

Abstract
Interactions between noradrenergic and cholinergic receptor signaling may be important in some forms of learning. To investigate whether noradrenergic and cholinergic receptor interactions regulate forms of synaptic plasticity thought to be involved in memory formation, we examined the effects of concurrent β-adrenergic and cholinergic receptor activation on the induction of long-term potentiation (LTP) in the hippocampal CA1 region. Low concentrations of the β-adrenergic receptor agonist isoproterenol (ISO) and the cholinergic receptor agonist carbachol had no effect on the induction of LTP by a brief train of 5 Hz stimulation when applied individually but dramatically facilitated LTP induction when coapplied. Although carbachol did not enhance ISO-induced increases in cAMP, coapplication of ISO and carbachol synergistically activated p42 mitogen-activated protein kinase (p42 MAPK). This suggests that concurrent β-adrenergic and cholinergic receptor activation enhances LTP induction by activating MAPK and not by additive or synergistic effects on adenylyl cyclase. Consistent with this, blocking MAPK activation with MEK inhibitors suppressed the facilitation of LTP induction produced by concurrent β-adrenergic and cholinergic receptor activation. Although MEK inhibitors also suppressed the induction of LTP by a stronger 5 Hz stimulation protocol that induced LTP in the absence of ISO and carbachol, they had no effect on LTP induced by high-frequency synaptic stimulation or low-frequency synaptic stimulation paired with postsynaptic depolarization. Our results indicate that MAPK activation has an important, modulatory role in the induction of LTP and suggest that coactivation of noradrenergic and cholinergic receptors regulates LTP induction via convergent effects on MAPK.
Keywords: long-term potentiation, β-adrenergic receptors, cholinergic receptors, adenylyl cyclase, mitogen-activated protein kinase, hippocampus
Numerous behavioral studies indicate that noradrenergic and cholinergic systems in the brain are involved in learning and memory (for review, see McGaugh, 1989; Blokland, 1996;Jerusalinsky et al., 1997) and that interactions between these two neurotransmitters may be important for some forms of learning (Decker et al., 1990; Decker and McGaugh, 1991; Ohno et al., 1997). Although norepinephrine and acetylcholine potentially could modulate memory formation via multiple effects occurring at different levels of CNS function, one possibility is that they act and interact at the cellular level to regulate activity-dependent changes in synaptic strength, such as long-term potentiation (LTP), that are thought to be involved in the storage of new information during learning (Izquierdo and Medina, 1995). Indeed, whereas the induction of LTP at excitatory synapses in the CA1 region of the hippocampus is primarily dependent on activation of NMDA-type glutamate receptors (Bliss and Collingridge, 1993), noradrenergic (Sah and Bekkers, 1996; Thomas et al., 1996;Katsuki et al., 1997; Izumi and Zorumski, 1999) and cholinergic (Blitzer et al., 1990; Huerta and Lisman, 1993) receptor activation strongly enhances the induction of LTP by some patterns of synaptic stimulation. Moreover, coactivation of these receptors synergistically enhances the induction of LTP in rat visual cortex (Brocher et al., 1992).
The cellular mechanisms responsible for the effects of concurrent noradrenergic and cholinergic receptor action on LTP induction are mainly unknown. One possibility is that coactivation of noradrenergic and cholinergic receptors modulates LTP induction via synergistic or additive effects on neuronal excitability that enhance NMDA receptor activation during synaptic activity (Brocher et al., 1992). Alternatively, coactivation of these receptors might influence LTP induction by modulating components of the LTP pathway that lie downstream of NMDA receptor activation. For instance, the induction of LTP by some patterns of synaptic stimulation requires a protein kinase A (PKA)-dependent suppression of protein phosphatases (Blitzer et al., 1995, 1998; Thomas et al., 1996), and coactivation of noradrenergic and muscarinic receptors thus might enhance LTP induction via synergistic or additive effects on adenylyl cyclase activity (Baumgold, 1992). Noradrenergic and cholinergic receptor agonists also activate the mitogen-activated protein kinase (MAPK) signaling cascade in hippocampal neurons (Roberson et al., 1999), whereas inhibitors of MAPK activation suppress LTP induction (English and Sweatt, 1997; Atkins et al., 1998; Coogan et al., 1999; Winder et al., 1999; Rosenblum et al., 2000). Thus, another possibility is that noradrenergic and cholinergic receptor signaling pathways modulate LTP induction via effects on MAPK.
To investigate the potential role of adenylyl cyclase and MAPK signaling in the noradrenergic and cholinergic modulation of LTP induction, we examined the effects of β-adrenergic and cholinergic receptor agonists on cAMP levels, MAPK activation, and LTP induction in the hippocampal CA1 region. Our results show that coactivation of β-adrenergic and cholinergic receptors facilitates the induction of LTP in a highly cooperative manner and suggest that coactivation of noradrenergic and cholinergic receptors regulates LTP induction via a synergistic activation of MAPK.
MATERIALS AND METHODS
Slice preparation and electrophysiology. Hippocampal slices used in both electrophysiological and biochemical experiments were obtained from 4- to 7-week-old male C57BL/6 mice. Animals were anesthetized with halothane and killed by cervical dislocation; then the brains were removed rapidly and placed in cold (4°C) artificial CSF (aCSF) consisting of (in mm) 124NaCl, 4.4 KCl, 25 NaHCO3, 1.0 NaH2PO4, 1.2 MgSO4, 2.0 CaCl2, and 10 glucose (gassed with 95% O2/5% CO2). Transverse slices (400 μm thick) were prepared by using standard techniques and then were transferred to an interface-type recording chamber (Fine Science Tools, Foster City, CA) where they were perfused continuously (1–3 ml/min) with warm (30°C) aCSF for at least 1 hr before electrophysiological experiments. Longer recovery times were used in the biochemical experiments (see below).
A bipolar, nichrome wire stimulating electrode placed in stratum radiatum of the CA1 region of the slice was used to activate Schaffer collateral/commissural fiber synapses onto CA1 pyramidal cells. In extracellular recording experiments the field EPSPs (fEPSPs) evoked by 0.02 Hz presynaptic fiber stimulation were recorded by using an aCSF-filled glass microelectrode placed in stratum radiatum (electrode resistance ranged from 5 to 10 MΩ). At the start of an experiment we determined the maximal fEPSP amplitude that could be generated in each slice and then set the intensity of presynaptic fiber stimulation to evoke baseline fEPSPs that were ∼50% of the maximal fEPSP amplitude.
In some experiments whole-cell current-clamp recordings were used to record EPSPs from CA1 pyramidal cells in slices maintained in submerged slice recording chambers. Slices (with the CA3 region removed) were bathed in a modified aCSF containing elevated levels of CaCl2 and MgSO4 (4 mm each), reduced levels of KCl (2.2 mm), and 100 μm picrotoxin. Patch-clamp electrodes were fabricated from 1.5 mm outer diameter borosilicate glass and filled with a solution containing (in mm) 122.5 Cs-gluconate, 0.9 CsCl, 10 tetraethylammonium chloride (TEA-Cl), 0.2 EGTA, 2 Mg-ATP, 0.3 GTP, 10 HEPES, and 0.2% dimethylsulfoxide (DMSO), pH 7.2 (osmolarity, 290–295 mOsm; electrode resistance, 2.5–5.0 MΩ). A modified electrode-filling solution containing an elevated concentration of CsCl (17.5 mm) and no DMSO was used in experiments in which MEK inhibitors were bath-applied. Current injected through the recording electrode was used to hyperpolarize cells to between −80 and −85 mV, and 50-msec-long pulses of hyperpolarizing current (100 pA) were delivered once every 20 sec to monitor access and input resistance. Only cells with resting membrane potentials more negative than −60 mV and an input resistance >140 MΩ were used. Access resistances, determined from the bridge balance circuit of the intracellular amplifiers (Dagan IX2–700, Axon Instruments Axoprobe 1A, Foster City, CA), ranged from 15 to 34 MΩ. At the start of each experiment the intensity of presynaptic stimulation was adjusted to evoke postsynaptic EPSPs between 4 and 7 mV in amplitude (stimulation rate, 0.05 Hz). To induce LTP, we paired 100 EPSPs evoked at 2 Hz with a constant injection of current through the recording electrode to depolarize the postsynaptic membrane potential to between 0 and +20 mV. Whole-cell recordings were maintained for 17–20 min before we attempted to induce LTP.
Data acquisition and analysis were performed with programs written with the Experimenter's Workbench and Common Processing software package (Data Wave Technologies). All values are reported as mean ± SEM. Synaptic responses were normalized to average values measured over a baseline period that was recorded before any experimental manipulation. The average size of EPSPs (obtained from measurements of the slope) recorded between 40 and 45 min after 5 Hz stimulation (in extracellular recordings) or 25–30 min after pairing EPSPs with low-frequency stimulation (in intracellular recordings) was used for statistical comparisons (paired and unpaired Student's t tests or ANOVAs, followed by Dunnett's test comparisons with control). Complex spike bursting during 5 Hz stimulation was determined by visually inspecting each synaptic response during the 5 Hz train and counting the number of negative spikes evoked by each EPSP (see Thomas et al., 1998).
cAMP assay. Hippocampal slices from the same animal were maintained in the same interface chambers as those used for electrophysiological experiments and were allowed to recover for at least 2 hr. Slices (two at a time) then either were left untreated (control slices) or were exposed to a 10 min bath application of 200 nm isoproterenol (ISO), 200 nm carbachol, or ISO plus carbachol (200 nm each). At the end of each treatment the slices were removed rapidly from the chamber and immediately homogenized in ice-cold 3% perchloric acid (PCA). The pH of the samples was neutralized with 15% KHCO3and, after centrifugation (5 min at 16,000 × g) the supernatants were assayed for cAMP by using a commercially available kit (Diagnostic Products, Los Angeles, CA). cAMP levels were normalized to the amount of protein in the PCA pellet that was determined with a modified Lowry assay (Sigma, St. Louis, MO) (Peterson, 1977).
Protein preparations and Western immunoblotting analysis. To examine the effects of β-adrenergic and cholinergic receptor activation on MAPK activation, we allowed hippocampal slices from the same animal to recover for 3–4 hr and then either left them untreated (control slices) or exposed them to 10 min bath applications of ISO alone, carbachol alone, or ISO plus carbachol. Then the slices (two to three in each treatment condition) were frozen quickly on a glass plate on dry ice, and the CA1 regions were isolated by microdissection. The isolated CA1 regions were transferred to a microcentrifuge tube kept in a dry ice/ethanol bath and subsequently were homogenized by sonication (Micron Ultrasonic Cell Disruptor) in 90 μl of an ice-cold lysis buffer containing (in mm) 50 Tris-HCl, pH 7.5, 50 NaCl, 10 EGTA, 10 EDTA, 5 sodium pyrophosphate, 1 sodium orthovanadate, 4para-nitrophenyl phosphate, and 1 phenylmethylsulfonyl fluoride plus 20 μg/ml of leupeptin, 4 μg/ml of aprotinin, and 0.01% Triton X-100. After centrifugation at 12,000 ×g (for 5 min at 4°C) aliquots of the supernatant were saved for protein determination, and denaturing loading buffer [0.5m Tris-HCl, pH 6.8, 4.4% (w/v) SDS, 20% (v/v) glycerol, 2% β-mercaptoethanol, and bromophenol blue] was added immediately to the rest of the sample. Protein concentrations were determined by using a Bio-Rad Protein Assay Kit (Hercules, CA) based on the Bradford method (Bradford, 1976).
Samples containing equivalent amounts of protein (15–40 μg) were boiled for 3 min, separated on 12% SDS-PAGE gels, transferred onto nitrocellulose membranes, and blocked for 3 hr in 5% dry milk in PBS with 0.05% Tween-20 (1 μm microcystin LR was also included for phospho-MAPK blots). Then the blots were incubated overnight with an antiserum that specifically recognizes the Thr202 and Tyr204 dually phosphorylated, activated forms of p42/p44 MAPK (1:2000; New England Biolabs, Beverly, MA). Another antibody that recognizes both phosphorylated and unphosphorylated forms of p42/p44 MAPK (1:1000; New England Biolabs) was used to measure total MAPK levels. After three washes (for 10 min each) with PBS containing 0.05% Tween-20, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies for 2 hr. Protein signals were visualized with enhanced chemiluminescence (ECL Western Blotting Analysis system, Amersham, Arlington Heights, IL) and quantified with a Molecular Dynamics Personal Densitometer SI with ImageQuaNT software (Molecular Dynamics, Sunnyvale, CA). Digital resolution was set at 12 bits per pixel (50 μm pixel size), and areas from a single experiment (untreated control slices plus slices treated with ISO, carbachol, and ISO plus carbachol; all slices from the same animal) were scanned as a single data set. Western blots were developed to be in a linear range for densitometry. For each experiment both total MAPK and phospho-MAPK levels were normalized relative to that seen in untreated, control slices. ANOVAs followed by Dunnett's test comparisons to untreated control groups were used to determine statistical significance in all biochemical assays.
Materials. Salts in the aCSF were from Sigma.R(−)-isoproterenol (+)-bitartrate, carbachol, and atropine were purchased from Research Biochemicals (RBI, Natick, MA). The MEK inhibitors U0126 and PD98059 were obtained from Promega (Madison, WI) and New England Biolabs, respectively. SL327 was a generous gift from DuPont Pharmaceuticals (Wilmington, DE). All three MEK inhibitors were prepared as concentrated stock solutions in DMSO and then diluted into aCSF before each experiment (maximal final DMSO concentration, 0.33%). Slices were pretreated with MEK inhibitors for at least 60 min before we attempted to induce LTP, and the inhibitors remained in the bath for the duration of the experiment. In experiments investigating the effects of U0126 and PD98059, aCSF containing the inhibitors (117–133 ml total volume) was recycled through the recording chambers (no more than twice) during the course of an experiment. Interleaved vehicle control experiments (0.2% and 0.33% DMSO) were performed in the same manner.
RESULTS
Coactivation of cholinergic and β-adrenergic receptors enables the induction of LTP by short trains of 5 Hz stimulation
To examine the potential role of β-adrenergic and cholinergic receptor interactions in the induction of LTP, we examined the effects of ISO and carbachol on the induction of LTP by a 5-sec-long train of 5 Hz stimulation that by itself induced only a small potentiation of synaptic transmission (Fig.1A). The amount of LTP induced by 5 Hz stimulation was not enhanced when 5 Hz stimulation was delivered at the end of a 10 min bath application of either 200 nm ISO or 200 nm carbachol alone (Fig. 1B), indicating that the induction of LTP is not modulated significantly by relatively low levels of β-adrenergic or cholinergic receptor activation. In contrast, the induction of LTP by 5 Hz stimulation was facilitated dramatically when β-adrenergic and cholinergic receptors were coactivated by a 10 min bath application of aCSF containing both ISO and carbachol (200 nm each; Fig. 1A,B). In control experiments we found that a 10 min bath application of ISO and carbachol by itself (no 5 Hz stimulation) had no lasting effect on synaptic transmission (n = 3; data not shown). Thus, the potentiation induced by 5 Hz stimulation in the presence of ISO plus carbachol is not attributable to an activity-independent enhancement of synaptic transmission induced by coactivation of β-adrenergic and cholinergic receptors. The induction of LTP by 5 Hz stimulation in the presence of ISO plus carbachol was blocked significantly, however, by the NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (d,l-APV, 100 μm; fEPSPs were 114.4 ± 10.8% of baseline after 5 Hz stimulation; n = 5), suggesting that coactivation of β-adrenergic and cholinergic receptors facilitates the induction of NMDA receptor-dependent LTP. Finally, the muscarinic receptor antagonist atropine (50 μm) completely blocked the enhancement of LTP induction by coapplication of ISO plus carbachol (n = 3; data not shown), indicating that the effects of carbachol are attributable to muscarinic receptor activation.
Fig. 1.
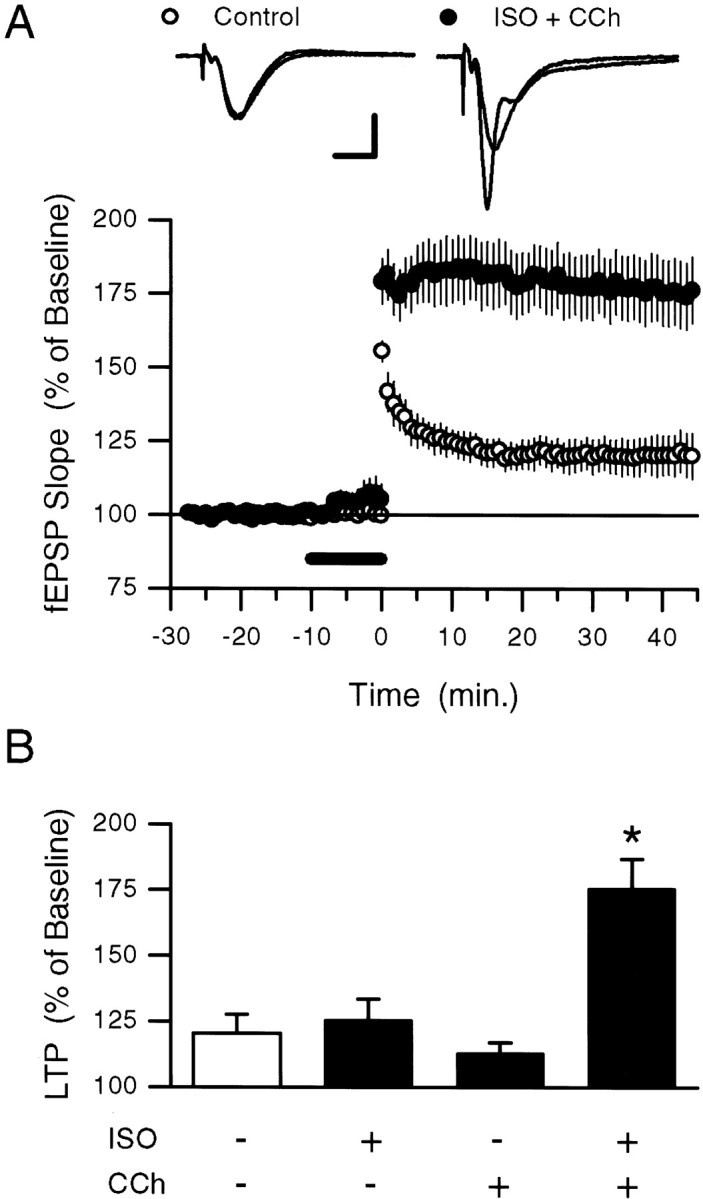
Coactivation of β-adrenergic and cholinergic receptors enhances the induction of LTP by a short train of 5 Hz stimulation. A, A 5 sec train of 5 Hz stimulation (delivered at time 0) by itself induced only a small potentiation of synaptic transmission (open symbols; fEPSPs potentiated to 120.5 ± 7.12% of baseline;n = 6; p < 0.05, pairedt test comparison to pre-5 Hz baseline) but induced robust LTP (filled symbols; fEPSPs potentiated to 175.6 ± 11.2% of baseline; n = 6) when delivered at the end of a 10 min bath application of ISO plus carbachol (CCh) (200 nm each; presence in bath indicated by the bar). Traces show fEPSPs recorded during baseline and 45 min after 5 Hz stimulation in a control experiment (5 Hz stimulation alone, lefttraces) and in an experiment in which 5 Hz stimulation was delivered in the presence of ISO plus carbachol (right traces). Calibration: 2 mV, 5 msec. B, Summary of the effects of 5 Hz stimulation on synaptic strength when 5 Hz stimulation was delivered alone (open bars; data from the experiment shown in A) or at the end of a 10 min application of ISO (n = 6), carbachol (n = 6), or ISO plus carbachol (data from the experiment shown in A). Although coapplication of ISO plus carbachol significantly enhances the amount of LTP induced by 5 Hz stimulation (*p < 0.05), neither ISO alone nor carbachol alone significantly enhances LTP induction (fEPSPs were potentiated to 125.4 ± 7.9% and 112.9 ± 4.1% of baseline, respectively).
Carbachol modulation of ISO-induced increases in cAMP does not account for the enhancement of LTP induction by coactivation of β-adrenergic and cholinergic receptors
Although a low concentration of ISO alone failed to enhance the induction of LTP by 5 sec of the 5 Hz stimulation, we found that the amount of potentiation induced by 5 Hz stimulation was increased significantly by a higher concentration of ISO (1.0 μm;n = 7; data not shown). This suggests that stronger activation of Gs-linked β-adrenergic receptors alone, and by extension larger increases in cellular levels of cAMP, can enhance the induction of LTP by a brief train of 5 Hz stimulation. The diverse regulation of different adenylyl cyclase isoforms by increases in intracellular Ca2+, G-protein βγ subunits, and protein kinase C phosphorylation as well as αs subunits of heterotrimeric G-proteins (for review, see Cooper et al., 1995; Mons and Cooper, 1995) provides a number of potential signaling pathways through which coactivation of Gs-coupled β-adrenergic and Gq/Gi/o-coupled muscarinic receptors might activate adenylyl cyclase synergistically (Baumgold, 1992). We thus investigated whether coapplication of ISO and carbachol might facilitate the induction of LTP by 5 Hz stimulation via additive or synergistic effects on adenylyl cyclase activity. In these experiments we measured cAMP levels in hippocampal slices that either were untreated or were exposed to 10 min bath applications of 200 nm ISO alone, 200 nmcarbachol alone, or a combination of ISO plus carbachol (Fig.2). Compared with basal levels measured in untreated control slices, cAMP levels were increased significantly in slices exposed to ISO (levels were 159 ± 17.1% of that measured in control slices; n = 5). In contrast, cAMP levels were not elevated significantly in slices treated with carbachol alone (cAMP levels were 106 ± 9.1% of control; n= 5). Carbachol also had no significant effect on ISO-induced increases in cAMP (cAMP levels in slices simultaneously exposed to both carbachol and ISO were 152.6 ± 19.9% of control; n = 5). In control experiments we found that a 10 min application of the adenylyl cyclase activator forskolin (50 μm) induced a more than sevenfold increase in cAMP levels (cAMP levels in forskolin-treated slices were 799 ± 39.2% of control;n = 4). This indicates that 200 nm ISO does not induce a saturating level of adenylyl cyclase activation that masks a modulatory effect of carbachol on ISO-induced increases in cAMP. On the basis of these observations it seems unlikely that coactivation of cholinergic and β-adrenergic receptors facilitates the induction of LTP by 5 Hz stimulation via additive or synergistic effects on cAMP production.
Fig. 2.

Carbachol does not modulate ISO-induced increases in cAMP. The histogram shows average ± SEM cAMP levels (in picomoles per milligram of protein) from five separate experiments in which hippocampal slices (from the same animal) either were untreated (open bar) or were exposed to a 10 min bath application of 200 nm ISO alone, 200 nm carbachol (CCh) alone, or ISO plus carbachol (filled bars). Compared with basal levels of cAMP seen in untreated control slices, only ISO alone and ISO plus carbachol induced significant increases in cAMP levels (*p < 0.05). In four of these experiments a pair of slices also was exposed to a 10 min bath application of 50 μm forskolin. In these slices cAMP levels were increased to 92.5 ± 13.1 pmol/mg of protein.
MEK inhibitors suppress 5 Hz stimulation-induced LTP
Numerous studies have demonstrated that a variety of neurotransmitters can activate signaling cascades that lead to MAPK activation in neurons (Bading and Greenberg, 1991; Kurino et al., 1995;Baron et al., 1996; English and Sweatt, 1996; Fukunaga and Miyamoto, 1998; Otani et al., 1999; Roberson et al., 1999; Winder et al., 1999;Rosenblum et al., 2000). Indeed, Roberson et al. (1999) recently have shown that, in the hippocampus, both muscarinic and β-adrenergic receptors are linked to the MAPK signaling cascade (see also Winder et al., 1999; Rosenblum et al., 2000). This, along with the results from studies showing that MAPK activity is required for the induction of NMDA receptor-dependent LTP by high-frequency stimulation (English and Sweatt, 1997; Atkins et al., 1998; Coogan et al., 1999, Jones et al., 1999; Rosenblum et al., 2000), led us to investigate whether MAPK activation has an important role in either the induction of 5 Hz stimulation-induced LTP or its modulation by concurrent β-adrenergic and cholinergic receptor activation. In these experiments we first examined the effects of MEK inhibitors on the induction of LTP by a longer-duration 5 Hz stimulation train (30 sec) that induces robust levels of LTP even in the absence of ISO and carbachol (Thomas et al., 1996, 1998). Although 30 sec of 5 Hz stimulation induced significant LTP in vehicle control-treated slices (0.33% DMSO), it had little effect on synaptic strength in slices incubated in aCSF containing the MEK inhibitor PD98059 (50 μm) for 60 min before the 5 Hz stimulation (Fig. 3B). Although this suggests that MAPK activation is required for the induction of LTP by 5 Hz stimulation, PD98059 not only prevents high-frequency stimulation-induced activation of MAPK but also can inhibit activation of the Ca2+/calmodulin-dependent kinase CaMKII in hippocampal slices (Liu et al., 1999). Thus, some of the effects of PD98059 on the induction of LTP might be attributable to indirect effects on CaMKII activation. We therefore also examined the effects of two additional MEK inhibitors, U0126 (Favata et al., 1998) and SL327, which have been shown to block MAPK activation potently at concentrations that have little effect on PKA, protein kinase C (PKC), and CaMKII activity (Atkins et al., 1998; Roberson et al., 1999). As shown in Figure 3, A and B, both U0126 (20 μm) and SL327 (10 μm) inhibited the induction of LTP by a 30-sec-long train of 5 Hz stimulation. Consistent with the recent report by Winder et al. (1999), we found that the same concentration of U0126 that inhibited 5 Hz stimulation-induced LTP had no significant effect on the amount of LTP present 60 min after high-frequency stimulation (Fig. 3C), suggesting that MAPK activation may be particularly important for the induction of LTP by low-frequency synaptic stimulation.
Fig. 3.

MEK inhibitors suppress low-frequency, but not high-frequency, stimulation-induced LTP. A, The amount of LTP induced by 30 sec of 5 Hz stimulation (delivered at time0) in slices continuously bathed in 20 μmU0126 (filledsymbols; fEPSPs were 122.2 ± 5.1% of baseline; n = 6) was reduced significantly (p < 0.01) as compared with that seen in interleaved vehicle control experiments (open symbols; 0.2% DMSO; fEPSPs were potentiated to 167.7 ± 9.9% of baseline; n = 5). B, Summary of the effects of three different MEK inhibitors on 5 Hz stimulation-induced LTP. Values show the amount of LTP present 40–45 min after 5 Hz stimulation. Similar amounts of LTP were induced by 5 Hz stimulation in interleaved vehicle control experiments (0.1–0.33% DMSO; n = 17), and the combined results are shown by the open bar. Results for U0126 are from the same experiments shown in A. fEPSPs in PD98059 (PD)-treated slices were 109.9 ± 9% of baseline (n = 5) after 5 Hz stimulation (**p < 0.01 compared with 0.33% DMSO control experiments), whereas fEPSPs were 118.4 ± 13.4% of baseline (n = 7) after 5 Hz stimulation in SL327 (SL)-treated slices (*p < 0.05 compared with 0.1% DMSO control experiments). In interleaved control experiments the fEPSPs were 155.1 ± 5.9% of baseline (0.33% DMSO; n = 5) and 165.1 ± 17.4% of baseline (0.1% DMSO; n = 7), respectively.C, High-frequency stimulation-induced LTP is not inhibited by U0126. Two 1-sec-long trains of 100 Hz stimulation (intertrain interval, 10 sec; delivered at time 0) induced similar amounts of LTP in vehicle control experiments (0.2% DMSO; open symbols; n = 6) and in slices continuously bathed in 20 μm U0126 (filled symbols; n = 5). In control experiments 55–60 min after 100 Hz stimulation the fEPSPs were potentiated to 185.9 ± 20.5% of baseline, whereas in U0126-treated slices the fEPSPs were 188.7 ± 43.2% of baseline.D, Summary of the effects of MEK inhibitors on 5 Hz stimulation-induced complex spike bursting. The plot shows the percentage of EPSPs during the 5 Hz stimulation train that evoked complex spike bursts (CSB, defined as two or more negative-going spikes after EPSP onset; see Thomas et al., 1998) in vehicle control experiments (left) and in the presence of MEK inhibitors (right). Measurements were taken from the experiments shown in B. Although complex spike bursting tended to be reduced in slices pretreated with MEK inhibitors, none of the inhibitors produced a statistically significant suppression of complex spike bursting when compared with interleaved vehicle control experiments (PD98059 vs 0.33% DMSO controls:t(8) = 1.78, p = 0.11; U0126 vs 0.2% DMSO controls:t(9) = 1.16, p = 0.28; SL327 vs 0.1% DMSO controls:t(11) = 1.12, p = 0.29).
How might MAPK activation be involved in the induction of LTP by low-frequency stimulation? Unlike high-frequency stimulation, the induction of LTP by 5 Hz trains of synaptic stimulation in the hippocampal CA1 region requires EPSP-evoked, complex spike-like bursts of postsynaptic action potentials (Thomas et al., 1998). Recently,Winder and colleagues (1999) found that both PD98059 and U0126 suppress complex spike bursting during 5 Hz stimulation, suggesting that MEK inhibitors may inhibit 5 Hz stimulation-induced LTP by suppressing the EPSP-evoked complex spike bursting required for LTP induction by this protocol. Although we did observe a tendency for EPSP-evoked complex spike bursting to be reduced in PD98059- and SL327-treated slices, none of the MEK inhibitors used in our experiments produced a statistically significant inhibition of EPSP-evoked complex spike bursting during 5 Hz stimulation (Fig. 3D). Although this suggests that a suppression of complex spike bursting is unlikely to account for the effects of MEK inhibitors on 5 Hz stimulation-induced LTP, extracellular recordings of complex spike bursting may lack the resolution needed to detect modest changes in excitability that still could influence LTP induction substantially. Moreover, effects of MEK inhibitors on excitability that are near threshold for detection in extracellular recordings also might be more or less obvious depending on experimental conditions and thus also could account for the different effects of MEK inhibitors on complex spike bursting observed in our experiments and those of Winder et al. (1999). We thus examined whether blocking MAPK activation inhibits LTP under conditions in which changes in postsynaptic excitability should have little effect on LTP induction. In these experiments we used whole-cell current-clamp techniques to record EPSPs from individual CA1 pyramidal cells and applied SL327 either in the bath or selectively to the postsynaptic CA1 pyramidal cells via the recording electrode. To minimize potential effects of SL327 on LTP induction that might arise from changes in postsynaptic excitability, we used a cesium-based intracellular solution containing TEA to block potassium conductances and induced LTP by pairing low-frequency presynaptic stimulation with depolarization of the postsynaptic cell to near 0 mV (see Materials and Methods). Although bath-applied SL327 inhibited the induction of LTP by 5 Hz stimulation, neither bath application nor postsynaptic injection of SL327 inhibited pairing-induced LTP (Fig.4). Because SL327 does not inhibit the induction of LTP by a low-frequency stimulation protocol under conditions designed to minimize potential effects on LTP induction attributable to changes in neuronal excitability, our results suggest that MEK inhibitors may suppress the induction of LTP by low-frequency stimulation-induced complex spike bursting via effects on membrane excitability that reduce NMDA receptor activation.
Fig. 4.
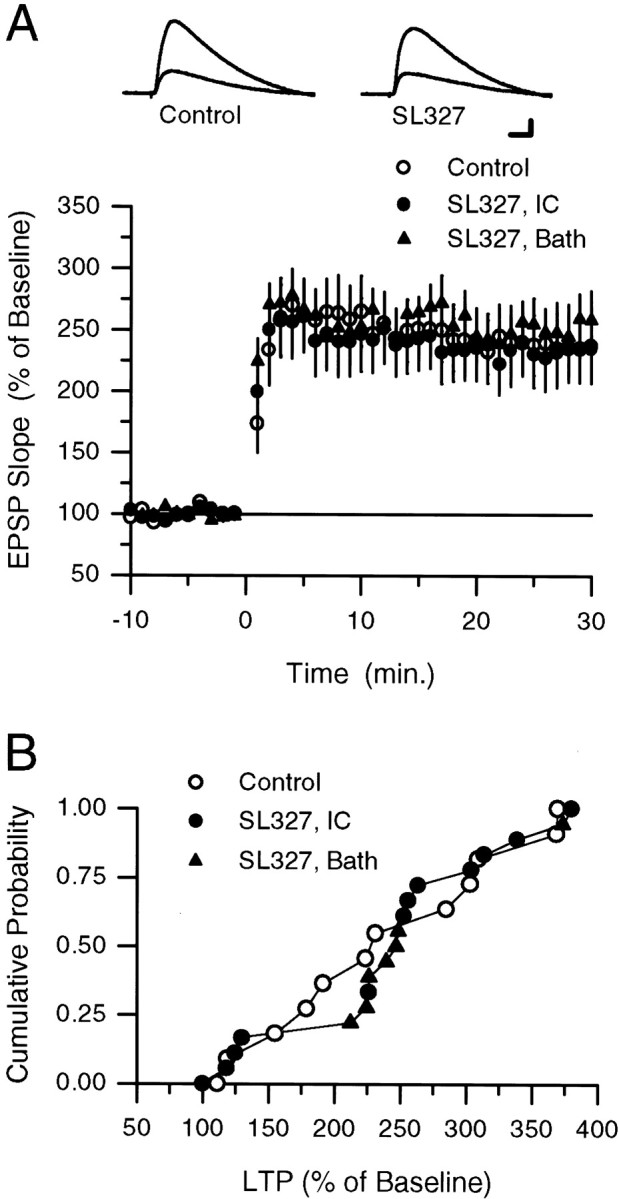
SL327 does not inhibit the induction of LTP by low-frequency synaptic stimulation paired with postsynaptic depolarization. A, One hundred EPSPs (evoked at 2 Hz) were paired with tonic postsynaptic depolarization to between 0 and +20 mV at time 0 in vehicle control experiments (open circles; 0.2% DMSO in electrode-filling solution;n = 12) and in experiments in which SL327 was present either in the bath (filled circles; 10 μm, 0.1% DMSO; n = 7) or in the recording electrode-filling solution (filled triangles; 100 μm, 0.2% DMSO;n = 12). The 25–30 min postpairing EPSPs were potentiated to 241.5 ± 26.9% of baseline in control experiments, 255.5 ± 28.7% of baseline in experiments in which SL327 was bath-applied, and 234.1 ± 27.5% of baseline in experiments in which SL327 was injected into CA1 pyramidal cells via the recording electrode. The inset shows EPSPs (average of three responses) recorded during baseline and 30 min postpairing (larger response) in a control experiment (lefttraces) and in an experiment in which 100 μm SL327 was present in the recording electrode (righttraces). Calibration: 5 mV, 15 msec. B, Cumulative probability distribution showing results from all of the experiments depicted in A. Results from experiments in which SL327 was bath-applied (filled circles) and applied via the recording electrode (filled triangles) have been combined into one distribution.
Coactivation of β-adrenergic and cholinergic receptors synergistically activates p42 MAPK
Because our experiments with MEK inhibitors suggested that MAPK activation has a role in the induction of LTP by low-frequency stimulation protocols, we investigated whether concurrent activation of β-adrenergic and cholinergic receptors enhances the induction of LTP by a brief train of 5 Hz stimulation by activating MAPK. First, this hypothesis predicts that the potentiation induced by 5 sec of 5 Hz stimulation in the presence of ISO and carbachol should be suppressed by inhibitors of MAPK activation. Consistent with this, U0126 had little effect on the change in synaptic strength induced by 5 Hz stimulation alone (fEPSPs were 116.0 ± 8.8% of baseline 45 min after 5 Hz stimulation alone in U0126-treated slices; n= 6) but significantly inhibited the induction of LTP by 5 Hz stimulation in the presence of ISO and carbachol (Fig.5A). The MEK inhibitor PD98059 (50 μm) also significantly (p < 0.05) suppressed the induction of LTP by 5 Hz stimulation in ISO plus carbachol (fEPSPs were potentiated to 110.9 ± 7.2% of baseline, n = 4, compared with 163.9 ± 9.9% of baseline in DMSO control experiments,n = 5). Second, this hypothesis indicates that ISO and carbachol should have at least additive effects on MAPK activation in hippocampal slices. To examine this possibility, we used Western blotting analysis with antibodies that specifically recognize dually phosphorylated p42/p44 MAPK to examine levels of MAPK activation in hippocampal slices exposed to a 10 min bath application of either ISO alone, carbachol alone, or ISO plus carbachol. At the low concentrations of ISO and carbachol used in our experiments, neither ISO nor carbachol alone produced a significant increase in phospho-MAPK levels in seven separate experiments (Fig. 5B1,B2). In contrast, levels of dually phosphorylated p42-MAPK were increased >10-fold, whereas total MAPK levels were not changed significantly, in slices in which ISO and carbachol were coapplied (Fig.5B1,B2).
Fig. 5.

MAPK activation may underlie the effects of ISO plus carbachol on 5 Hz stimulation-induced LTP. A, U0126 inhibits the induction of LTP by 5 sec of 5 Hz stimulation delivered during the coapplication of ISO and carbachol. A 5 sec train of 5 Hz stimulation delivered at the end of a 10 min bath application of ISO plus carbachol (200 nm each; the presence of agonists in the bath indicated by the bar) induced robust LTP in vehicle (0.2% DMSO) control experiments (open symbols; fEPSPs were potentiated to 163.9 ± 6.8% of baseline;n = 5) but had little effect on synaptic strength in slices continuously bathed in 20 μm U0126 (filled symbols; fEPSPs were 112.8 ± 7.3% of baseline; n = 5). The traces show superimposed fEPSPs recorded during baseline and 45 min post-5 Hz stimulation in a control experiment (left) and in a slice bathed in U0126 (right). Calibration: 1 mV, 5 msec. B, Synergistic activation of MAPK by coactivation of β-adrenergic and cholinergic receptor agonists. B1, Representative Western immunoblots showing protein bands visualized with antibodies to dually phosphorylated p42/44 MAPK (PP) and total p42/44 MAPK (Total) in control, untreated slices (Con), and slices bathed for 10 min in aCSF containing 200 nm ISO, 200 nm carbachol (CCh), or ISO plus carbachol (ISO + CCh). B2, Average results ± SEM from seven separate experiments like that shown inB1. Only coapplication of ISO plus carbachol induced a statistically significant (*p < 0.05) increase in phospho-p42 MAPK levels.
DISCUSSION
Coactivation of β-adrenergic and cholinergic receptors has synergistic effects on both LTP induction and MAPK activation in the CA1 region of the hippocampus
Behavioral studies indicate that interactions between noradrenergic and cholinergic receptor signaling have an important role in learning and memory (Decker and Gallagher, 1987; McGaugh, 1989;Decker et al., 1990; Harrell et al., 1990; Riekkinen et al., 1990;Decker and McGaugh, 1991; Ohno et al., 1993, 1997). Although the behavioral expression of these interactions potentially could arise from effects of norepinephrine and acetylcholine at multiple levels of CNS function, one possibility is that these transmitters interact at the cellular level to regulate activity-dependent changes in synaptic strength involved in the storage of new information during learning. We thus examined whether coactivation of cholinergic and β-adrenergic receptors modulates LTP induction in the hippocampal CA1 region. Our results show that, whereas low concentrations of carbachol and ISO by themselves have little effect on LTP, coapplication of carbachol and ISO dramatically facilitates the induction of LTP by low-frequency trains of synaptic stimulation. This effect does not appear to arise from synergistic or additive effects on adenylyl cyclase activity, because cAMP levels in slices exposed to both ISO and carbachol were not different from those seen in slices exposed to ISO alone. Instead, coapplication of ISO and carbachol synergistically activated p42 MAPK, suggesting that the MAPK signaling pathway may have an important role in the modulation of 5 Hz stimulation-induced LTP by concurrent β-adrenergic and cholinergic receptor activation.
MAPK signaling is required for the induction of LTP by 5 Hz trains of synaptic stimulation
Emerging evidence indicates that the MAPK signaling cascade has an important role in learning (Atkins et al., 1998; Berman et al., 1998;Crow et al., 1998; Blum et al., 1999) and in forms of synaptic plasticity, such as LTP, that are thought to be involved in learning (English and Sweatt, 1997; Rosenblum et al., 2000) (see also Martin et al., 1997; Atkins et al., 1998; Coogan et al., 1999; Winder et al., 1999). Although many of the molecular details of the role of MAPK in synaptic plasticity are unknown, it seems likely that MAPK is involved in multiple aspects of synaptic plasticity. First, MAPK appears to contribute to the CREB-dependent activation of gene expression required for the long-lasting, protein synthesis-dependent stages of long-term memory and synaptic potentiation in both the hippocampus (Impey et al., 1998) and in Aplysia (Martin et al., 1997). Second, recent findings suggest that MAPK also acts on cytoplasmic targets required for the induction and/or early stages of LTP maintenance (English and Sweatt, 1997; Atkins et al., 1998; Coogan et al., 1999; Jones et al., 1999; Winder et al., 1999; Rosenblum et al., 2000). Consistent with this, we found that three different inhibitors of MAPK activation strongly suppressed LTP induced by 5 Hz trains of synaptic stimulation. Although our results do not rule out a role for MAPK in the maintenance phase of LTP, the effects of MEK inhibitors on 5 Hz stimulation-induced LTP were evident almost immediately after 5 Hz stimulation, suggesting that inhibiting MAPK activation interferes with LTP induction (see also English and Sweatt, 1997).
Although all three MEK inhibitors used in our experiments strongly suppressed the induction of LTP by 5 Hz stimulation, U0126 and SL327 had no effect on high-frequency stimulation-induced and pairing-induced LTP, respectively. Unlike high-frequency stimulation-induced LTP, the induction of LTP by trains of 5 Hz stimulation requires EPSP-evoked complex spike-like bursts of postsynaptic action potentials to provide the depolarization needed for NMDA receptor activation (Thomas et al., 1998). Thus, the selective effects of MEK inhibitors on 5 Hz stimulation-induced LTP seen in our experiments might be attributable to changes in postsynaptic excitability that inhibit complex spike bursting during 5 Hz stimulation (see Winder et al., 1999). Although we did not observe a strong suppression of complex spike bursting during 5 Hz stimulation in slices treated with MEK inhibitors, there was a nonsignificant trend toward less bursting in the presence of MEK inhibitors. Thus, to examine further whether changes in excitability might contribute to the effects of MEK inhibitors on low-frequency stimulation-induced LTP, we examined the effects of SL327 on the induction of LTP by low-frequency presynaptic stimulation paired with postsynaptic depolarization, a protocol in which changes in postsynaptic excitability should have little effect on LTP induction. Although blocking MAPK activation with SL327 strongly inhibited the induction of LTP in experiments in which postsynaptic bursting during low-frequency stimulation is required for the induction of LTP, it did not inhibit the induction of LTP by low-frequency synaptic stimulation paired with depolarizing current injections delivered through the intracellular recording electrode. On the basis of these findings it seems likely that MEK inhibitors suppress the induction of LTP by low-frequency trains of synaptic stimulation via effects on postsynaptic excitability. One possibility is that a NMDA receptor-dependent activation of the MAPK signaling pathway during 5 Hz stimulation enables LTP induction by a positive feedback mechanism that amplifies NMDA receptor activation via a MAPK-dependent modulation of ion channels that facilitates EPSP-induced postsynaptic depolarization. Indeed, NMDA receptor activation leads to activation of the MAPK signaling cascade in hippocampal neurons (Bading and Greenberg, 1991;Kurino et al., 1995; English and Sweatt, 1996), and MAPK may phosphorylate and inhibit the activity of some types of K+ channels in hippocampal neurons (Adams et al., 1997).
MAPK activation may underlie the effects of modulatory neurotransmitters on LTP induction
Because MEK inhibitors suppress the induction of LTP by stronger trains of 5 Hz stimulation that induce LTP in the absence of ISO and carbachol, it is difficult to determine whether MAPK activation mediates the modulatory effects of coactivation of β-adrenergic and cholinergic receptors on LTP induction by weaker 5 Hz stimulation trains or whether inhibiting MAPK activation simply blocks the ability of synapses to undergo LTP. MEK inhibitors do not, however, block the induction of LTP by low-frequency synaptic stimulation paired postsynaptic depolarization or by high-frequency stimulation, suggesting that MAPK activation is not a necessary component of the pathways responsible for LTP. Instead, our results suggest that MAPK activation is part of a modulatory process important for LTP induction by some patterns of synaptic stimulation. We thus favor the idea that coactivation of β-adrenergic and cholinergic receptors enhances the induction of LTP via convergent effects on MAPK activation. Importantly, synergistic activation of MAPK also appears to have a role in the induction of long-term depression after coactivation of G-protein-linked receptors for dopamine and glutamate at excitatory synapses in prefrontal cortex (Otani et al., 1999). Thus, the MAPK signaling cascade may represent a common mechanism by which a variety of neurotransmitters may act to regulate activity-dependent changes in synaptic strength.
In the hippocampus the activation of MAPK after β-adrenergic receptor activation is blocked by PKA inhibitors, whereas MAPK activation induced by carbachol is prevented by inhibitors of PKC (Roberson et al., 1999). Because direct activation of adenylyl cyclase with forskolin and of PKC with phorbol esters synergistically activates MAPKs in some cells (Frodin et al., 1994; Yamazaki et al., 1997), the synergistic effects of β-adrenergic and cholinergic receptor agonists on MAPK activation in the hippocampus observed in our experiments most likely arise from interactions occurring downstream of adenylyl cyclase and PKC activation. One possibility is that this occurs at the level of MEK activation via the combined effects of a PKC-mediated activation of Raf-1 (Kolch et al., 1993; Marais et al., 1998) and a cAMP-dependent activation of B-Raf (Vossler et al., 1997; de Rooij et al., 1998;Kawasaki et al., 1998). Recent evidence suggests, however, that B-Raf and Raf-1 are localized to different cellular compartments (dendritic vs somatic, respectively) in CA1 pyramidal cells (Morice et al., 1999). Moreover, muscarinic receptor activation also can activate the MAPK signaling pathway in neurons via a PKC-independent mechanism involving Src family kinases and phosphoinositide-3 kinase (Rosenblum et al., 2000). Finally, some of the effects of β-adrenergic receptor activation on LTP induction appear to be attributable to a PKA-dependent suppression of protein phosphatase activity (Thomas et al., 1996), and protein phosphatase inhibition can lead to MAPK activation in hippocampal neurons (Runden et al., 1998). Thus, interactions at several points in the MAPK signaling cascade potentially might contribute to the synergistic effects of ISO and carbachol on MAPK activation in the CA1 region of the hippocampus. Our results suggest, however, that understanding the actions and interactions of G-protein-coupled receptors on MAPK signaling may provide important insights into how modulatory neurotransmitters regulate activity-dependent changes in synaptic strength that contribute to memory formation during learning.
Footnotes
This work was supported by grants from the National Institute of Mental Health, the Pew Charitable Trusts, the UCLA Center on Aging, and Dr. P. Gail Mahoney to T.J.O. P.A.Z. was supported by a Howard Hughes Medical Institute Predoctoral Fellowship. T.J.O. is a member of the UCLA Brain Research Institute. We are grateful to Mark Barad, Holly Carlisle, and Kelsey Martin for comments on an earlier version of this manuscript and to Chris Evans and Joseph Watson for use of equipment and reagents for biochemical assays. We thank James Trzaskos, Janice Hytrek, James Piecara, Christine Tabaka, and Jia-Sheng Yan of DuPont Pharmaceuticals for a generous gift of SL327.
Correspondence should be addressed to Dr. Thomas O'Dell, Department of Physiology, University of California Los Angeles School of Medicine, 53-231 Center for the Health Sciences, 10833 Le Conte Avenue, Los Angeles, CA 90095. E-mail: todell@mednet.ucla.edu.
REFERENCES
- 1.Adams JP, Anderson AE, Johnston D, Pfaffinger PJ, Sweatt JD. Kv4.2: a novel substrate for MAP kinase phosphorylation. Soc Neurosci Abstr. 1997;23:1176. [Google Scholar]
- 2.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 3.Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 4.Baron C, Benes C, Tan HV, Fagard R, Roisin M-P. Potassium chloride pulse enhances mitogen-activated protein kinase activity in rat hippocampal slices. J Neurochem. 1996;66:1005–1010. doi: 10.1046/j.1471-4159.1996.66031005.x. [DOI] [PubMed] [Google Scholar]
- 5.Baumgold J. Muscarinic receptor-mediated stimulation of adenylyl cyclase. Trends Pharmacol Sci. 1992;13:339–340. doi: 10.1016/0165-6147(92)90105-f. [DOI] [PubMed] [Google Scholar]
- 6.Berman DE, Hazvi S, Rosenblum K, Seger R, Yadin D. Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J Neurosci. 1998;18:10037–10044. doi: 10.1523/JNEUROSCI.18-23-10037.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 8.Blitzer RD, Gil O, Landau EM. Cholinergic stimulation enhances long-term potentiation in the CA1 region of rat hippocampus. Neurosci Lett. 1990;118:207–210. doi: 10.1016/0304-3940(90)90835-w. [DOI] [PubMed] [Google Scholar]
- 9.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in the hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 10.Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1943. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 11.Blokland A. Acetylcholine: a neurotransmitter for learning and memory? Brain Res Rev. 1996;21:285–300. doi: 10.1016/0165-0173(95)00016-x. [DOI] [PubMed] [Google Scholar]
- 12.Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–252. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 14.Brocher S, Artola A, Singer W. Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Res. 1992;573:27–36. doi: 10.1016/0006-8993(92)90110-u. [DOI] [PubMed] [Google Scholar]
- 15.Coogan AN, O'Leary DM, O'Connor JJ. p42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- 16.Cooper DMF, Mons N, Karpen JW. Adenylyl cyclases and the interaction between calcium and cAMP signaling. Nature. 1995;374:421–424. doi: 10.1038/374421a0. [DOI] [PubMed] [Google Scholar]
- 17.Crow T, Xue-Bian J-J, Siddiqi V, Kang Y, Neary JT. Phosphorylation of mitogen-activated protein kinase by one-trial and multi-trial classical conditioning. J Neurosci. 1998;18:3480–3487. doi: 10.1523/JNEUROSCI.18-09-03480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decker MW, Gallagher M. Scoploamine-disruption of radial arm maze performance: modification by noradrenergic depletion. Brain Res. 1987;417:59–69. doi: 10.1016/0006-8993(87)90179-x. [DOI] [PubMed] [Google Scholar]
- 19.Decker MW, McGaugh JL. The role of interactions between the cholinergic system and other neuromodulatory systems in learning and memory. Synapse. 1991;7:151–168. doi: 10.1002/syn.890070209. [DOI] [PubMed] [Google Scholar]
- 20.Decker MW, Gill TM, McGaugh JL. Concurrent muscarinic and β-adrenergic blockade in rats impairs place-learning in a water maze and retention of inhibitory avoidance. Brain Res. 1990;513:81–85. doi: 10.1016/0006-8993(90)91091-t. [DOI] [PubMed] [Google Scholar]
- 21.de Rooij J, Zwartkruis FJT, Verheijen MHG, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 22.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long-term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 23.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 24.Favata M, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 25.Frodin M, Peraldi P, Van Obberghen E. Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J Biol Chem. 1994;269:6207–6214. [PubMed] [Google Scholar]
- 26.Fukunaga K, Miyamoto E. Role of MAP kinase in neurons. Mol Neurobiol. 1998;16:79–95. doi: 10.1007/BF02740604. [DOI] [PubMed] [Google Scholar]
- 27.Harrell LE, Peagler A, Parson DS. Adrenoreceptor antagonist treatment influences recovery of learning following medial septal lesions and hippocampal sympathetic ingrowth. Pharmacol Biochem Behav. 1990;35:21–28. doi: 10.1016/0091-3057(90)90198-q. [DOI] [PubMed] [Google Scholar]
- 28.Huerta PT, Lisman JE. Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature. 1993;364:723–725. doi: 10.1038/364723a0. [DOI] [PubMed] [Google Scholar]
- 29.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 30.Izquierdo I, Medina JH. Correlation between the pharmacology of long-term potentiation and the pharmacology of memory. Neurobiol Learn Mem. 1995;53:19–32. doi: 10.1006/nlme.1995.1002. [DOI] [PubMed] [Google Scholar]
- 31.Izumi Y, Zorumski CF. Norepinephrine promotes long-term potentiation in the adult rat hippocampus in vitro. Synapse. 1999;31:196–202. doi: 10.1002/(SICI)1098-2396(19990301)31:3<196::AID-SYN4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 32.Jerusalinsky D, Kornisiuk E, Izquierdo I. Cholinergic neurotransmission and synaptic plasticity concerning memory processing. Neurochem Res. 1997;22:507–515. doi: 10.1023/a:1027376230898. [DOI] [PubMed] [Google Scholar]
- 33.Jones MW, French PJ, Bliss TVP, Rosenblum K. Molecular mechanisms of long-term potentiation in the insular cortex in vivo. J Neurosci. 1999;19:RC361–RC368. doi: 10.1523/JNEUROSCI.19-21-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsuki H, Izumi Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- 35.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 36.Kolch W, Heldecker G, Kochs G, Hummel R, Vahldl H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Protein kinase C-α activates raf-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- 37.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Fukunaga K, Yamamoto H, Nishi K, Miyamoto E. Differential roles of Ca2+/calmodulin-dependent protein kinase II and mitogen-activated protein kinase activation in hippocampal long-term potentiation. J Neurosci. 1999;19:8292–8299. doi: 10.1523/JNEUROSCI.19-19-08292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science. 1998;280:109–112. doi: 10.1126/science.280.5360.109. [DOI] [PubMed] [Google Scholar]
- 40.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 41.McGaugh JL. Involvement of hormonal and neuromodulatory systems in the regulation of memory storage. Annu Rev Neurosci. 1989;12:255–287. doi: 10.1146/annurev.ne.12.030189.001351. [DOI] [PubMed] [Google Scholar]
- 42.Mons N, Cooper DMF. Adenylate cyclases: critical foci in neuronal signaling. Trends Neurosci. 1995;18:536–542. doi: 10.1016/0166-2236(95)98375-9. [DOI] [PubMed] [Google Scholar]
- 43.Morice C, Nothias F, Konig S, Vernier P, Baccarini M, Vincent J-D, Barnier JV. Raf-1 and B-Raf proteins have similar regional distributions but differential subcellular localization in adult rat brain. Eur J Neurosci. 1999;11:1995–2006. doi: 10.1046/j.1460-9568.1999.00609.x. [DOI] [PubMed] [Google Scholar]
- 44.Ohno M, Yamamoto T, Kobayashi M, Watanabe S. Impairment of working memory induced by scopolamine in rats with noradrenergic DSP-4 lesions. Eur J Pharmacol. 1993;238:117–120. doi: 10.1016/0014-2999(93)90514-i. [DOI] [PubMed] [Google Scholar]
- 45.Ohno M, Yoshimatsu A, Kobayashi M, Watanabe S. Noradrenergic DSP-4 lesions aggravate impairment of working memory produced by hippocampal muscarinic blockade in rats. Pharmacol Biochem Behav. 1997;57:257–261. doi: 10.1016/s0091-3057(96)00353-x. [DOI] [PubMed] [Google Scholar]
- 46.Otani S, Auclair N, Desce J-M, Roisin M-P, Crepel F. Dopamine receptors and Groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinase. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 48.Riekkinen P, Sirvio J, Valjakka A, Pitkanen A, Partanen J, Riekkinen P. The effects of concurrent manipulations of cholinergic and noradrenergic systems on neocortical EEG and spatial learning. Behav Neural Biol. 1990;54:204–210. doi: 10.1016/0163-1047(90)91436-f. [DOI] [PubMed] [Google Scholar]
- 49.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenblum K, Futter M, Jones M, Hulme EC, Bliss TVP. ERKI/II regulation by the muscarinic acetylcholine receptors in neurons. J Neurosci. 2000;20:977–985. doi: 10.1523/JNEUROSCI.20-03-00977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Runden E, Seglen PO, Haug F-M, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas MJ, Moody T, Makhinson M, O'Dell TJ. Activity-dependent β-adrenergic modulation of low-frequency stimulation-induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 54.Thomas MJ, Watabe AW, Moody TD, Makhinson M, O'Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7118–7126. doi: 10.1523/JNEUROSCI.18-18-07118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vossler MR, Yao H, York RD, Pan M-G, Rim CS, Stork PJS. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 56.Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by β-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 57.Yamazaki T, Komuro I, Zou Y, Kudoh S, Mizuno T, Hiroi Y, Shiojima I, Takano H, Kinugawa K-I, Kohmoto O, Takahashi T, Yazaki Y. Protein kinase A and protein kinase C synergistically activate the Raf-1 kinase/mitogen-activated protein kinase cascade in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 1997;29:2491–2501. doi: 10.1006/jmcc.1997.0488. [DOI] [PubMed] [Google Scholar]