence values are normalized to the initial (resting) values. Thus, a normal [Ca2+]i at rest in combination with higher normalized peak fluorescence values during stimulation would signify higher stimulus-evoked cytosolic Ca2+levels.
We addressed this question by measuring resting values of [Ca2+]i using the ratiometric indicator fura-2 AM. This indicator was much more difficult to use than the selected non-ratiometric indicators because of less efficient loading of the terminals and a higher background signal in the muscle. Nevertheless, resting values for [Ca2+]i were obtained in several experiments (Fig. 8). Estimates of [Ca2+]i were made in 1 mm[Ca2+]e. No significant differences in resting [Ca2+]i were found between control (170 ± 6.1 nm) and csp mutant boutons (195 ± 15.0 nm) at room temperature (24°C) (Fig. 8A). However, at 34°C, csp mutant terminals had a significantly higher resting level of [Ca2+]i (361.4 ± 35.72 nm) than control terminals (230 ± 6.7 nm;p = 0.01, df = 5, Student's ttest).
Fig. 8.

Estimated [Ca2+]ifor Drosophila type Ib boutons in 1 mm[Ca2+]e. Values of resting [Ca2+]i estimated with ratiometric imaging using fura-2 AM are shown for control andcspU1 boutons (4 animals, 14 boutons). No difference was found between control and csp boutons at room temperature (23°C). However, at 34°C, [Ca2+]i was significantly higher incsp boutons than in controls (p = 0.01, Student's t test).
Our results suggest that, at room temperature, cytosolic Ca2+ attains significantly higher values incsp mutant than in control boutons during nerve stimulation. This is indicated by normal resting levels of [Ca2+]i and increased evoked fluo-4 signals (ΔF/Frest) during stimulation in csp mutant boutons. At temperatures above 30°C, Ca2+ during stimulation also appears to reach mostly higher levels in mutant than in control boutons, because resting levels of [Ca2+]i are increased, whereas evoked fluo-4 signals are similar. Thus, it is most likely that the loss of neurotransmitter release in cspmutants is primarily caused by defect of a direct step in Ca2+-regulated exocytosis and not by a defect of Ca2+ entry as originally suggested.
Frequency dependence of presynaptic Ca2+ entry and Ca2+ clearance
An additional set of experiments was performed to examine the effect of stimulation frequency on peak Ca2+ signals and their recovery after stimulation at high temperatures (34°C). In controls, Ca2+ signals increased progressively with frequency, as found previously (Karunanithi et al., 1997), and declined rapidly to baseline after stimulation at frequencies below 20 Hz. At frequencies of 20 Hz and above, a persistent “tail” of elevated Ca2+ was sometimes observed in controls (Fig.9A). In two differentcsp mutant alleles (cspU1 andcspX1), an aberrant time course of Ca2+ signals was observed at 34°C (Table1). In thecspU1 mutant, Ca2+increased progressively with frequency, but recovery was slower than in controls, even at frequencies below 20 Hz (Fig. 9C). A more extreme situation was found in the cspX1mutant; Ca2+ increased proportionately less with higher stimulation frequency, and recovery after stimulation was very slow, even at 5 Hz (Fig. 9B). The slight difference between the two deletion mutants is probably attributable to differences in the genetic background because neither allele expresses any detectable CSP protein at NMJs (Zinsmaier et al., 1994; Eberle et al., 1998).
Fig. 9.

Ca2+ clearance incsp mutant and control boutons at 34°C. A–C, Preparations were stimulated in HL3 solution for 5 sec (horizontal bar). Symbols indicate mean ± SEM values at each time. A, Control preparations show abrupt increases in Ca2+ signals with a plateau for all frequencies. B, cspX1mutants exhibit an exaggerated slowing of Ca2+ decay after stimulation and relative small increases in peak Ca2+ signals when the stimulus frequency is increased. C, cspU1preparations show an increase in Ca2+ signals with increased stimulus frequency and prolonged recovery, especially at high frequencies. A significant difference was found betweencspU1 and control preparations at 15, 20, and 40 Hz (ANOVA, df = 5, p < 0.05).D, Relative changes of cytosolic Ca2+ in type Ib boutons of cspU1 Drosophilalarvae before, during, and after a 120 sec stimulus at 10 Hz (horizontal bar) at two different temperatures, 20 and 34°C. Symbols indicate mean ± SEM values, andasterisks indicate significant differences. Experiments were performed in standard Drosophila (Jan and Jan, 1976). During stimulation at 20°C, Ca2+ signals plateau at a relative fluorescence change of ∼80%, whereas at 34°C, Ca2+ signals increase up to 163.7 ± 10.53% (ANOVA, df = 5, p < 0.05). Thirty seconds after the stimulus, the Ca2+ signal returns to resting levels at 20°C; however, at 34°C, the Ca2+signal decreases only to 136.84 ± 14.76% (ANOVA, df = 7,p < 0.05) during that time, and the preparations were often refractory to subsequent stimulation.
Table 1.
Decay kinetics of Ca2+ signals incspU1 mutants and controls
| Control (6) | cspU1 (6) | p Value | |
|---|---|---|---|
| 5 Hz | 2.78 ± 0.35 | 2.60 ± 0.62 | 0.803 |
| 15 Hz | 2.99 ± 0.52 | 6.80 ± 0.51 | <0.001 |
| 20 Hz | 1.51 ± 0.08 | 5.19 ± 0.94 | 0.002 |
| 25 Hz | 2.04 ± 0.81 | 3.40 ± 0.62 | 0.053 |
| 40 Hz | 3.80 ± 0.75 | 10.08 ± 0.83 | <0.001 |
Time constants (in seconds) were measured at five different frequencies in both cspU1 and control preparations at 34°C. Values represent the mean ± SD. At all frequencies above 5 Hz, Ca2+ signals ofcspU1 preparations have a significantly longer time constant of decay than those of controls (p < 0.05, Student's t test).
The slower decay of Ca2+ signals after stimulation indicates a possible defect of Ca2+ extrusion incsp mutants at elevated temperatures. A final set of experiments was conducted to ascertain the temperature dependence of this phenotype. Relative changes of Ca2+ in type Ib boutons of cspU1 Drosophila larvae were recorded before, during, and after a 120 sec stimulus at 10 Hz at two different temperatures, 20 and 34°C (Fig. 9D). The relatively prolonged stimulation in the standard Drosophilasolution duplicates the conditions used for calcium imaging by Umbach et al. (1998). During stimulation at 20°C, Ca2+levels reached a plateau at a relative fluorescence change of ∼80%, whereas at 34°C, Ca2+ changes increased up to ∼165%. At 20°C, the Ca2+ signal returned to its resting value after the stimulus in ∼30 sec. In contrast, at 34°C, the Ca2+ signal decreased only to ∼136% of the initial resting fluorescence in that time and did not reach its initial resting value during the next 100 sec (Fig. 1B). Often, the preparations were refractory to subsequent stimulation.
These observations indicate a deficit in Ca2+clearance in both mutant alleles, with a more extreme impairment of Ca2+ extrusion in thecspX1 allele. The effect is exacerbated at high temperatures (Fig. 9D). The slower clearance of Ca2+ probably contributes to the build-up of Ca2+ in the boutons and to the distinctive properties of the Ca2+ signals generally observed in the mutants (slower time course, and greater variability in amplitude and time course).
DISCUSSION
Our observations suggest that the loss of neurotransmitter release in csp mutants is primarily caused by a defect in a direct step of Ca2+-regulated exocytosis and not by inactivation of presynaptic Ca2+ channels as suggested previously (Umbach et al., 1998). In mutant boutons lacking CSP, a stimulus-evoked Ca2+ signal occurs at all temperatures tested, which is frequently larger than in controls. The relatively large Ca2+ signals between 24 and 30°C are coupled with severely reduced transmitter release; this disjunction does not support the previous hypothesis that lack of Ca2+ entry causes reduced neurotransmitter release in csp mutants (Umbach et al., 1998). This conclusion accords with other studies that found no evidence for regulation of presynaptic Ca2+ channels by CSP in slow secretion systems, including peptidergic boutons ofDrosophila (Brown et al., 1998; Chamberlain and Burgoyne, 1998; Zhang et al., 1998, 1999; Morales et al., 1999).
Our results support the hypothesis that CSP may regulate a direct step in exocytosis (Brown et al., 1998; Chamberlain and Burgoyne, 1998;Zhang et al., 1998, 1999), because transmitter release is much reduced in csp mutants, even under conditions that permit substantial presynaptic Ca2+ entry. This idea is consistent with the observation that the loss of neurotransmitter release elicited by single stimuli at csp mutant terminals (Umbach et al., 1994; Zinsmaier et al., 1994; Umbach and Gundersen, 1997) can be restored by increasing the frequency of stimulation or by raising extracellular Ca2+. Extending a previous study that showed increased paired-pulse facilitation in cspnull mutants at 16–18°C (Heckmann et al., 1997), we found that facilitation of release persists at 30°C and can act to restore release when it cannot be elicited by single stimuli. Consistently, raising extracellular Ca2+ to 8 mm fully restores neurotransmitter release elicited by single stimuli at 23°C in csp mutants, suggesting that the absence of CSP reduces the efficiency of Ca2+ in triggering neurotransmitter release. Thus, extra calcium is needed to compensate for the deficiencies caused by a lack of CSP.
The regulatory or protective effects of CSP on cellular mechanisms are apparently more widespread than previously thought. CSP may act on multiple synaptic mechanisms as indicated by the variety of defects observed at csp mutant boutons. Mutant terminals exhibit relatively slow Ca2+ clearance after repetitive stimulation, in particular at high temperatures. Consistently, cytosolic Ca2+ at rest is elevated in mutant boutons at high but not at low temperatures. Although a slower Ca2+ clearance would increase the build-up of cytosolic Ca2+ in the terminal (Tank et al., 1995), it does not rule out an additional defect in Ca2+entry, as suggested by the large drop in normalized Ca2+ signals above 32°C. The slow clearance of intracellular Ca2+ may also contribute to the neurodegeneration observed in adult csp mutant flies and their ultimate death (Zinsmaier, et al., 1994).
In addition to altered Ca2+ homeostasis, presynaptic Ca2+ entry appears to be less firmly regulated incsp mutants than in controls, although it is not the primary cause for the loss of neurotransmitter release. Evidence for abnormal Ca2+ entry was apparent in the unusual temperature dependence of stimulus-evoked intraterminal Ca2+signals and in spontaneous flashes of cytosolic Ca2+. In principle, these could be caused by a variety of defects, including accumulation of cytosolic Ca2+, increased numbers of Ca2+channels, prolonged Ca2+ channel opening times, and/or a defect in Ca2+-activated K+ conductance.
Ca2+ entry can be compromised at high temperatures in csp mutants, particularly under adverse or stressful physiological conditions. For example, use of the standard recording solution (Jan and Jan, 1976), which is quite different in ionic composition from Drosophila haemolymph (Stewart et al., 1994), appeared to exacerbate the problems of Ca2+signaling in csp mutants. Frequently, standard solution produced an unresponsive preparation, especially when repetitive stimulation was delivered for a long period. Such findings may help to explain the differences between our results and those of a previous study of csp mutants that reported that stimulus-evoked intraterminal Ca2+ signals are absent after extensive stimulation above 29°C (Umbach et al., 1998).
CSP appears to protect cellular mechanisms controlling intracellular Ca2+ signaling and homeostasis. These functions may involve known interactions with heat-shock proteins and heat-shock cognate proteins (Braun et al., 1996; Chamberlain and Burgoyne, 1997b;Zhang et al., 1999). Mammalian CSP binds to the 70 kDa heat-shock cognate protein (Hsc70) (synonym, clathrin-uncoating ATPase) in vitro and activates its intrinsic ATPase activity 12-fold (Braun et al., 1996; Chamberlain and Burgoyne, 1997b; Zhang et al., 1999). Because studies of Drosophila csp null mutations revealed no defects in synaptic vesicle recycling (Ranjan et al., 1998), the CSP–Hsc70 interaction may play a major role in neurotransmitter exocytosis. The prevention of protein aggregation by CSP itself (Chamberlain and Burgoyne, 1997a) and more efficiently in cooperation with Hsc70 (Braun et al., 1996) is consistent with the deterioration of neurotransmitter release in csp deletion mutants at high temperatures, which suggests that target proteins of CSP action must be destabilized in the absence of CSP.
A direct role of CSP in exocytosis was suggested by several studies using slow secretion systems. Although overexpression of bovine CSP in neuroendocrine PC12 cells had no effect on Ca2+entry, it enhanced Ca2+-dependent dopamine release in permeabilized PC12 cells and GTPγS-induced release in the absence of Ca2+ (Chamberlain and Burgoyne, 1998). Similarly, overexpression of CSP or CSP antibody injections showed no affect on Ca2+ channel activity in insulin-secreting cell lines (Brown et al., 1998) but significantly decreased insulin release (Brown et al., 1998; Zhang et al., 1999). Expression of antisense mRNA also reduced induced insulin release in intact and in permeabilized β-cell lines (Zhang et al., 1998). Because all effects on exocytosis persisted in permeabilized cells, these studies suggest a direct role of CSP in exocytosis independent of transmembrane Ca2+ fluxes (Chamberlain and Burgoyne, 1998; Zhang et al., 1998, 1999). More recently, CSP has been found to act close to fusion pore opening during Ca2+-regulated exocytosis. Overexpression of CSP in chromaffin cells inhibited not only the extent of exocytosis but also slowed the kinetics of individual release events (Graham and Burgoyne, 2000).
The original hypothesis of CSP function at nerve terminals proposed that CSP may promote neurotransmitter release by increasing Ca2+ channel activity (Mastrogiacomo et al., 1994). This idea has been mostly supported by the coexpression of CSP mRNA, which modulated ectopically expressed N-type Ca2+channel currents (Gundersen and Umbach, 1992). Although several studies failed to demonstrate binding of CSP to native Ca2+channels (Martin-Moutot et al., 1996; Pupier et al., 1997; Leveque et al., 1998), CSP has been shown to bind the “synprint site” containing cytoplasmic loop of P/Q-type channels (Leveque et al., 1998). The synprint site mediates interactions of multiple synaptic proteins with Ca2+ channels, including syntaxin, synaptotagmin, and SNAP25 (for review, see Sheng et al., 1998; Seagar et al., 1999). Syntaxin binding downregulates Ca2+channel activity by prolonging an inactivated state (Bezprozvanny et al., 1995; Wiser et al., 1996). Because CSP is an effective competitor of the syntaxin–synprint site interaction in vitro (Wu et al., 1999), it has been suggested that CSP may dissociate syntaxin from Ca2+ channels to promote channel activity (Bezprozvanny et al., 1995; Umbach et al., 1995; Seagar et al., 1999;Wu et al., 1999). However, this possibility is not consistent with ourin vivo studies revealing increased stimulus-evoked cytosolic Ca2+ levels in csp mutants.
The present study suggests similar functions of CSP in evoked neurotransmitter and peptidergic exocytosis. We suggest multiple synaptic functions for CSP in nerve terminals. The most significant one for neurotransmitter release appears to increase the Ca2+ sensitivity of a direct step in exocytosis, as proposed for slow peptidergic exocytosis. This regulatory action may include steps of Ca2+ signaling located between the postulated Ca2+ sensor of vesicle fusion and the fusion machinery itself. In addition, CSP appears to stabilize Ca2+ entry and Ca2+ clearance, although these functions may only be significant for evoked release at high temperatures. The recently demonstrated in vitro andin vivo interaction of CSP with syntaxin (Nie et al., 1999;Wu et al., 1999) is compatible with a role of CSP in regulating SNARE complex-associated protein interactions of syntaxin or, alternatively, Ca2+ channel–syntaxin interactions. Further work, however, is necessary to clarify the most likely steps through which CSP increases the Ca2+ sensitivity of exocytosis.
Footnotes
This work was supported in part by grants from the National Science Foundation and the National Institute of Neurological Disorders and Stroke to K.E.Z., a National Research Service award to P.B., and grants from the Medical Research Council of Canada to H.L.A. We thank Gowan Tervo (National Sciences and Engineering Research Council of Canada summer student) for his contributions to initial trials of fura-2 AM in Drosophila boutons and Andrew Millar for helping with the measurements of resting Ca2+reported here.
K.D.-S and P.B. contributed equally to this work.
Correspondence should be addressed to Konrad E. Zinsmaier, Department of Neuroscience, 234d Stemmler Hall, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6974. E-mail:zinsmaie@mail.med.upenn.edu.
REFERENCES
- 1.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 2.Braun J, Wilbanks SM, Scheller RH. The cysteine string secretory vesicle protein activates Hsc70 ATPase. J Biol Chem. 1996;271:25989–25993. doi: 10.1074/jbc.271.42.25989. [DOI] [PubMed] [Google Scholar]
- 3.Brown H, Larsson O, Branstrom R, Yang SN, Leibiger B, Leibiger I, Fried G, Moede T, Deeney JT, Brown GR, Jacobsson G, Rhodes CJ, Braun JE, Scheller RH, Corkey BE, Berggren PO, Meister B. Cysteine string protein (CSP) is an insulin secretory granule-associated protein regulating beta-cell exocytosis. EMBO J. 1998;17:5048–5058. doi: 10.1093/emboj/17.17.5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchner E, Gundersen CB. The DnaJ-like cysteine string protein and exocytotic neurotransmitter release. Trends Neurosci. 1997;20:223–227. doi: 10.1016/s0166-2236(96)10082-5. [DOI] [PubMed] [Google Scholar]
- 5.Chamberlain LH, Burgoyne RD. The molecular chaperone function of the secretory vesicle cysteine string proteins. J Biol Chem. 1997a;272:31420–31426. doi: 10.1074/jbc.272.50.31420. [DOI] [PubMed] [Google Scholar]
- 6.Chamberlain LH, Burgoyne RD. Activation of the ATPase activity of heat-shock proteins Hsc70/Hsp70 by cysteine-string protein. Biochem J. 1997b;322:853–858. doi: 10.1042/bj3220853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain LH, Burgoyne RD. Cysteine string protein functions directly in regulated exocytosis. Mol Biol Cell. 1998;9:2259–2267. doi: 10.1091/mbc.9.8.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney KR, Zucker RS, Tank DW. Calcium in motor nerve terminals associated with posttetanic potentiation. J Neurosci. 1989;9:3558–3567. doi: 10.1523/JNEUROSCI.09-10-03558.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eberle KK, Zinsmaier KE, Buchner S, M. G, Jenni M, Arnold C, Leibold C, Reisch D, Walter N, Hafen E, Hofbauer A, Pflugfelder GO, Buchner E. Wide distribution of cysteine string protein in Drosophila tissues revealed by targeted mutagenesis. Cell Tissue Res. 1998;294:203–217. doi: 10.1007/s004410051170. [DOI] [PubMed] [Google Scholar]
- 10.Graham ME, Burgoyne RD. Comparison of cysteine string protein (Csp) and mutant a-SNAP overexpression reveals a role for Csp in late steps of membrane fusion in Dense-Core granule exocytosis in adrenal chromaffin cells. J Neurosci. 2000;20:1281–1289. doi: 10.1523/JNEUROSCI.20-04-01281.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 12.Gundersen CB, Umbach JA. Suppression cloning of the cDNA for a candidate subunit of a presynaptic calcium channel. Neuron. 1992;9:527–537. doi: 10.1016/0896-6273(92)90190-o. [DOI] [PubMed] [Google Scholar]
- 13.Heckmann M, Adelsberger H, Dudel J. Evoked transmitter release at neuromuscular junctions in wild type and cysteine string protein null mutant larvae of Drosophila. Neurosci Lett. 1997;228:167–170. doi: 10.1016/s0304-3940(97)00390-x. [DOI] [PubMed] [Google Scholar]
- 14.Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol (Lond) 1976;262:189–214. doi: 10.1113/jphysiol.1976.sp011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karunanithi S, Georgiou J, Charlton MP, Atwood HL. Imaging of calcium in Drosophila larval motor nerve terminals. J Neurophysiol. 1997;78:3465–3467. doi: 10.1152/jn.1997.78.6.3465. [DOI] [PubMed] [Google Scholar]
- 16.Kurdyak P, Atwood HL, Stewart BA, Wu CF. Differential physiology and morphology of motor axons to ventral longitudinal muscles in larval Drosophila. J Comp Neurol. 1994;350:463–472. doi: 10.1002/cne.903500310. [DOI] [PubMed] [Google Scholar]
- 17.Leveque C, Pupier S, Marqueze B, Geslin L, Kataoka M, Takahashi M, De WM, Seagar M. Interaction of cysteine string proteins with the alpha1A subunit of the P/Q-type calcium channel. J Biol Chem. 1998;273:13488–13492. doi: 10.1074/jbc.273.22.13488. [DOI] [PubMed] [Google Scholar]
- 18.Martin-Moutot N, Charvin N, Leveque C, Sato K, Nishiki T, Kozaki S, Takahashi M, Seager M. Interaction of SNARE complexes with P/Q-type calcium channels in rat cerebellar synaptosoms. J Biol Chem. 1996;271:6567–6570. doi: 10.1074/jbc.271.12.6567. [DOI] [PubMed] [Google Scholar]
- 19.Mastrogiacomo A, Parsons SM, Zampighi GA, Jenden DJ, Umbach JA, Gundersen CB. Cysteine string proteins—a potential link between synaptic vesicles and presynaptic Ca2+ channels. Science. 1994;263:981–982. doi: 10.1126/science.7906056. [DOI] [PubMed] [Google Scholar]
- 20.Morales M, Ferrus A, Martinez PM. Presynaptic calcium-channel currents in normal and csp mutant Drosophila peptidergic terminals. Eur J Neurosci. 1999;11:1818–1826. doi: 10.1046/j.1460-9568.1999.00604.x. [DOI] [PubMed] [Google Scholar]
- 21.Msghina M, Millar AG, Charlton MP, Govind CK, Atwood HL. Calcium entry related to active zones and differences in transmitter release at phasic and tonic synapses. J Neurosci. 1999;19:8419–8434. doi: 10.1523/JNEUROSCI.19-19-08419.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nie Z, Ranjan R, Wenniger JJ, Hong SN, Bronk P, Zinsmaier KE. Overexpression of cysteine string protein in Drosophila reveals interaction with syntaxin. J Neurosci. 1999;19:10270–10279. doi: 10.1523/JNEUROSCI.19-23-10270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poage RE, Meriney SD, Gundersen CB, Umbach JA. Antibodies against cysteine string proteins inhibit evoked neurotransmitter release at Xenopus neuromuscular junctions. J Neurophysiol. 1999;82:50–59. doi: 10.1152/jn.1999.82.1.50. [DOI] [PubMed] [Google Scholar]
- 24.Pupier S, Leveque C, Marqueze B, Kataoka M, Takahashi M, Seagar MJ. Cysteine string proteins associated with secretory granules of the rat neurohypophysis. J Neurosci. 1997;17:2722–2727. doi: 10.1523/JNEUROSCI.17-08-02722.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ranjan R, Bronk P, Zinsmaier KE. Cysteine string protein is required for calcium secretion coupling of evoked neurotransmission in Drosophila but not for vesicle recycling. J Neurosci. 1998;18:956–964. doi: 10.1523/JNEUROSCI.18-03-00956.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravin R, Spira ME, Parnas H, Parnas I. Simultaneous measurement of intracellular Ca2+ and asynchronous transmitter release from the same crayfish bouton. J Physiol (Lond) 1997;501:251–262. doi: 10.1111/j.1469-7793.1997.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seagar M, Leveque C, Charvin N, Marqueze B, Martin MN, Boudier JA, Boudier JL, Shoji KY, Sato K, Takahashi M. Interactions between proteins implicated in exocytosis and voltage-gated calcium channels. Phil Trans R Soc London B Biol Sci. 1999;354:289–297. doi: 10.1098/rstb.1999.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheng ZH, Westenbroek RE, Catterall WA. Physical link and functional coupling of presynaptic calcium channels and the synaptic vesicle docking/fusion machinery. J Bioenerg Biomembr. 1998;30:335–345. doi: 10.1023/a:1021985521748. [DOI] [PubMed] [Google Scholar]
- 29.Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 30.Tank DW, Regehr WG, Delaney KR. A quantitative analysis of presynaptic calcium dynamics that contribute to short-term enhancement. J Neurosci. 1995;15:7940–7952. doi: 10.1523/JNEUROSCI.15-12-07940.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Umbach JA, Gundersen CB. Evidence that cysteine string proteins regulate an early step in the Ca2+-dependent secretion of neurotransmitter at Drosophila neuromuscular junctions. J Neurosci. 1997;17:7203–7209. doi: 10.1523/JNEUROSCI.17-19-07203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Umbach JA, Zinsmaier KE, Eberle KK, Buchner E, Benzer S, Gundersen CB. Presynaptic dysfunction in Drosophila csp mutants. Neuron. 1994;13:899–907. doi: 10.1016/0896-6273(94)90255-0. [DOI] [PubMed] [Google Scholar]
- 33.Umbach JA, Mastrogiacomo A, Gundersen CB. Cysteine string proteins and presynaptic function. J Physiol (Paris) 1995;89:95–101. doi: 10.1016/0928-4257(96)80556-0. [DOI] [PubMed] [Google Scholar]
- 34.Umbach JA, Saitoe M, Kidokoro Y, Gundersen CB. Attenuated influx of calcium ions at nerve endings of csp and shibire mutant Drosophila. J Neurosci. 1998;18:3233–3240. doi: 10.1523/JNEUROSCI.18-09-03233.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiser O, Trus M, Tobi D, Halevi S, Giladi E, Atlas D. The alpha 2/delta subunit of voltage sensitive Ca2+ channels is a single transmembrane extracellular protein which is involved in regulated secretion. FEBS Lett. 1996;379:15–20. doi: 10.1016/0014-5793(95)01475-6. [DOI] [PubMed] [Google Scholar]
- 36.Wu MN, Fergestad T, Lloyd TE, He Y, Broadie K, Bellen HJ. Syntaxin 1A interacts with multiple exocytic proteins to regulate neurotransmitter release in vivo. Neuron. 1999;23:593–605. doi: 10.1016/s0896-6273(00)80811-9. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Kelley WL, Chamberlain LH, Burgoyne RD, Wollheim CB, Lang J. Cysteine-string proteins regulate exocytosis of insulin independent from transmembrane ion fluxes. FEBS Lett. 1998;437:267–272. doi: 10.1016/s0014-5793(98)01233-2. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Kelley WL, Chamberlain LH, Burgoyne RD, Lang J. Mutational analysis of cysteine-string protein function in insulin exocytosis. J Cell Sci. 1999;112:1345–1351. doi: 10.1242/jcs.112.9.1345. [DOI] [PubMed] [Google Scholar]
- 39.Zinsmaier KE, Hofbauer A, Heimbeck G, Pflugfelder GO, Buchner S, Buchner E. A cysteine-string protein is expressed in retina and brain of Drosophila. J Neurogenet. 1990;7:15–29. doi: 10.3109/01677069009084150. [DOI] [PubMed] [Google Scholar]
- 40.Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]