

Abstract
Experimental absence seizures are associated with perturbations in the presynaptic release of GABA and glutamate within thalamocortical circuitry. The release of both glutamate and GABA is regulated by group III metabotropic glutamate receptors (mGluRs). Therefore, we examined the susceptibility of mice lacking the mGluR4 subtype of mGluR (mGluR4−/−) versus their wild-type controls (mGluR4+/+) to absence seizures induced either by γ-hydroxybutyrate (GHB) or the GABABagonist (−) baclofen or by low doses of the GABAA receptor (GABAAR) antagonists pentylenetetrazole, bicuculline, or picrotoxin. There was no difference between mGluR4−/− and mGluR4+/+ mice in threshold to absence seizures induced by either GHB or (−) baclofen. In contrast, the mGluR4−/− mice were markedly resistant to absence seizures induced by low doses of GABAAR antagonists. No differences were observed between mGluR4−/− and mGluR4+/+ mice in threshold to clonic or tonic seizures induced by higher doses of GABAAR antagonists, strychnine, or electroshock, indicating that seizure resistance in the mGluR4−/− mice was restricted solely to absence seizures. The resistance of mGluR4−/− mice to absence seizures induced by GABAAR antagonists was mimicked by bilateral administration of a mGluR4 antagonist into the nucleus reticularis thalami (nRT) of mGluR4+/+ mice. Conversely, intra-nRT administration of a mGluR4 agonist in mGluR4+/+ mice exacerbated GABAAR-induced absence seizures. These data indicate that the presence of mGluR4 within nRT is critical to GABAergic modulation of thalamocortical synchronization in normal and pathological states, such as generalized absence epilepsy.
Keywords: group III metabotropic glutamate receptors, thalamus, thalamocortical, mGluR4, absence seizures, GABA
Burst firing between reciprocally interconnected glutamatergic thalamic relay neurons in the ventral basal thalamus and neocortical pyramidal neurons is synchronized by GABAergic neurons in the nucleus reticularis thalami (nRT) (Steriade et al., 1993; Oh et al., 1995; Cox et al., 1997; Kim et al., 1997;McCormick and Bal, 1997). The nRT-driven synchronization that drives the phasic oscillatory activity within this circuitry generates normal rhythms, such as sleep spindles, and pathological phenomena, such as bilaterally synchronous spike-and-wave discharges (SWD), that characterize generalized absence seizures (Snead, 1995; McCormick and Bal, 1997; Danober et al., 1998; Snead et al., 1999).
Perturbations in the presynaptic release of glutamate and GABA within thalamocortical circuitry have been demonstrated in several animal models of absence seizures (Banerjee and Snead, 1995; Lin et al., 1995;Richards et al., 1995). The release of glutamate and GABA in thalamocortical circuitry is modulated by presynaptic metabotropic glutamate receptors (mGluRs) (East et al., 1995; Salt et al., 1996;Cochilla and Alford, 1998; Schaffhauser et al., 1998), giving rise to the hypothesis that mGluRs also may play a role in the pathogenesis of absence seizures. The eight mGluR subtypes (mGluR1–mGluR8) have been classified into three groups on the basis of sequence homology, signal transduction mechanisms, and pharmacological profiles (Nakanishi, 1994;Pin and Duvoisin, 1995; Conn and Pin, 1997). Activation of group III mGluRs inhibits the release of glutamate and GABA from nerve terminals (East et al., 1995; Pin and Duvoisin, 1995; Neugebauer et al., 1997;Schaffhauser et al., 1998). This subgroup of mGluRs is activated selectively by the glutamate analogl-amino-4-phosphonobutyrate (l-AP4).
Among the group III mGluRs ultrastructural studies using mGluR4-specific antibodies consistently have shown a presynaptic localization for this receptor (Kinoshita et al., 1996; Shigemoto et al., 1997; Bradley et al., 1999). mGluR4 is highly expressed in the rodent (Thomsen and Hampson, 1999) and human thalamus (Makoff et al., 1996). The presence of mGluR4 mRNA in the thalamic relay neurons (Ohishi et al., 1995) indicates that the receptor protein is expressed on the terminals of these neurons, which make synaptic contacts with neocortical pyramidal neurons and the GABAergic neurons in the nRT. On the basis of the strategic position of mGluR4 in thalamocortical circuitry, together with evidence for the involvement of GABA and glutamate release in the mechanisms of absence seizures, we sought to test the hypothesis that animals lacking the mGluR4 subtype of metabotropic glutamate receptor have an altered sensitivity to absence seizures.
MATERIALS AND METHODS
Production, maintenance, and phenotype of mGluR4 knock-out mice.CD-1 and 129svj mice were obtained from Jackson Laboratories (Bar Harbor, ME). The mGluR4 knock-out mice are a hybrid between these two strains of mice. The mGluR4+/+ mouse line is also a hybrid between CD-1 and 129svj mice. Gene targeting was performed by inserting a pgk-1/neoexpression cassette into anEcoRV site in an exon located in the first one-third of the putative extracellular N-terminal domain of the mouse mGluR4 gene (Pekhletski et al., 1996). Homozygous (mGluR4−/−) knock-out mice are devoid of mGluR4 protein (Pekhletski et al., 1996; Shigemoto et al., 1997;Bradley et al., 1999). mGluR4−/− mice and their wild-type controls (mGluR4+/+) were housed in a pathogen-free facility on a 12 hr light/dark schedule. All analyses were conducted on animals 2–3 months of age. Although mGluR4−/− mice show deficits in motor learning (Pekhletski et al., 1996) and spatial learning and memory (Gerlai et al., 1998), they do not differ from wild-type littermates on measures of general motor activity (open field test) or motor coordination (bar cross test), and they show no detectable abnormalities in neuroanatomy (Pekhletski et al., 1996).
Drugs. γ-Butyrolactone (GBL), pentylenetetrazole (PTZ), bicuculline methiodide (BMI), picrotoxin (PXN), and strychnine were obtained from Sigma (St. Louis, MO).l-2-Amino-4-phosphonobutyric acid (l-AP4) and (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG) were obtained from Tocris Cookson (Ballwin, MO). All other chemicals were obtained from standard commercial sources and were of the highest available purity.
Surgeries and recordings. Permanent epidural electrodes were implanted in mGluR4−/− and mGluR4+/+ mice under halothane anesthesia to allow for continuous recording of the electrocorticogram (ECoG). Then 7 d were allowed for recovery before commencement of the experiments. All ECoG recordings were made with animals in the freely moving state within shielded, heated, clear Plexiglas containers so that the behavioral response to drug could be observed and correlated with any drug-induced ECoG event. The ECoG was recorded continuously for 60 min before and for 3 hr after the administration of any drug.
Seizure models. Absence seizures were induced by administering either γ-butyrolactone (Snead, 1988), (−) baclofen (Aizawa et al., 1997), or one of the GABAAR antagonists PTZ, bicuculline, or picrotoxin (Depaulis et al., 1989;Matejovska et al., 1998). γ-Butyrolactone was given in a dose of 100 mg/kg as the pure drug. In previous work standardizing the γ-hydroxybutyric acid (GHB) model of generalized seizures, γ-butyrolactone has been shown to be biologically inactive; however, after parenteral administration γ-butyrolactone is converted rapidly and irreversibly to its active metabolite, GHB, by a circulating lactonase (Lettieri and Fung, 1978; Snead, 1991). γ-Butyrolactone has been used to induce the absence-like seizure because γ-butyrolactone produces the same progression of EEG and behavioral events in the rat as GHB (Snead et al., 1980) but with a more rapid onset of action and predictable dose–response (Bearden et al., 1980). (−) Baclofen was given in a dose of 20 mg/kg (Aizawa et al., 1997).
Low doses of GABAAR antagonists in rodents induce absence-like seizures that are similar to those induced by γ-butyrolactone (GBL) (Depaulis et al., 1989; Matejovska et al., 1998). The absence seizures induced by both GBL and low doses of GABAAR antagonists are characterized by 7–9 Hz bilaterally synchronous SWD. These epileptiform discharges may be recorded from depth, cortical, or epidural electrodes in freely moving animals and are associated with behavioral arrest, facial myoclonus, and vibrissal twitching. The SWD originate in, and are restricted to, thalamocortical circuitry (Snead et al., 1999). Rats and mice are unable to generate the 3 Hz SWD seen in human absence seizures; however, the electrographic and behavioral seizures in both γ-butyrolactone-treated and low-dose GABAAR antagonist-treated rodents are abolished by antiepileptic drugs specific for absence epilepsy, such as ethosuximide, as well as by GABAB antagonists (Snead, 1988, 1992; Snead et al., 1999).
A dose–response curve was constructed to determine the dose of PTX, bicuculline, and picrotoxin required to induce absence-like seizures in mGluR+/+ and mGluR−/− mice. CD100 was defined as the minimal dose to induce absence seizures in 100% of the mGluR+/+mice tested. The dose range used to define the CD100 of these GABAAR antagonists for absence seizures was 15–35 mg/kg of PTZ, 2–4 mg/kg of bicuculline, and 1–1.5 mg/kg of picrotoxin. All drugs were given intraperitoneally. After γ-butyrolactone, (−) baclofen, PTZ, bicuculline, or picrotoxin administration the experimental absence seizures were quantitated as described below. Paired trials with mGluR4−/−and mGluR4+/+ mice were used in all experiments.
Seizure model experiments. Chemoconvulsive. A dose–response curve was constructed in mGluR+/+ and mGluR−/− mice to determine the dose of PTX, bicuculline, and picrotoxin needed to induce clonic and tonic seizures. CD95 was defined as the dose of chemoconvulsant that resulted in clonic or tonic seizures in 95% of the animals that were tested. The dose range used to determine the CD95 for clonic seizures was 35–60 mg/kg of PTZ, 3.5–5 mg/kg of bicuculline, and 1.5–2 mg/kg of picrotoxin. The dose range used to determine the CD95 for tonic seizures was 55–80 mg/kg of PTZ, 5–6 mg/kg of bicuculline, and 2–2.5 mg/kg of picrotoxin. In addition, the susceptibility of mGluR4+/+ and mGluR4−/− mice to tonic seizures induced by the glycine antagonist strychnine was tested by using a dose range of 1–1.75 mg/kg to determine the CD95. Strychnine was chosen as a non-GABAAR antagonist chemoconvulsant in this series of experiments to determine whether there was a generic or specific GABAAR-mediated alteration in chemoconvulsive threshold. Paired trials with mGluR4−/− and mGluR4+/+ mice were used in all experiments.
Seizure model experiments. Electroconvulsive shock (ECS).ECS was administered via corneal electrodes by using a purpose-built stimulator. Sixty Hertz sine-wave constant current was used with a train duration of 0.2 sec. Five different intensities, ranging from 5 to 50 mA, were tested in descending order, with a minimum of 48 hr between tests. Before each stimulation the electrodes were dipped in 0.9% saline solution to improve electrical contact. The CD95 was defined as the dose, in mA, that resulted in seizures in 95% of the animals that were tested.
Intracerebral microinjection experiments. Microinjection cannulae were implanted in the nRT bilaterally along with epidural electrodes in mGluR4−/− and mGluR4+/+ mice under halothane anesthesia, using the atlas of Franklin and Paxinos (1997). The coordinates were −1.82 mm from bregma and 1.86 mm interaural. At 7 d after surgery, 5 nmol of the mGluR4 antagonist CPPG (Jane et al., 1996), the mGluR4 agonist l-AP4, or vehicle was infused in a volume of 0.5 μl into the nRT bilaterally 10 min before the intraperitoneal administration of either γ-butyrolactone or PTZ. The dose of PTZ used in the microinjection experiments was the CD100from the absence seizure dose–response curves. The accuracy of injection was determined by lesioning, using stimulator-generated (Grass Instruments, Quincy, MA) cathodal current (2mA/2 sec) through the indwelling cannulae to produce a localized lesion at the cannulae tips. The placement of cannulae was verified with reference to Franklin and Paxinos (1997), and only those animals with proven cannulae placement in nRT were used for analysis.
Data analysis. The experimental absence seizures induced by γ-butyrolactone, PTZ, bicuculline, or picrotoxin were quantitated objectively by measuring the duration of the bilaterally synchronous SWD induced by these compounds. Latency was defined as the time in minutes from administration of drug to the onset of SWD as recorded on ECoG. SWD duration was measured and expressed as seconds per 20 min epoch of time (Depaulis et al., 1989). γ-Butyrolactone, PTZ, bicuculline, or picrotoxin-induced SWD duration was compared in mGluR4−/− and mGluR4+/+ mice. Means and SE were calculated for all SWD data. The data were expressed in latency of SWD onset (in min). The data were subjected to analysis by two-way ANOVA, with time treated as a repeated measure. In the absence model experiments there were 10 mGluR4−/− and 10 mGluR4+/+ mice in each group. For the chemoconvulsive and ECS experiments there were 20 mGluR4−/− and 20 mGluR4+/+ mice in each group.
RESULTS
mGluR4 knock-out mice were resistant to absence seizures induced by low doses of GABAA receptor antagonists, but not to absence seizures induced by GHB or (−) baclofen
There was no difference between mGluR4+/+ and mGluR4−/− mice in the baseline ECoG recordings (Fig. 1A). Administration of γ-butyrolactone resulted in absence seizures in all mGluR 4+/+ (Fig. 1B), mGluR4−/− (Fig. 1C),CD-1, and 129svj mice (data not shown) that were tested. These absence seizures were characterized by bilaterally synchronous SWD on the ECoG (Fig. 1B,C). The behavior associated with these epileptiform discharges consisted of facial myoclonus, vibrissal twitching, and arrest of motor activity. Administration of the low doses of GABAAR antagonists (Fig. 1D) and (−) baclofen (data not shown) resulted in ECoG and behavioral changes similar to those induced by γ-butyrolactone. There were no significant differences between the mGluR4+/+ and mGluR4−/− mice in the latencies from administration of any of the drugs used to the onset of SWD. Similarly, there were no significant differences between the mGluR4+/+ and mGluR4−/− mice in SWD duration induced by GBL and (−) baclofen (Fig. 2).
Fig. 1.
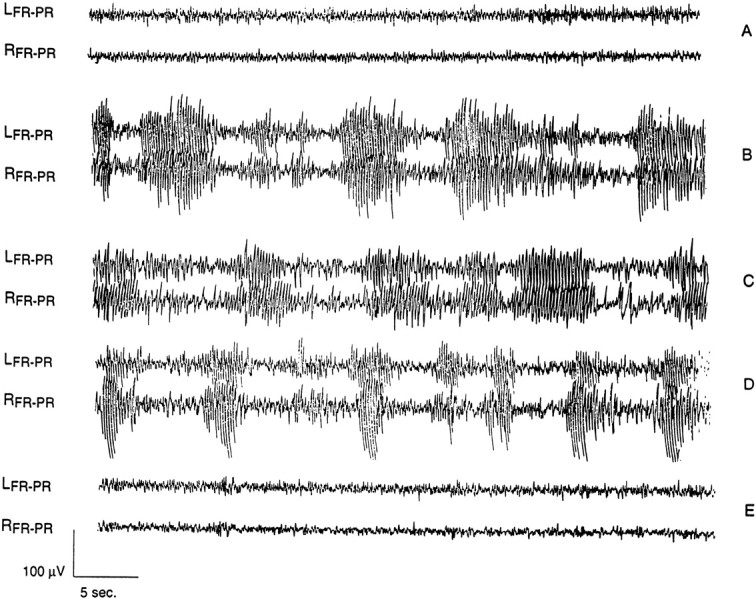
Electrocorticography (ECoG) of mGluR4−/− and mGluR4+/+ mice given either GBL or PTZ. In all ECoG figuresLFR-PR andRFR-PR represent the ECoG recording from the left and right frontoparietal electrodes, respectively. A , Baseline ECoG in a mGluR4−/− mouse. There was no difference between mGluR4+/+ and mGluR4−/− mice in baseline recordings.B, ECoG in a mGluR4+/+ mouse 20 min after 100 mg/kg of GBL. C, ECoG in a mGluR4−/− mouse 20 min after 100 mg/kg of GBL.D, ECoG in an mGluR4+/+ mouse 20 min after 30 mg/kg of PTZ. E, ECoG in an mGluR4−/− mouse 20 min after 30 mg/kg of PTZ. The dose of PTZ was the CD100 in the absence seizure dose–response studies. All drugs were given intraperitoneally. Administration of PTZ and GBL to mGluR4+/+ mice and of GBL to mGluR4−/− mice resulted in the bilaterally synchronous SWD shown. These paroxysms were associated with absence-like behavior in all of the mGluR4−/− mice that were tested, namely facial myoclonus, vibrissal twitching, and arrest of motor activity. Administration of (−) baclofen (20 mg/kg, i.p.) produced EEG and behavioral findings similar to those shown for PTZ and GBL in both mGluR4−/− and mGluR4+/+ mice. In addition to being refractory to the PTZ-induced absence seizures as shown in E, mGluR4−/− mice also were resistant to bicuculline- and picrotoxin-induced absence seizures.
Fig. 2.

Mean SWD duration ± SEM of mGluR4−/− and mGluR4+/+ mice given either GHB or (−) baclofen. A, Mean SWD duration ± SEM in mGluR4−/− and mGluR4+/+ mice (n = 10 for each group) that received 100 mg/kg of GBL, the prodrug of GHB. There was no significant difference between mGluR4−/−and mGluR4+/+ mice in the duration of GHB-induced absence seizures (p > 0.1, ANOVA).B, Mean SWD duration ± SEM in mGluR4−/− and mGluR4+/+ mice (n = 10 for each group) that received 20 mg/kg of (−) baclofen. There was no significant difference between mGluR4−/− and mGluR4+/+ mice in the duration of (−) baclofen-induced absence seizures (p > 0.1, ANOVA).
The dose–response curve for GABAAR antagonist-induced absence seizures indicated that the CD100 in mGluR4+/+mice was 30 mg/kg for PTZ, 3 mg/kg for bicuculline, and 1.5 mg/kg for picrotoxin. However, the mGluR4−/− mice were resistant to absence seizures induced by low doses of the GABAAR antagonists (Fig.3A). mGluR4−/− mice treated with low-dose PTZ were completely resistant to GABAAR antagonist-induced absence seizures (Figs. 1E,3B). Furthermore, the SWD induced by the bicuculline (Fig.3C) and picrotoxin (Fig. 3D) in mGluR4−/− mice were uniformly and markedly decreased in duration as compared with the SWD induced in mGluR4+/+ mice (p< 0.001, ANOVA).
Fig. 3.

Dose–response curves and mean SWD duration ± SEM for GABAAR antagonist-induced absence seizures in mGluR4−/− and mGluR4+/+ mice.A, The dose–response curve for GABAAR antagonist-induced absence seizures in mGluR4−/−and mGluR4+/+ mice (n = 10 for each group). The CD100 for pentylenetetrazole (PTZ), bicuculline (BMI), and picrotoxin (PXN) was 30, 3, and 1.5 mg/kg, respectively, in the mGluR4+/+ mice. The CD100 of PTZ for mGluR4+/+ mice failed to induce absence seizures in any mGluR4−/− mice. The CD100 of bicuculline and picrotoxin for mGluR4+/+ mice induced short-lived absence seizures in only 10% of mGluR4−/− mice that were tested. SEM was <10% for all data points and is not shown. ●, mGluR4+/+ (PXN); ♦, mGluR4+/+(BMI); ▪, mGluR4+/+ (PTZ); ▿, mGluR4−/− (PXN); ⋄, mGluR4−/− (BMI); ■, mGluR4−/− (PTZ). B, Mean SWD duration ± SEM for PTZ-induced absence seizures in mGluR4−/− and mGluR4+/+ mice (n = 10 for each group) that received 30 mg/kg of PTZ, the CD100 for mGluR4+/+ mice. The mGluR4−/− mice were completely resistant to absence seizures induced by low doses of this GABAAR antagonist. C, Mean SWD duration ± SEM in mGluR4−/− and mGluR4+/+ mice (n = 10 for each group) that received 3 mg/kg of bicuculline, the CD100 for mGluR4+/+mice. The SWD duration in the absence seizure induced by low-dose GABAAR antagonists was significantly (p < 0.001, ANOVA) shorter in the mGluR4−/− than in the mGluR4+/+mice. D, Mean SWD duration ± SEM in mGluR4−/− and mGluR4+/+ mice (n = 10 for each group) that received 1.5 mg/kg of picrotoxin, the CD100 for mGluR4+/+mice. The SWD duration in the absence seizures induced by low-dose GABAAR antagonists was significantly (p < 0.001, ANOVA) shorter in the mGluR4−/− than in the mGluR4+/+mice.
Because mGluR 4−/− mice are a hybrid between CD-1 and 129svj mice, it is conceivable that any observed alteration in sensitivity of the mGluR 4−/− mice to absence seizures could be caused by genetic differences in the two strains of mice. To test this possibility, we repeated the GABAAR antagonist-induced absence seizure experiments on the two parent strains of mice, using the CD100 for mGluR4+/+ mice (Fig. 3A). There were no significant differences between CD-1 and 129svj mice in SWD duration in absence seizures induced by PTZ (Fig. 4), bicuculline, or picrotoxin (data not shown). These data indicate that differences in the genetic makeup of the two strains are unlikely to account for the resistance of the mGluR4−/− mice to GABAAR antagonist-induced absence seizures.
Fig. 4.

Mean SWD duration ± SEM of mGluR4+/+, CD-1, or 129svj mice treated with 30 mg/kg of PTZ, the CD100 for mGluR4+/+ mice. This dose of PTZ induced absence-like seizures in all three strains. There were no significant differences among the three groups in latency or SWD.
mGluR4 knock-out mice and wild-type mice do not differ in threshold to convulsive seizures
The dose–response curves indicated that the EEG and behavior changes induced by GABAAR antagonists in mice represent a highly dose-specific continuum (Fig.5A). Low doses induce absence-like seizures as described above. As the dosage of GABAAR antagonists is increased, clonic seizures appear. With a further increase in dosage tonic seizures emerge. There was no temporal progression from one seizure type to another with a specific dose of GABAAR antagonist. The CD95 for the induction of clonic seizures with PTZ, bicuculline, and picrotoxin in the mGluR4+/+ mice was 45, 4.5, and 1.8 mg/kg, respectively. By comparison, the CD95 for the induction of clonic seizures with PTZ, bicuculline, and picrotoxin in the mGluR4−/− mice was 40, 5.0, and 1.9 mg/kg, respectively (Fig. 5B). This was not significantly different (p > 0.1, ANOVA; n = 20/group). There was no significant difference in the CD95 for the induction of tonic seizures with PTZ, bicuculline, picrotoxin (Fig. 5C), and strychnine (data not shown). In the mGluR4+/+ mice the CD95 was 74, 5.5, 2.2, and 1.5 mg/kg, respectively, whereas the CD95 for the induction of tonic seizures with PTZ, bicuculline, picrotoxin, PTZ, and strychnine in the mGluR4−/− mice was 70, 5.5, 2.4, and 1.5 mg/kg, respectively (p > 0.1, ANOVA; n = 20/group). There also was no significant difference between mGluR4+/+ and mGluR4−/− mice in the ECS seizure model experiments. The CD95 was 10 mA for both (dose–response curve not shown).
Fig. 5.

Dose–response curves for GABAAR antagonist-induced convulsive seizures in mGluR4−/− and mGluR4+/+ mice.A, Dose specificity of seizures induced by GABAAR antagonists in mGluR4+/+ mice.Seizure Grade refers to the type of seizure that was induced. One is absence, two is clonic, and three is tonic seizure. The dosage shown for PTZ is the CD100 rather than CD95 because only 10 animals were used to generate the absence seizure dose–response curve. B, The dose–response curve for GABAAR antagonist-induced clonic seizures in mGluR4−/− and mGluR4+/+ mice (n = 20 for each group). There was no significant difference between the mGluR4−/− and mGluR4+/+ mice in the CD95 of the GABAAR antagonists (p > 0.1, ANOVA; n = 20/group). The CD95 for the induction of clonic seizures with PTZ, bicuculline (BMI), and picrotoxin (PXN) in the mGluR4+/+ mice was 45, 4.5, and 1.8 mg/kg, respectively. The CD95 for the induction of clonic seizures with PTZ, bicuculline, and picrotoxin in the mGluR4−/− mice was 40, 5.0, and 1.9 mg/kg, respectively. SEM was <10% for all data points and is not shown. ●, mGluR4+/+ (PXN); ♦, mGluR4+/+(BMI); ▪, mGluR4+/+ (PTZ); ▿, mGluR4−/− (PXN); ⋄, mGluR4−/− (BMI); ■, mGluR4−/− (PTZ). C, The dose–response curve for GABAAR antagonist-induced tonic seizures in mGluR4−/− and mGluR4+/+ mice (n = 20 for each group). There was no significant difference between the mGluR4−/− and mGluR4+/+ mice in the CD95 of the GABAAR antagonists (p > 0.1, ANOVA; n = 20/group). In the mGluR4+/+ mice the CD95 for PTZ, bicuculline, and picrotoxin was 74, 5.5, and 2.2 mg/kg, respectively, whereas the CD95 for the induction of tonic seizures with PTZ, bicuculline, and picrotoxin in the mGluR4−/− mice was 70, 5.5, and 2.4, respectively. SEM was <10% for all data points and is not shown.
Microinjection of an mGluR4 antagonist into the nRT of mGluR4+/+ mice mimicked the phenotype of the mGluR4−/− mice
Bilateral microinjection of the mGluR4 antagonist CPPG into the nRT had no effect on baseline ECoG and induced no ictal behaviors in the mGluR4+/+ mice. In addition, bilateral intra-nRT infusion of CPPG had no effect on GBL-induced absence seizures in mGluR4+/+ mice (Fig.6A). However, bilateral intra-nRT infusion of CPPG in mGluR4+/+mice resulted in a marked reduction in the duration of absence seizures induced by low doses of PTZ (Fig. 6B). Thus, microinjection of the mGluR4 antagonist CPPG into the nRT of mGluR4+/+ mice conferred resistance to PTZ-induced absence seizures and mimicked the phenotype of the mGluR4−/− mice.
Fig. 6.

Mean SWD duration ± SEM of mGluR4+/+ mice treated with CPPG orl-AP4 given intra-nRT before induction of absence seizures with either PTZ or GHB. A, Mean SWD duration ± SEM for mGluR4+/+ mice (n = 10 for each group) that received 100 mg/kg of GBL 10 min after bilateral intra-nRT infusion of 5 nmol of the mGluR4 antagonist CPPG, the mGluR4 agonist l-AP4, or vehicle control. There was no significant (p > 0.1, ANOVA) difference in SWD duration among any of the nRT treatment groups. B, Mean SWD duration ± SEM for mGluR4+/+ mice (n = 10 for each group) that received 30 mg/kg of PTZ 10 min after bilateral intra-nRT infusion of 5 nmol of the mGluR4 antagonist CPPG or vehicle control. Intra-nRT infusion of CPPG conferred significant (p < 0.001, ANOVA) resistance to absence seizures induced by low doses of PTZ.C, Mean SWD duration ± SEM for mGluR4+/+ mice (n = 10 for each group) that received 30 mg/kg of PTZ 10 min after bilateral intra-nRT infusion of 5 nmol of the mGluR4 agonist l-AP4 or vehicle control. l-AP4 administration into the nRT significantly (p < 0.001, ANOVA) exacerbated absence seizures induced by low doses of PTZ.
Bilateral microinjection of the mGluR4 agonist l-AP4 into the nRT had no effect on baseline ECoG and induced no ictal behaviors in the mGluR4+/+ mice. Bilateral intra-nRT infusion of l-AP4 in mGluR4+/+before the intraperitoneal administration of PTZ mice had an effect opposite to that of CPPG. Bilateral intra-nRT infusion of CPPG resulted in resistance to PTZ-induced absence seizures, but bilateral intra-nRT infusion of l-AP4 was associated with a significant (p < 0.001, ANOVA) prolongation of PTZ-induced SWD duration (Fig. 6C) (i.e., exacerbation of PTZ-induced absence seizures). However, neither CPPG norl-AP4 given into the nRT affected the ability of GBL to induce SWD in mGluR4+/+ mice (Fig.6A).
DISCUSSION
We have hypothesized that mice lacking the mGluR4 gene would have an altered sensitivity to pharmacologically induced absence seizures. The experimental observations that prompted the formulation of this hypothesis were based on two findings. First, group III mGluRs are located presynaptically and modulate both GABA and glutamate release (East et al., 1995; Pin and Duvoisin, 1995; Salt et al., 1996;Neugebauer et al., 1997). Second, glutamate and GABA release are altered in thalamocortical circuitry in absence seizures (Banerjee and Snead, 1995; Lin et al., 1995; Richards et al., 1995; Hu et al., 2000). Therefore, we compared the sensitivity of mGluR4−/− and mGluR4+/+ mice to absence seizures induced by GBL, the GABABR agonist (−) baclofen, or low doses of the three GABAAR antagonists PTZ, bicuculline, and picrotoxin. In these experiments there was no significant difference between the mGluR4+/+ and mGluR4−/− mice in the sensitivity to absence seizures induced by GHB or (−) baclofen. However, mGluR4−/− mice were highly resistant to absence seizures induced by low doses of GABAAR antagonists.
GHB is a naturally occurring metabolite of GABA and has the ability to induce absence seizures in a number of animal species, including rats and mice (Snead et al., 1999). The precise mechanism by which GHB induces absence seizures is not known, but there is some evidence that GHB may be a weak GABABR agonist (Bernasconi et al., 1999; Lingenhoehl et al., 1999). However, other studies suggest that GHB exerts its effect at unique GHB-specific binding sites (Snead, 1996; Maitre, 1997). Therefore, to be certain that we were examining GABABR-mediated absence seizures in the mGluR4−/− mice, we also induced absence seizures with the specific GABABR agonist (−) baclofen. GABABR agonists cause absence seizures by acting on thalamocortical relay neurons in the ventrobasal thalamus (Liu et al., 1992; McCormick and Bal, 1997; Snead et al., 1999). The observation that both GHB-induced and GABABR agonist-induced absence seizures are equally robust in mGluR4−/− and mGluR4+/+ mice suggests that GABAB-mediated inhibition within the thalamocortical circuitry is unaffected by the absence of mGluR4.
We then determined whether the resistance of mGluR4−/− mice to absence seizures induced by low doses of GABAAR antagonists extended to convulsive seizures. Convulsive seizures were induced by both chemoconvulsant drugs and ECS. No significant differences were observed between mGluR4−/− and mGluR4+/+ mice in any of the models that were used for convulsive seizures. The threshold of mGluR4−/− mice to chemoconvulsant- and electroconvulsant-induced clonic and tonic seizures is an important issue because there is increasing interest in the therapeutic potential of mGluR modulation in epileptogenesis (Thomsen and Dalby, 1998;Meldrum et al., 1999; Lie et al., 2000). Activation of group I mGluRs appears to produce convulsive seizures, whereas some group I antagonists are reported to be anticonvulsant in rodent models of limbic or convulsive seizures (Tizzano et al., 1995; Chapman et al., 1999). However, the effect of group III mGluR agonists and antagonists on seizures is less clear. Selective activation of group III mGluRs has been reported to decrease epileptiform activity in rat neocortex or hippocampus (Burke and Hablitz, 1994; Tizzano et al., 1995; Abdul-Ghani et al., 1997), but administration of l-AP4 intracerebroventricularly reportedly causes seizures and exacerbates sound-induced seizures in mice (Ghauri et al., 1996). The proconvulsant activity of l-AP4 in some studies could be the result of activation of NMDA receptors at higher concentrations of this amino acid (Thomsen and Dalby, 1998). In our experiments no alterations in EEG were seen, nor were any ictal behaviors induced in mGluR4+/+ mice after bilateral intra-nRT infusion of either l-AP4 or CPPG.
If the hypothesis that selective activation of mGluR4 results in an elevated threshold for limbic and convulsive seizure activity (Burke and Hablitz, 1994; Tizzano et al., 1995; Abdul-Ghani et al., 1997) is correct, the convulsive threshold for clonic and tonic seizures would be predicted to be lower in animals devoid of mGluR4. However, in our experiments the CD95 for clonic and tonic seizures induced by higher doses of GABAAR antagonists, the glycine antagonist strychnine, or ECS in the mGluR4−/− mice was not significantly different from that observed for the mGluR4+/+ mice. Given the evidence for the exclusive involvement of thalamus and cortex in the genesis of absence seizures (Snead, 1995; McCormick and Bal, 1997; Danober et al., 1998;Snead et al., 1999), these findings implicate thalamocortical circuitry as the neuronal network involved in the resistance of mGluR4−/− to absence seizures induced by GABAAR antagonists and indicate the circuitry-dependent nature of any pro- or anticonvulsant action that might result from mGluR4 activation. An explanation for the dramatic resistance to absence seizures in the mGluR4 knock-out mice may be that other group III mGluRs are unable to compensate for the lack of mGluR4 in the thalamus. mGluR6 is expressed only in the retina, and within the thalamus the expression of mGluR8 is restricted to the reticular neurons (Saugstad et al., 1997; Corti et al., 1998). mGluR7, which has very low affinity for glutamate and l-AP4, is expressed at low levels in several thalamic relay nuclei of the rat (Ohishi et al., 1995; Neto et al., 2000). However, a preliminary report has indicated that mGluR7 knock-out mice are not resistant to seizures but instead develop convulsant seizures several weeks after birth (Sansig et al., 1999).
Physiological activation of group III mGluR is reported to depress glutamate-mediated excitation in corticothalamic neurons and, thus, to decrease corticothalamic input to thalamus (Turner and Salt, 1999). However, a different part of the thalamocortical circuitry is involved in the mechanism of GABAAR antagonist-induced absence seizures. Low doses of GABAAR antagonists induce absence seizures by blocking intra-nRT GABAAR-mediated inhibition (Huguenard and Prince, 1994; Ulrich and Huguenard, 1997) rather than acting on corticothalamic pathways. Therefore, we tested and confirmed the hypothesis that the nRT is critical to the resistance of mGluR4−/− mice to GABAAR antagonist-induced absence seizures. In the low-dose PTZ model of absence seizures, microinjection of the mGluR4 antagonist CPPG into the nRT of wild-type mGluR4+/+ mice mimicked the phenotype of the mGluR4−/− mice. Conversely, bilateral intra-nRT administration of the mGluR4 agonistl-AP4 in mGluR4−/− mice resulted in exacerbation of absence seizures induced by PTZ, but not by GHB.
Recurrent GABAergic collateral connections provide intranuclear GABAAR-mediated inhibition within nRT that regulates nRT output during thalamic oscillations. The intra-nRT recurrent GABAergic inhibitory activity acts as a desynchronizer to prevent hypersynchrony and the induction of absence seizures (Huguenard and Prince, 1994; Ulrich and Huguenard, 1997; Huntsman and Huguenard, 1999). Suppression of intra-nRT GABAAR-mediated inhibition, either by disruption of the gene coding for the β3 subunit of the GABAAR (Homanics et al., 1997) or by administration of low doses of GABAAR antagonists, results in a marked increase in oscillatory synchrony that is characteristic of absence seizures (Huntsman et al., 1999; Sohal et al., 2000). Therefore, intra-nRT GABAAR-mediated inhibition appears to be required for preventing the pathological hypersynchrony of absence seizures. We suggest that in mGluR4−/− mice the absence of mGluR4 in presynaptic glutamatergic terminals that project from thalamic relay neurons to nRT may cause an increase in glutamate release (East et al., 1995) on GABAergic neurons within the nRT. This results in an enhancement of intra-nRT GABAAR-mediated inhibition leading to increased desynchronization within the thalamocortical circuitry and a resistance to absence seizures induced by low doses of GABAAR antagonists. This effect is opposite to that seen in mice devoid of the β3 subunit of the postsynaptic GABAAR (β3−/−). This mutant is of particular relevance to the question of the role of the nRT in modulation of neuronal synchrony and absence seizures because the β3 subunit is restricted mainly to the nRT. In the β3−/− mice, GABAAR-mediated inhibition was nearly abolished in nRT but was unaffected in thalamocortical relay nuclei. Further, in the β3−/− mice oscillatory synchrony was intensified dramatically, leading to the conclusion that recurrent inhibitory connections within nRT act as desynchronizers (i.e., decreased intra-nRT GABAAR-mediated inhibition causes hypersynchrony) (Huntsman et al., 1999).
Recent computer modeling experiments have extended these findings by showing that intra-nRT inhibition restricts intrathalamic oscillatory activity to particular spatiotemporal patterns to allow for focal recurrent activity that is relevant for normal thalamocortical function while preventing the pathological synchronization that results in absence seizures (Sohal et al., 2000). The selective resistance of mGluR4−/− mice to absence seizures induced by GABAAR antagonists indicates that the absence of mGluR4 reinforces the intra-nRT GABAAR-mediated control of thalamocortical synchrony. These findings suggest that there is a tight regulation between mGluR4 and GABAAR within intra-nRT inhibitory circuitry. Presynaptic glutamatergic mechanisms at the level of the nRT may play a key role in the maintenance of the fine balance between synchronization and desynchronization within thalamocortical circuitry and, therefore, in the pathogenesis of absence seizures. Our data indicate that mGluR4-mediated perturbation of GABAergic function within the nRT of mGluR4−/− mice causes a shift of the synchronization/desynchronization equilibrium toward desynchronization. Thus, an increase in desynchronization in this circuit produces resistance to absence seizures induced by GABAAR antagonists.
The findings that mGluR4−/− mice are resistant to absence seizures induced by low doses of GABAAR antagonists and that this phenotype is mimicked by the intra-nRT administration of an mGluR4 antagonist suggest that mGluR4 antagonist drugs may be potentially useful in the treatment of absence epilepsy. There is precedent for drugs that act in a similar circuitry-specific manner having clinical utility in this disorder. The therapeutic efficacy of benzodiazepines in generalized absence seizures may be attributed to the fact that these compounds enhance GABAAR-mediated inhibition within the nRT and enhance desynchronization, with a resultant decrease in hypersynchrony and absence seizures (Huguenard and Prince, 1994; Gibbs et al., 1996; Snead et al., 1999).
Footnotes
This work was supported in part by Medical Research Council Canada and by the Bloorview Children's Hospital Foundation. We are grateful to Mr. Chun Che Liu for superb technical support.
Correspondence should be addressed to Dr. O. Carter Snead III, University of Toronto, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada. E-mail: csnead@sickkids.on.ca.
REFERENCES
- 1.Abdul-Ghani AS, Attwell PJ, Singh N, Bradford HF, Croucher MJ, Jane DE. Anti-epileptogenic and anticonvulsant activity of l-2-amino-4-phosphonobutyrate, a presynaptic glutamate receptor agonist. Brain Res. 1997;755:202–212. doi: 10.1016/s0006-8993(97)00098-x. [DOI] [PubMed] [Google Scholar]
- 2.Aizawa M, Ito Y, Fukuda H. Roles of γ-amino butyric acidB and γ-hydroxybutyric acid receptors in hippocampal long-term potentiation and pathogenesis of absence seizures. Biol Pharm Bull. 1997;10:1066–1070. doi: 10.1248/bpb.20.1066. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee PK, Snead OC. Presynaptic γ-hydroxybutyric acid (GHB) and GABAB receptor-mediated release of GABA and glutamate in rat thalamic ventrobasal nucleus: a possible mechanism for the generation of absence-like seizures induced by GHB. J Pharmacol Exp Ther. 1995;273:1534–1543. [PubMed] [Google Scholar]
- 4.Bearden LJ, Snead OC, Healey CT, Pegram VG. Antagonism of γ-hydroxybutyric acid-induced frequency shifts in the cortical EEG of rats by dipropyl acetate. Electroencephalogr Clin Neurophysiol. 1980;49:181–183. doi: 10.1016/0013-4694(80)90365-x. [DOI] [PubMed] [Google Scholar]
- 5.Bernasconi R, Mathivet P, Bischoff S, Marescaux C. γ-Hydroxybutyric acid: an endogenous neuromodulator with abuse potential? Trends Pharmacol Sci. 1999;20:135–141. doi: 10.1016/s0165-6147(99)01341-3. [DOI] [PubMed] [Google Scholar]
- 6.Bradley SR, Standaert DA, Rhodes KJ, Rees HD, Testa CM, Levey AI, Conn PJ. Immunohistochemical localization of subtype 4a metabotropic glutamate receptor in the rat and mouse basal ganglia. J Comp Neurol. 1999;407:33–46. [PubMed] [Google Scholar]
- 7.Burke JP, Hablitz JJ. Metabrotropic glutamate receptor activation decreases epileptiform activity in rat neocortex. Neurosci Lett. 1994;174:29–33. doi: 10.1016/0304-3940(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 8.Chapman AG, Yip PK, Yap JS, Quinn LP, Tang E, Harris JR, Meldrum BS. Anticonvulsant actions of LY 367385 [(+)-2-methyl-4-carboxyphenylglycine] and AIDA [(RS)-1-aminoindan-1,5-dicarboxylic acid]. Eur J Pharmacol. 1999;368:17–24. doi: 10.1016/s0014-2999(99)00014-x. [DOI] [PubMed] [Google Scholar]
- 9.Cochilla AJ, Alford S. Metabotropic glutamate receptor-mediated control of neurotransmitter release. Neuron. 1998;20:1007–1016. doi: 10.1016/s0896-6273(00)80481-x. [DOI] [PubMed] [Google Scholar]
- 10.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 11.Corti C, Restituito S, Rimland JM, Brabet I, Corsi M, Pin J-P, Ferraguti F. Cloning and characterization of alternative mRNA forms for the rat metabotropic glutamate receptors mGluR7 and mGluR8. Eur J Neurosci. 1998;10:3629–3641. doi: 10.1046/j.1460-9568.1998.00371.x. [DOI] [PubMed] [Google Scholar]
- 12.Cox CL, Huguenard JR, Prince DA. Nucleus reticularis neurons mediate diverse inhibitory effects in thalamus. Proc Natl Acad Sci USA. 1997;94:8854–8859. doi: 10.1073/pnas.94.16.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol. 1998;55:27–57. doi: 10.1016/s0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- 14.Depaulis A, Snead OC, Marescaux C, Vergnes M. Suppressive effects of intranigral injection of muscimol in three models of generalized nonconvulsive epilepsy induced by chemical agents. Brain Res. 1989;498:64–72. doi: 10.1016/0006-8993(89)90399-5. [DOI] [PubMed] [Google Scholar]
- 15.East SJ, Hill MP, Brotchie JM. Metabotropic glutamate receptor agonists inhibit endogenous glutamate release from rat striatal synaptosomes. Eur J Pharmacol. 1995;277:117–121. doi: 10.1016/0014-2999(95)00119-6. [DOI] [PubMed] [Google Scholar]
- 16.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 17.Gerlai R, Roder JC, Hampson DR. Altered spatial learning and memory in mice lacking the mGluR subtype of metabotropic glutamate receptor. Behav Neurosci. 1998;112:525–532. doi: 10.1037//0735-7044.112.3.525. [DOI] [PubMed] [Google Scholar]
- 18.Ghauri M, Chapman AG, Meldrum BS. Convulsant and anticonvulsant actions of agonists and antagonists of group III mGluRs. NeuroReport. 1996;7:1469–1474. doi: 10.1097/00001756-199606170-00005. [DOI] [PubMed] [Google Scholar]
- 19.Gibbs JW, Schroder GB, Coulter DA. GABAA receptor function in developing rat thalamic reticular neurons: whole-cell recordings of GABA-mediated currents and modulation by clonazepam. J Neurophysiol. 1996;76:2568–2579. doi: 10.1152/jn.1996.76.4.2568. [DOI] [PubMed] [Google Scholar]
- 20.Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Makela R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW. Mice devoid of γ-aminobutyrate type A receptor β3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad USA. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu RQ, Banerjee PK, Snead OC. Regulation of presynaptic γ-amino butyric acid (GABA) and glutamate release mediated by the GABAB/γ-hydroxybutyric acid (GHB) receptor complex in rat cerebral cortex in the GHB model of absence seizures in rat. Neuropharmacology. 2000;39:427–439. doi: 10.1016/s0028-3908(99)00152-5. [DOI] [PubMed] [Google Scholar]
- 22.Huguenard JR, Prince DA. Clonazepam suppresses GABAB-mediated inhibition in thalamic relay neurons through effects in nucleus reticularis. J Neurophysiol. 1994;71:2576–2581. doi: 10.1152/jn.1994.71.6.2576. [DOI] [PubMed] [Google Scholar]
- 23.Huntsman MM, Huguenard JR. Differential pharmacology of fast and slow inhibition in thalamic reticular nucleus: implications for the treatment of absence epilepsy. Epilepsia. 1999;40[Suppl 7]:23. [Google Scholar]
- 24.Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- 25.Jane DE, Thomas NK, Tse HW, Watkins JC. Potent antagonists at the l-AP4- and (1S,3S)-ACPD-sensitive presynaptic metabotropic glutamate receptors in the neonatal rat spinal cord. Neuropharmacology. 1996;35:1029–1035. doi: 10.1016/s0028-3908(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 26.Kim U, Sachez-Vives V, McCormick DA. Functional dynamics of GABAergic inhibition in the thalamus. Science. 1997;278:130–134. doi: 10.1126/science.278.5335.130. [DOI] [PubMed] [Google Scholar]
- 27.Kinoshita A, Ohishi H, Nomura S, Shigemoto R, Nakanishi S, Mizuno N. Presynaptic localization of a metabotropic glutamate receptor, mGluR4a, in the cerebellar cortex: a light and electron microscopic study of the rat. Neurosci Lett. 1996;207:199–202. doi: 10.1016/0304-3940(96)12519-2. [DOI] [PubMed] [Google Scholar]
- 28.Lettieri J, Fung JL. Improved pharmacological activity via prodrug modifications: comparative pharmacokinetics of sodium γ-hydroxybutyrate and γ-butyrolactone. Res Commun Chem Pathol Pharmacol. 1978;22:107–118. [PubMed] [Google Scholar]
- 29.Lie A, Becker A, Behle K, Beck H, Malitschek B, Conn PJ, Kuhn R, Nitsch R, Plaschke M, Schramm J, Elger CE, Wiestler OD, Blumcke I. Up-regulation of the metabotropic glutamate receptor mGluR4 in hippocampal neurons with reduced seizure vulnerability. Ann Neurol. 2000;47:26–35. [PubMed] [Google Scholar]
- 30.Lin F-H, Wang Y, Lin S, Cao Z, Hosford DA. GABAB receptor-mediated effects in synaptosomes of lethargic (lh/lh) mice. J Neurochem. 1995;65:2087–2095. doi: 10.1046/j.1471-4159.1995.65052087.x. [DOI] [PubMed] [Google Scholar]
- 31.Lingenhoehl K, Brom R, Heid J, Beck P, Froestl W, Kaupmann K, Bettler B, Mosbacher J. γ-Hydroxybutyrate is a weak agonist at recombinant GABAB receptors. Neuropharmacology. 1999;38:1667–1674. doi: 10.1016/s0028-3908(99)00131-8. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Vergnes M, Depaulis A, Marescaux C. Involvement of intrathalamic GABAB neurotransmission in the control of absence seizures in the rat. Neuroscience. 1992;48:87–93. doi: 10.1016/0306-4522(92)90340-8. [DOI] [PubMed] [Google Scholar]
- 33.Maitre M. The γ-hydroxybutyrate signaling system in brain: organization and functional implications. Prog Neurobiol. 1997;51:337–361. doi: 10.1016/s0301-0082(96)00064-0. [DOI] [PubMed] [Google Scholar]
- 34.Makoff A, Lelchuk R, Oxer M, Harrington K, Emson P. Molecular characterization and localization of human metabotropic glutamate receptor type 4. Mol Brain Res. 1996;37:239–248. doi: 10.1016/0169-328x(95)00321-i. [DOI] [PubMed] [Google Scholar]
- 35.Matejovska I, Veliskova J, Velisek L. Bicuculline-induced rhythmic EEG episodes as a model of absence seizures: gender differences and the effects of ethosuximide and baclofen treatment. Epilepsia. 1998;39:1243–1252. doi: 10.1111/j.1528-1157.1998.tb01321.x. [DOI] [PubMed] [Google Scholar]
- 36.McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- 37.Meldrum BS, Akbar MT, Chapman AG. Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Res. 1999;36:189–204. doi: 10.1016/s0920-1211(99)00051-0. [DOI] [PubMed] [Google Scholar]
- 38.Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 39.Neto FL, Schadrack J, Berthele A, Zieglgansberger W, Tolle TR, Castro-Lopez JM. Differential distribution of the metabotropic glutamate receptor subtype mRNAs in the thalamus of the rat. Brain Res. 2000;854:93–105. doi: 10.1016/s0006-8993(99)02326-4. [DOI] [PubMed] [Google Scholar]
- 40.Neugebauer V, Keele NB, Shinnick-Gallagher P. Epileptogenesis in vivo enhances the sensitivity of inhibitory presynaptic metabotropic glutamate receptors in basolateral amygdala neurons in vitro. J Neurosci. 1997;17:983–995. doi: 10.1523/JNEUROSCI.17-03-00983.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oh K-S, Lee C-J, Gibbs JW, Coulter DA. GABAA receptor function in developing rat thalamic reticular neurons: whole-cell recordings of GABA-mediated currents and modulation by clonazepam. J Neurophysiol. 1995;76:2568–2579. doi: 10.1152/jn.1996.76.4.2568. [DOI] [PubMed] [Google Scholar]
- 42.Ohishi H, Akazawa C, Shigemoto R, Nakanishi S, Mizuno N. Distributions of the mRNAs for l-AP4-sensitive mGluRs mGluR4 and mGluR7 in the rat brain. J Comp Neurol. 1995;360:555–570. doi: 10.1002/cne.903600402. [DOI] [PubMed] [Google Scholar]
- 43.Pekhletski R, Gerlai R, Overstreet LS, Huang X-P, Agopyan N, Slater NT, Abramow-Newerly W, Roder JC, Hampson DR. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J Neurosci. 1996;16:6364–6373. doi: 10.1523/JNEUROSCI.16-20-06364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pin J-P, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 45.Richards DA, Lemos T, Whitton PS, Bowery NG. Extracellular GABA in the ventrolateral thalamus of rats exhibiting spontaneous absence epilepsy: a microdialysis study. J Neurochem. 1995;65:1674–1680. doi: 10.1046/j.1471-4159.1995.65041674.x. [DOI] [PubMed] [Google Scholar]
- 46.Salt TE, Eaton SA, Turner JP. Characterization of the metabotropic glutamate receptors (mGluRs) which modulate GABA-mediated inhibition in the ventrobasal thalamus. Neurochem Int. 1996;29:317–322. doi: 10.1016/0197-0186(95)00146-8. [DOI] [PubMed] [Google Scholar]
- 47.Sansig G, Bushnell TJ, Clarke VRJ, Rozov A, Burnashev N, Portet C, Gaspirini F, Schmutz M, Klebs K, Shigemoto R, Flor PJ, Kuhn R, Knopful T, Schroeder M, Collet V, Collingridge GL, van der Putten H. Epilepsy in mice lacking the metabotropic glutamate receptor mGluR7. Neuropharmacology. 1999;38:A39. doi: 10.1523/JNEUROSCI.21-22-08734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saugstad JA, Kinzie JM, Shinohara MM, Segerson TP, Westbrook GL. Cloning and expression of rat metabotropic glutamate receptor 8 reveals a distinct pharmacological profile. Mol Pharmacol. 1997;51:119–125. doi: 10.1124/mol.51.1.119. [DOI] [PubMed] [Google Scholar]
- 49.Schaffhauser H, Knoflach F, Pink JR, Bleul Z, Cartmell J, Goepfgert F, Kemp JA, Richards JG, Adam G, Mutel V. Multiple pathways for regulation of the KCl-induced [3H]GABA release by metabotropic glutamate receptors in primary rat cortical cultures. Brain Res. 1998;782:91–104. doi: 10.1016/s0006-8993(97)01271-7. [DOI] [PubMed] [Google Scholar]
- 50.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snead OC. The γ-hydroxybutyrate model of absence seizures: further characterization and comparison with other absence models. Epilepsia. 1988;29:361–368. doi: 10.1111/j.1528-1157.1988.tb03732.x. [DOI] [PubMed] [Google Scholar]
- 52.Snead OC. The γ-hydroxybutyrate model of absence seizures: correlation of regional brain levels of γ-hydroxybutyric acid and γ-butyrolactone with spike wave discharges. Neuropharmacology. 1991;30:161–167. doi: 10.1016/0028-3908(91)90199-l. [DOI] [PubMed] [Google Scholar]
- 53.Snead OC. Evidence for GABAB-mediated mechanisms in experimental generalized absence seizures. Eur J Pharmacol. 1992;213:343–349. doi: 10.1016/0014-2999(92)90623-c. [DOI] [PubMed] [Google Scholar]
- 54.Snead OC. Basic mechanisms of generalized absence seizures. Ann Neurol. 1995;37:146–157. doi: 10.1002/ana.410370204. [DOI] [PubMed] [Google Scholar]
- 55.Snead OC. Relation of the [3H] γ-hydroxybutyric acid (GHB) binding site to the γ-aminobutyric acidB (GABAB) receptor in rat brain. Biochem Pharmacol. 1996;52:1235–1243. doi: 10.1016/0006-2952(96)00477-7. [DOI] [PubMed] [Google Scholar]
- 56.Snead OC, Bearden LJ, Healey CT, Pegram V. Effect of acute and chronic anticonvulsant administration on endogenous γ-hydroxybutyrate in rat brain. Neuropharmacology. 1980;19:47–56. doi: 10.1016/0028-3908(80)90165-3. [DOI] [PubMed] [Google Scholar]
- 57.Snead OC, Depaulis A, Vergnes M, Marescaux C. Absence epilepsy: advances in experimental animal models. In: Delgado-Escueta AV, Wilson W, Olsen RW, Porter RJ, editors. Jasper's basic mechanisms of the epilepsies, 3rd Ed. Raven; New York: 1999. pp. 253–278. [PubMed] [Google Scholar]
- 58.Sohal VS, Huntsman MM, Huguenard JR. Reciprocal inhibitory connections regulate the spatiotemporal properties of intrathalamic oscillations. J Neurosci. 2000;20:1735–1745. doi: 10.1523/JNEUROSCI.20-05-01735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 60.Thomsen C, Dalby NO. Roles of metabotropic glutamate receptor subtypes in modulation of pentylenetetrazole-induced seizure activity in mice. Neuropharmacology. 1998;37:1465–1473. doi: 10.1016/s0028-3908(98)00138-5. [DOI] [PubMed] [Google Scholar]
- 61.Thomsen C, Hampson DR. Contribution of metabotropic glutamate receptor mGluR4 to l-2-[3H]amino-4-phosphonobutyrate binding in mouse brain. J Neurchem. 1999;72:835–840. doi: 10.1046/j.1471-4159.1999.0720835.x. [DOI] [PubMed] [Google Scholar]
- 62.Tizzano JP, Griffey KI, Schoepp DD. Induction or protection of limbic seizures in mice by mGluR subtype selective agonists. Neuropharmacology. 1995;34:1063–1067. doi: 10.1016/0028-3908(95)00083-i. [DOI] [PubMed] [Google Scholar]
- 63.Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J Physiol (Lond) 1999;519:481–491. doi: 10.1111/j.1469-7793.1999.0481m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ulrich D, Huguenard JR. GABAA receptor-mediated rebound burst firing and burst shunting in thalamus. J Neurophysiol. 1997;78:1748–1751. doi: 10.1152/jn.1997.78.3.1748. [DOI] [PubMed] [Google Scholar]