

Abstract
Amyloid plaques are a neuropathological hallmark of Alzheimer's disease (AD), but their relationship to neurodegeneration and dementia remains controversial. In contrast, there is a good correlation in AD between cognitive decline and loss of synaptophysin-immunoreactive (SYN-IR) presynaptic terminals in specific brain regions. We used expression-matched transgenic mouse lines to compare the effects of different human amyloid protein precursors (hAPP) and their products on plaque formation and SYN-IR presynaptic terminals. Four distinct minigenes were generated encoding wild-type hAPP or hAPP carrying mutations that alter the production of amyloidogenic Aβ peptides. The platelet-derived growth factor β chain promoter was used to express these constructs in neurons. hAPP mutations associated with familial AD (FAD) increased cerebral Aβ1–42 levels, whereas an experimental mutation of the β-secretase cleavage site (671M→I) eliminated production of human Aβ. High levels of Aβ1–42 resulted in age-dependent formation of amyloid plaques in FAD-mutant hAPP mice but not in expression-matched wild-type hAPP mice. Yet, significant decreases in the density of SYN-IR presynaptic terminals were found in both groups of mice. Across mice from different transgenic lines, the density of SYN-IR presynaptic terminals correlated inversely with Aβ levels but not with hAPP levels or plaque load. We conclude that Aβ is synaptotoxic even in the absence of plaques and that high levels of Aβ1–42are insufficient to induce plaque formation in mice expressing wild-type hAPP. Our results support the emerging view that plaque-independent Aβ toxicity plays an important role in the development of synaptic deficits in AD and related conditions.
Keywords: Alzheimer's disease, amyloid, APP, neurodegeneration, synapses, transgenic mice
Alzheimer's disease (AD) is an age-dependent neurodegenerative disorder that causes a chronically progressive decline in cognitive functions. Because of the increasing longevity of many populations around the world, AD is a medical problem of mounting social and economic impact (Alloul et al., 1998). The disease is associated with a characteristic combination of morphological CNS alterations, including deposition of amyloid proteins in parenchymal plaques and cerebral blood vessels, intraneuronal formation of neurofibrillary tangles, loss of presynaptic terminals and neuronal subpopulations, and reactive gliosis (Terry et al., 1999). The severity of these alterations varies widely and specifically across different areas of the brain (Braak and Braak, 1998), suggesting that AD preferentially affects certain types of neural elements or that these elements are particularly susceptible to the disease.
Loss of synaptophysin-immunoreactive (SYN-IR) presynaptic terminals (Terry et al., 1991; Honer et al., 1992; Masliah et al., 1994; Dickson et al., 1995; Sze et al., 1997) and the number of neurofibrillary tangles (Gomez-Isla et al., 1997) in specific brain regions correlate well with cognitive decline in AD. In contrast, the relationship between amyloid plaques and clinical manifestations or neurodegenerative changes remains controversial (Cummings et al., 1996;Terry, 1996; Bartoo et al., 1997; Davis and Chisholm, 1997; Gomez-Isla et al., 1997; Lue et al., 1999; McLean et al., 1999). This is puzzling in light of different lines of evidence implicating the amyloid-β protein precursor (APP) and its metabolites in the pathogenesis of AD.
Mutations in genes encoding APP or presenilins 1 or 2 have been linked to autosomal dominant forms of familial AD (FAD), and these mutations increase the production of APP-derived Aβ peptides, either total Aβ or Aβ ending at residue 42 (Aβ42) (for review, see Younkin, 1995; Price and Sisodia, 1998; Storey and Cappai, 1999). A variety of Aβ preparations elicit neurotoxicity in cultures of neural cells or tissue sections (Yankner et al., 1989; Pike et al., 1993; Yankner, 1996; Lambert et al., 1998), and acute injections of fibrillar Aβ into the brain induce significant neuronal loss in aged rhesus monkeys (Geula et al., 1998).
Several transgenic mouse models have been developed to further elucidate the pathogenic role of APP/Aβ in vivo(Price and Sisodia, 1998). Although low-level neuronal expression of wild-type or FAD-mutant human APP (hAPP) did not result in the formation of typical AD-like amyloid plaques (Quon et al., 1991;Mucke et al., 1994), it did elicit age-related deficits in spatial learning and memory (Moran et al., 1995; D'Hooge et al., 1996). High-level neuronal expression of FAD-mutant hAPP resulted in the brain region-dependent development of several AD-like CNS alterations, including typical neuritic plaques, reactive gliosis, and loss of SYN-IR presynaptic terminals and neuronal subpopulations (Games et al., 1995; Masliah et al., 1996; Johnson-Wood et al., 1997; Hsia et al., 1999). Many of these findings have been confirmed and extended in independent models expressing FAD-mutant hAPP in the absence (Hsiao et al., 1996; Sturchler-Pierrat et al., 1997) or presence (Duff et al., 1996; Borchelt et al., 1997) of FAD-mutant presenilins.
In some hAPP transgenic models, behavioral impairments (Hsiao et al., 1996) (but see Routtenberg, 1997) or loss of neurons (Calhoun et al., 1998) correlated with the extent of amyloid deposition. In others, behavioral impairments (Holcomb et al., 1998; Moechars et al., 1999), synaptic transmission deficits, and loss of SYN-IR presynaptic terminals and microtubule-associated protein 2-IR neurons (Hsia et al., 1999) clearly preceded plaque formation, raising the possibility that hAPP or Aβ can induce structural and functional neuronal deficits independent of plaque formation. These discrepancies underline the need for a systematic comparison of Aβ levels, plaque formation, and neurodegeneration in transgenic lines expressing wild-type or FAD-mutant forms of hAPP at comparable levels. Here, we report the results of such an analysis.
MATERIALS AND METHODS
Animals. The platelet-derived growth factor (PDGF)-APP transgene (Games et al., 1995; Rockenstein et al., 1995) and the generation of PDGF-APPInd line H6 (Wyss-Coray et al., 1997) and PDGF-APPSw,Ind line J9 (Hsia et al., 1999) have been described previously. To generate PDGF-APPWt, the sequence of PDGF-APPInd was converted to wild type by PCR primer modification, essentially as described previously (Rockenstein et al., 1995). To generate PDGF-APPM-I, the EcoRI toSpeI fragment of PDGF-APPIndcontaining the 717V→F mutation was subcloned into analogous sites in pCMV695 M596I (Citron et al., 1995) to form pCMV695HaM596I. The 1.4 kb XhoI to SpeI fragment from pCMV695HaM596 was then ligated into the analogous sites of PDGF-APPInd to create the PDGF-APPM-I transgene. The correctness of PDGF-APPWt and PDGF-APPExp,Ind was confirmed by sequencing across modified regions.
Microinjection of transgenes into C57BL/6 × DBA/2 F2 one-cell embryos, identification of transgenic founders by slot-blot analysis of genomic DNA, and selection of lines with cerebral hAPP mRNA expression by RNase protection assay analysis were performed as described previously (Games et al., 1995; Rockenstein et al., 1995). For each construct, several transgenic founders (PDGF-APPWt, n = 7; PDGF-APPInd, n = 12; PDGF-APPSw,Ind, n = 7; and PDGF-APPExp,Ind, n = 19) were generated, and their offspring were screened for cerebral transgene expression. Transgenic expresser lines were maintained by crossing heterozygous transgenic mice with nontransgenic C57BL/6 × DBA/2 F1 breeders. All transgenic mice were heterozygous with respect to the transgene. Nontransgenic littermates served as controls.
Mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline. Brains were removed and divided sagittally. One hemibrain was post-fixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, at 4°C for 48 hr for vibratome sectioning; the other was snap frozen and stored at −70°C for RNA or protein analysis.
RNA analysis. RNA extraction and mRNA quantitation by solution hybridization RNase protection assay were performed as described previously (Rockenstein et al., 1995), using 10 μg of total RNA per sample in combination with the following32P-labeled antisense riboprobes [protected nucleotides (GenBank accession number)]: hAPP [nt2468–2657 (X06989) of hAPP fused via NotI linker with nt2532–2656 (M24914) of SV40]; actin [nt480–559 (X03672) of mouse β-actin].
Quantitation of Aβ. Snap-frozen hippocampi were homogenized in guanidine buffer, and human Aβ peptides were quantitated by ELISA as described previously (Johnson-Wood et al., 1997). The Aβ1–42 ELISA detects only Aβ1–42, whereas the Aβ1-x ELISA detects Aβ1–40, Aβ1–42, and Aβ1–43, as well as C-terminally truncated forms of Aβ containing amino acids 1–28.
Detection of Aβ deposits. Vibratome sections were incubated overnight at 4°C with biotinylated mouse monoclonal antibody 3D6 (diluted to 5 μg/ml), which specifically recognizes Aβ1–5 (Johnson-Wood et al., 1997; Wyss-Coray et al., 1997). Binding of primary antibody was detected with the Elite kit from Vector Laboratories (Burlingame, CA) using diaminobenzidine and H2O2 for development. Sections were counterstained with 1% hematoxylin and examined with a Vanox light microscope (Olympus Optical, Tokyo, Japan) using a 2.5× objective. The percent area of the hippocampus covered by 3D6-immunoreactive material (“plaque load”) was determined with a Quantimet 570C (Leica, Deerfield, IL). Three immunolabeled sections were analyzed per mouse, and the average of the individual measurements was used to calculate group means. Some sections were double-immunolabeled with a rabbit polyclonal antibody against Aβ (R1280; courtesy of Dr. Dennis Selkoe) and mouse monoclonal antibodies against phosphorylated neurofilaments (SMI312; Sternberger Monoclonals, Baltimore, MD) as described previously (Masliah et al., 1996).
Density of SYN-IR presynaptic terminals. Vibratome sections were incubated overnight with a monoclonal antibody against synaptophysin (1 μg/ml; Boehringer Mannheim, Indianapolis, IN), followed by incubation with fluorescein isothiocyanate-conjugated horse anti-mouse IgG (1:75; Vector Laboratories). Sections were then transferred to SuperFrost slides (Fisher Scientific, Tustin, CA), mounted under glass coverslips with antifading medium (Vector Laboratories), and imaged with a laser scanning confocal microscope (MRC1024; Bio-Rad, Hercules, CA) as described previously (Games et al., 1995; Masliah et al., 1996). For each experiment, we first determined the linear range of the fluorescence intensity of immunoreactive terminals in nontransgenic control sections. This setting was then used, as described previously (Buttini et al., 1999), to collect all images analyzed in the same experiment. For each mouse, 12 confocal images (four per section) of the molecular layer of the dentate gyrus, each covering an area of 7282 μm2, were obtained. Digitized images were transferred to a Macintosh computer (Apple Computers, Cupertino, CA) and analyzed with NIH Image software. The area occupied by SYN-IR presynaptic terminals was quantified and expressed as a percentage of the total image area as described previously (Masliah et al., 1992b; Games et al., 1995).
This method of quantitating SYN-IR presynaptic terminals has been used extensively to assess neurodegenerative alterations in diverse experimental models (Toggas et al., 1994; Games et al., 1995; Buttini et al., 1999) and in diseased human brains (Masliah et al., 1991b,1992a; Knowles et al., 1998). It has also been validated previously by comparisons with quantitative immunoblots (Alford et al., 1994; Mucke et al., 1994), quantitations of synaptic proteins by ELISA (Brown et al., 1998; Buttini et al., 1999), and the optical “disector” approach (Masliah et al., 1991a; Everall et al., 1999; Hsia et al., 1999). To ensure objective assessments and reliability of results, brain sections from mice to be compared in any given experiment were blind coded and processed in parallel. Codes were broken after the analysis was complete.
Statistical analyses. Statistical analyses were performed with the StatView 5.0 program (SAS Institute Inc., Cary, NC). Differences among means were assessed by one-way ANOVA followed by, Dunnett's or Tukey–Kramer post hoc test. Correlation studies were performed by simple regression analysis. The null hypothesis was rejected at the 0.05 level.
RESULTS
Generation of transgenic mice expressing wild-type and FAD-mutant hAPP at comparable levels
The PDGF β chain promoter was used to direct neuronal expression of alternatively spliced minigenes encoding hAPP695, hAPP751, and hAPP770, as described previously (Games et al., 1995; Rockenstein et al., 1995). Four types of hAPP were expressed individually in different lines of transgenic mice (Fig. 1): wild-type hAPP (APPWt), hAPP carrying the FAD-linked (Murrell et al., 1991) 717V→Fmutation (APPInd), hAPP carrying the 717V→F mutation plus the FAD-linked (Mullan et al., 1992) 670/671KM→NL double mutation (APPSw,Ind), and hAPP carrying the 717V→F mutation plus an experimental 671M→I mutation (APPExp,Ind) that inhibits Aβ production in cell culture (Citron et al., 1995). Several independent lines of transgenic mice were established for each construct: 7 for APPWt, 11 for APPInd, 7 for APPSw,Ind, and 15 for APPExp,Ind. The generation of APPInd line H6 (Wyss-Coray et al., 1997) and APPSw,Ind line J9 (Hsia et al., 1999) has been described previously.
Fig. 1.

Summary of transgenic lines. A, Diagram of hAPP indicating the mutations expressed in transgenic mice. FAD-linked mutations are commonly referred to by place of discovery or residence of affected kindred. The 670/671KM→NL double mutation affects a large pedigree in Sweden (Mullan et al., 1992), and the 717V→F mutation was identified in Indiana (Murrell et al., 1991) (numbers refer to amino acids in APP770). Mutations at position 717 are often collectively referred to as “London mutations” based on the first report of the FAD-linked 717V→I mutation (Goate et al., 1991); however, the latter mutation was not studied here. The sequence of Aβ is indicated inbold in single-letter amino acid code.KPI, Kunitz-type protease inhibitor domain. Elements are not drawn to scale. B, Relative levels of cerebral transgene expression (values in parentheses) were determined in different lines of PDGF-hAPP mice as illustrated in Figure 2. The expression level in line I63 was arbitrarily defined as 1.0.
The overall level of cerebral transgene expression in each line was determined by RNase protection assay (Fig.2). Based on this analysis, three groups of transgenic lines, each consisting of two or more lines expressing different hAPP constructs at comparable levels, were selected for further analysis (Fig. 1B). Cerebral hAPP mRNA levels in the highest expresser lines, APPWt I63 and APPInd H6, are similar to those in the APPInd line 109 described previously (Games et al., 1995; Rockenstein et al., 1995). Although we were able to generate lines representing a broad range of expression levels for APPWt, APPInd, and APPSw,Ind, all APPExp,Indlines in which hAPP mRNA could be detected in the brain (n = 15) had low levels of transgene expression (Fig.1B and data not shown). The reasons for this remain to be determined.
Fig. 2.

Identification of wild-type and FAD-mutant hAPP mice with matching levels of cerebral transgene expression.A, Representative autoradiograph showing results of an RNase protection assay. Total RNA was extracted from entire hemibrains. The left lane shows signals of undigested radiolabeled riboprobes; the other lanes contained the same riboprobes plus brain RNA samples, digested with RNases. Each samplelane contains RNA from a different mouse. The hAPP probe detects human but not mouse APP; it also recognizes an SV40 segment (S) of transgene-derived mRNAs.Non-tg, Nontransgenic. B, Phosphorimager quantitation of signals shown in A. Values represent group means ± SD.
In all PDGF-APP mice, cerebral expression of hAPP immunoreactivity was primarily neuronal and widespread across different brain regions, with maximal levels in the neocortex and hippocampus (data not shown), consistent with previous observations (Games et al., 1995; Johnson-Wood et al., 1997).
Effects of hAPP mutations on human Aβ levels
Neocortical and hippocampal levels of Aβ1-x, approximating total Aβ (Johnson-Wood et al., 1997; Gouras et al., 1998), and Aβ1–42were determined by ELISA. Because intraparenchymal Aβ deposits significantly increase the overall Aβ burden, as measured by ELISA (Johnson-Wood et al., 1997), the effects of hAPP mutations on cerebral Aβ production were evaluated at 2–4 months of age, when brains of transgenic mice from all lines were devoid of 3D6-immunoreactive Aβ deposits (see below).
For any given construct, levels of Aβ1-x and Aβ1–42 (Fig. 3) were dependent on overall transgene expression levels (Figs. 1, 2), with the highest Aβ levels seen in the highest hAPP expresser lines. In both the hippocampus (Fig. 3) and neocortex (data not shown), the 717V→F mutation increased the relative proportion of Aβ1–42 without increasing Aβ1-x levels, whereas the 670/671KM→NL double mutation significantly increased Aβ1-x levels, consistent with previous observations (Citron et al., 1992; Cai et al., 1993; Younkin, 1995). Consequently, for a given level of transgene expression, Aβ levels were lower in APPWt mice than in APPSw,Ind mice (Figs. 1-3). No human Aβ could be detected in brains of mice expressing hAPP carrying the experimental 671M→I mutation, confirming in vivothe effects this mutation has in vitro (Citron et al., 1995).
Fig. 3.

Comparison of human Aβ levels in hippocampi of mice expressing wild-type or FAD-mutant hAPP. Aβ1-x and Aβ1–42 were quantitated by ELISA in mice from different transgenic lines (n = 6–9 mice per line) at 2–4 months of age. Values represent group means ± SD. No plaques were detected in the opposite hemibrains of these mice by immunostaining with the 3D6 antibody (data not shown).
Effects of hAPP mutations on formation of amyloid plaques
To assess plaque formation, sections from transgenic and nontransgenic mice were immunolabeled with a monoclonal antibody against Aβ (3D6). At 5–7 months of age, amyloid deposition was detected only in APPSw,Ind mice (Fig.4). Diffuse amyloid immunoreactivity at this age was observed in a laminar pattern in the molecular layer of the dentate gyrus, and a few dense amyloid deposits 4–10 μm in diameter were detected in the deeper layers of the neocortex (data not shown). Both the diffuse plaques and the microplaques lacked a neuritic component. No plaques were detected at 5–7 months in transgenic mice from APPWt lines I5 and I63 or APPInd lines H6 and H40 (7–17 mice per line). At 8–10 months of age, APPSw,Ind lines also had the highest proportion of mice with plaques (Fig. 4), compared with hAPP expression-matched APPInd mice, and the highest hippocampal plaque loads (Fig. 5and data not shown). In both APPInd and APPSw,Ind mice, the onset and extent of plaque formation were influenced by levels of human Aβ, with mice expressing higher levels of Aβ showing earlier and more extensive amyloid deposition (Figs. 3-5), even among lines that were well matched for overall transgene expression (Figs. 1, 2). At 21–27 months of age, the proportion of APPInd mice with plaques increased to 93% in the high expresser H6 line and to 83% in the low expresser H40 line (Fig. 4). Plaques in adult mice were typically larger and denser than those in young mice and showed a prominent neuritic component when double-labeled with antibodies against Aβ and neurofilaments (Fig. 5E). No plaques were detected in nontransgenic mice at 2–27 months of age (n = 84).
Fig. 4.

Hippocampal plaque formation in different lines of FAD-mutant hAPP mice. Aβ deposits were detected by immunostaining of brain sections (n = 3 per mouse) with the 3D6 antibody as described in Materials and Methods. Six to 18 mice per line were analyzed at 2–4, 8–10, and 21–25 months of age, and 1–6 (mean = 4.3) mice per line were analyzed at 5–7 and 11–16 months of age.
Fig. 5.
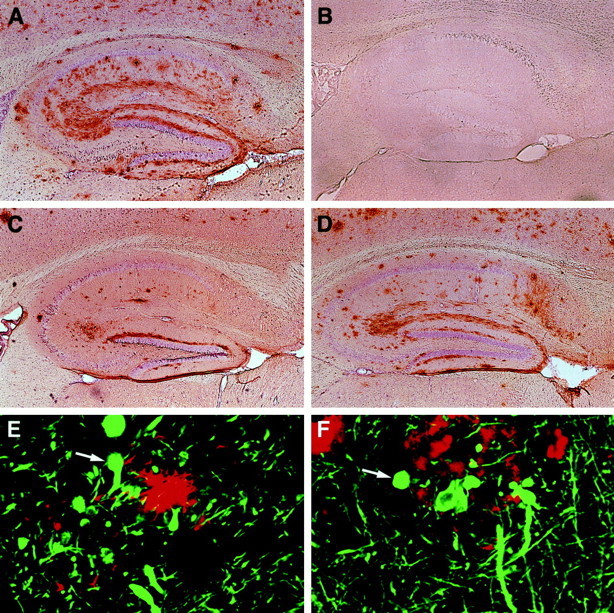
Age-related cerebral Aβ deposition occurs in mice expressing FAD-mutant hAPP but not in mice expressing wild-type hAPP. Brain sections were immunoperoxidase-stained for Aβ with the 3D6 antibody and imaged by light microscopy (A–D) or double-labeled with antibodies against Aβ (R1280; red) and monoclonal antibodies against phosphorylated neurofilaments (SMI312; green) and imaged by laser scanning confocal microscopy (E,F). Hippocampal sections of transgenic mice are shown: A, APPInd line H6 (18 months);B, APPWt line I63 (15 months);C, APPSw, Ind line J9 (10 months);D, APPSw,Ind line J20 (10 months);E, APPInd line H6 (10 months).F, Midfrontal gyrus from a human AD brain. Magnifications: A–D, 4×; E,F, 930×.
Decreased levels of SYN-IR presynaptic terminals are unrelated to plaque load
We showed previously that transgenic mice expressing FAD-mutant hAPP have a decreased density of SYN-IR presynaptic terminals in specific subfields of the hippocampus and that this decrease precedes plaque formation (Games et al., 1995; Hsia et al., 1999). Because diverse factors associated with aging could link parallel processes in time, simulating cause–effect relationships that may not exist, it is critical to compare the effects of plaque load on neurodegeneration within relatively narrow age ranges. To assess whether plaque formation in FAD-mutant hAPP lines exacerbates the decrease in SYN-IR presynaptic terminals in old mice, we compared hippocampal density of SYN-IR presynaptic terminals and plaque load in APPIndand APPSw,Ind mice at 21–27 months of age, when most of these mice have plaques (see above). No correlation was identified between SYN-IR presynaptic terminals and plaque load (Fig.6). At 8–10 months of age, when some mice have plaques and others do not (Fig. 4), the density of SYN-IR presynaptic terminals also did not correlate with plaque load in APPInd mice from line H6 (n = 24,r = 0.015, p = 0.94) or APPSw,Ind mice from lines J9 and J20 (n = 16, r = 0.28, p= 0.29).
Fig. 6.

Density of SYN-IR presynaptic terminals does not correlate with plaque load in mice from different FAD-mutant hAPP lines. Plaque load and density of SYN-IR presynaptic terminals in the hippocampus were determined in 31 transgenic mice from APPInd lines H6, H9, and H40 and APPSw, Indline J9 at 21–27 months of age. No correlation was found between the two variables.
APPWt mice with high Aβ levels do not develop plaques but have decreased levels of SYN-IR presynaptic terminals
Although Aβ1–42 levels in the APPWt line I63 were similar to those in the plaque-bearing APPInd line H6 (Fig.7A), no plaques were detected in mice from APPWt line I63 between 2 and 28 months of age (Figs. 5, 7B). Mice from APPWt line I5 also had no plaques at 8–10 (n = 9 mice) or 24 (n = 2) months of age (data not shown). Despite the lack of plaque formation, mice from APPWt line I63 showed decreases in SYN-IR presynaptic terminals similar to those in mice from APPInd line H6 (Fig. 7C). Simple regression analysis revealed a significant decline in SYN-IR presynaptic terminals with age (age range of 2–28 months) in mice from APPInd line H6 (n = 56,r = 0.30, p = 0.0243) and APPWt line I63 (n = 23,r = 0.53, p = 0.0095) but not in nontransgenic controls (n = 87, r = 0.07, p = 0.53). Because APPIndline H6 and APPWt line I63 are well matched not only for Aβ1–42 (Figs. 3, 7A) but also for overall transgene expression (Figs. 1, 2), their comparable decrease in SYN-IR presynaptic terminals could be attributable to neuronal overexpression of either Aβ1–42 or hAPP. To elucidate the relative importance of hAPP and Aβ in determining the density of SYN-IR presynaptic terminals in these models, we took advantage of the fact that hAPP expression-matched APPWt, APPInd, and APPSw,Ind mice have different levels of Aβ1–42 production. The density of SYN-IR presynaptic terminals and levels of hAPP and its products were assessed in 2- to 4-month-old mice, because at this age, decreases in SYN-IR presynaptic terminals are already detectable in transgenic mice (Fig.7C), and steady-state levels of Aβ can be assessed most reliably because none of the mice have plaques (Fig. 4). First, we examined the relationship between the density of SYN-IR presynaptic terminals and hAPP levels. We found no correlation between these variables across different lines of transgenic mice (Fig.8). Next, we examined whether there was evidence for dose-dependent synaptotoxicity of Aβ. A significant inverse correlation was identified between the density of SYN-IR presynaptic terminals and the levels of Aβ1-xor Aβ1–42 (Fig.9).
Fig. 7.

Comparison of hippocampal Aβ levels, plaque formation, and density of SYN-IR presynaptic terminals in the high expresser lines APPWt I63 and APPInd H6. Note that the cerebral hAPP mRNA levels in these lines are very well matched (Figs. 1, 2). A, Levels of human Aβ were determined at 2–4 months of age in 8–9 mice per line by ELISA.Circles represent values in individual mice;horizontal lines indicate group means. B, Proportion of mice in which 3D6-immunoreactive plaques were identified (black) at the ages indicated (n = 4–18 mice per line and age range). C, The density of SYN-IR presynaptic terminals was determined in 4–41 mice per genotype and age range. Data represent group means ± SD. *p < 0.05, **p < 0.01 versus nontransgenic controls (Tukey–Kramer post hoctest).
Fig. 8.

Density of SYN-IR presynaptic terminals does not correlate with hAPP levels across hAPP mice from different lines. Levels of full-length plus α-secreted hAPP and density of SYN-IR presynaptic terminals in the hippocampus were determined in 36 transgenic mice from APPWt lines I5, I7, and I63, APPInd lines H6 and H40, and APPSw, Ind line J9 at 2–4 months of age. No correlation was identified between the two variables. No plaques were detected in the opposite hemibrains of these mice by immunostaining with the 3D6 antibody (data not shown).
Fig. 9.
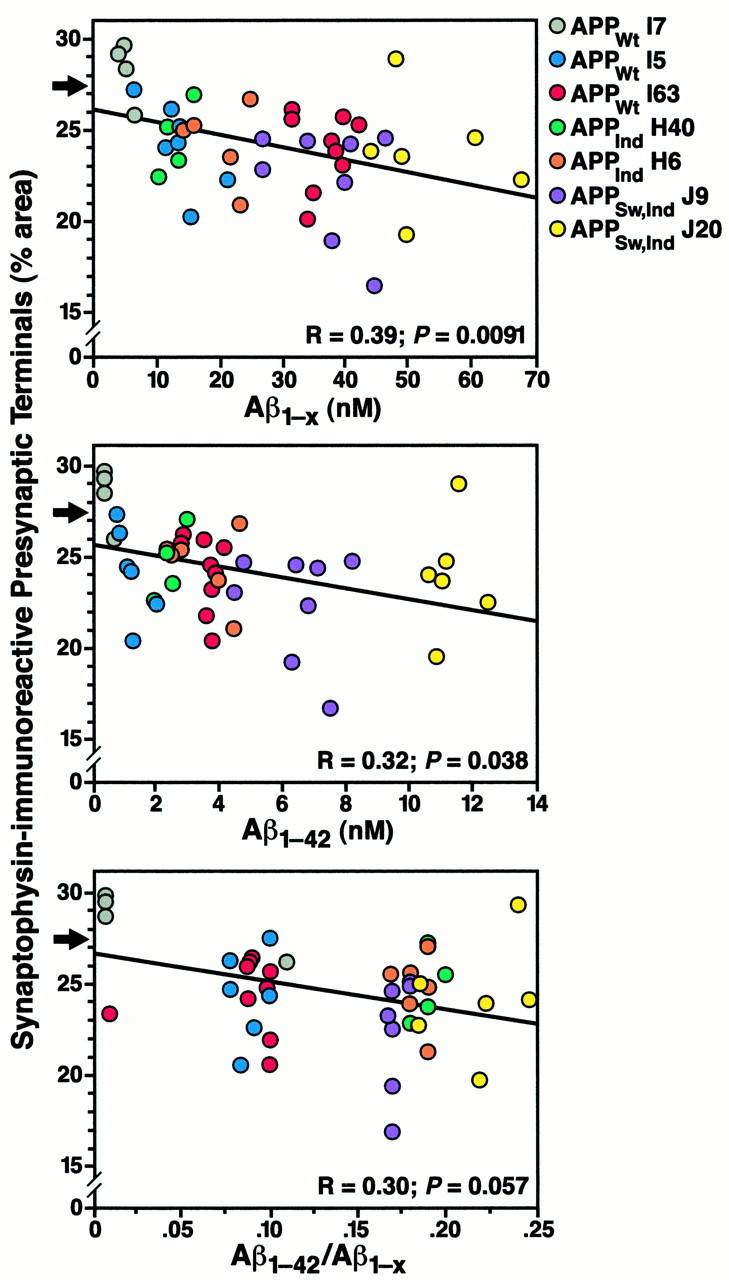
Inverse correlation between density of SYN-IR presynaptic terminals and levels of Aβ. At 2–4 months of age, hippocampal levels of Aβ1-x and Aβ1–42 in one hemibrain were correlated with the hippocampal density of SYN-IR presynaptic terminals in the opposite hemibrain in mice from lines expressing APPWt, APPInd, or APPSw, Ind at different levels (4–9 mice per line). None of these mice had plaques by immunostaining with the 3D6 antibody (data not shown). Arrows indicate the normal density of SYN-IR presynaptic terminals in age-matched nontransgenic controls (mean of 29 mice).
DISCUSSION
High-level neuronal production of Aβ1–42in mice expressing wild-type hAPP did not result in the formation of amyloid plaques but was associated with decreased levels of SYN-IR presynaptic terminals in the molecular layer of the dentate gyrus. Across different wild-type and FAD-mutant hAPP transgenic lines, decreases in SYN-IR presynaptic terminals correlated with Aβ levels but not with hAPP levels or plaque load. These results support a plaque-independent role for Aβ in AD-related synaptic toxicity.
Plaque formation depends on both absolute levels of Aβ1–42 and Aβ1–42/Aβ1–40ratio
In transgenic lines carrying FAD mutations, the onset and progression of plaque formation were closely related to levels of Aβ1–42 expression measured before the development of plaque pathology. In hAPP expression-matched lines containing the 717V→F mutation, plaque formation was accelerated and intensified by the 670/671KM→NL double mutation, which increases Aβ production (Citron et al., 1992; Cai et al., 1993; Younkin, 1995). These findings suggest that critical levels of Aβ1–42 in vulnerable brain regions are necessary for the development of plaques. However, the absence of plaques in line I63 demonstrates that high levels of Aβ1–42 are not sufficient for plaque formation. Line I63 is, to our knowledge, the first APPWt transgenic line that produces Aβ1–42 levels comparable with those in FAD-mutant hAPP mice that develop plaques. The close match in hAPP and Aβ1–42 levels in APPWtline I63 and APPInd line H6 was fortuitous but exceptional, because in other APPInd lines the 717V→F mutation increased Aβ1–42 levels over those identified in APPWt lines. We cannot exclude the possibility that the difference in plaque formation between wild-type and FAD-mutant hAPP lines involves Aβ-independent factors. However, for the following reasons, we favor the hypothesis that differences in Aβ1–42/Aβ1-x ratios play a key role. For unknown reasons, Aβ1-xlevels were higher in APPWt line I63 than in APPInd line H6, resulting in a lower Aβ1–42/Aβ1-x ratio in line I63 (Figs. 3, 7). Compared with APPWt line I63, APPInd line H40 had lower Aβ1-x levels but comparable Aβ1–42 levels (Fig. 3). The higher Aβ1–42/Aβ1-x ratio in line H40 was associated with plaque formation, whereas the lower Aβ1–42/Aβ1-x ratio in line I63 was not. Aβ1–40 accounts for most of the Aβ1-x that does not end at Aβ residue 42 (Gouras et al., 1998). It is conceivable that Aβ1–40 interferes with Aβ1–42 aggregation in vivo, as it does in vitro (Snyder et al., 1994), and that the lack of plaque formation in line I63 reflects an antiamyloidogenic effect of Aβ1–40. If confirmed in future studies, this effect could be exploited therapeutically.
Synaptotoxicity depends on Aβ levels but not hAPP levels, plaque load, or presence of FAD mutations
Decreases in synaptophysin immunoreactivity in specific brain regions correlate well with the severity of cognitive deficits in AD (Terry et al., 1991; Honer et al., 1992; Masliah et al., 1994; Dickson et al., 1995; Sze et al., 1997), highlighting the clinical relevance of this marker. Compared with the decreases in SYN-IR presynaptic terminals in late stages of AD (40%) (Masliah et al., 1994), the decreases we found in hAPP transgenic mice (10–30%) may seem relatively subtle. However, the decreases in SYN-IR presynaptic terminals in hAPP mice were not only statistically significant but were also associated with major synaptic transmission deficits (Hsia et al., 1999), supporting their pathophysiological relevance.
In all transgenic models in which Aβ is expressed from the full-length precursor molecule, overexpression of Aβ is inseparably linked to overexpression of hAPP itself. Because hAPP could affect neuronal function through a number of mechanisms (Milward et al., 1992;Mattson et al., 1993; Greenberg et al., 1994; Multhaup et al., 1996;Okamoto et al., 1996; Masliah et al., 1998), it is important to determine whether hAPP per se is responsible for the neuropathological alterations in these models. In transgenic mice expressing hAPP from the relatively weak neuron-specific enolase promoter, levels of SYN-IR presynaptic terminals were increased in mice with lower levels of hAPP expression but not in mice with higher levels of hAPP expression (Mucke et al., 1994). Those results led us to postulate a bell-shaped dose–response curve for synaptotrophic effects of hAPP at near-physiological levels of hAPP expression (Mucke et al., 1994). The relatively high density of SYN-IR presynaptic terminals in the low expresser APPWt line I7 (Fig. 9) may be consistent with this hypothesis. At higher levels of hAPP expression, the density of SYN-IR presynaptic terminals did not correlate with hAPP levels, suggesting that hAPP overexpression per se is not responsible for the decreased density of these structures in hAPP mice.
Although the high expresser APPWt line I63 did not develop plaques, it showed significant decreases in SYN-IR presynaptic terminals that worsened with age. Thus, FAD mutations are not required for the decrease in SYN-IR presynaptic terminals in hAPP mice, consistent with the loss of these structures in humans with sporadic AD, who also lack FAD mutations. The decreased levels of SYN-IR presynaptic terminals in line I63 also demonstrate that plaques are not required for this deficit to occur. Moreover, across different lines of aged plaque-bearing mice, plaque load did not correlate with the density of SYN-IR presynaptic terminals. If not extracellular deposits of fibrillar Aβ, what is causing the synaptic deficits? Possibilities include neurotoxic effects induced by the intraneuronal accumulation of Aβ or by diffusible forms of extracellular Aβ (Masliah et al., 1996; Turner et al., 1996; Lambert et al., 1998; Lee et al., 1998; Hartley et al., 1999; Hsia et al., 1999; Wilson et al., 1999). Consistent with either of these possibilities and with recent findings in AD (Lue et al., 1999; McLean et al., 1999), decreases in SYN-IR presynaptic terminals in transgenic lines expressing wild-type or FAD-mutant hAPP correlated inversely and plaque-independently with levels of Aβ1-x and Aβ1–42.
In conclusion, our findings suggest that plaque formation is influenced not only by absolute but also by relative levels of Aβ1–42 and Aβ1–40, with relatively high concentrations of Aβ1–40being potentially antiamyloidogenic. Decreases in SYN-IR presynaptic terminals were critically dependent on Aβ levels but not on hAPP levels, plaque formation, or presence of FAD mutations, suggesting that plaque-independent Aβ toxicity could play a key role in the pathogenesis of AD-related neurodegeneration.
Footnotes
This work was supported by National Institutes of Health Grants AG11385 (L. Mucke), AG10869 (E.M.), and AG5131 (E.M.). We thank D. Selkoe for the pCMV695 M596I plasmid and the R1280 antibody; the Transgenic Animal Core Facility of the J. David Gladstone Institutes for generating transgenic founder mice; M. Wogules, I. Lieberburg, D. Schenk, and R. Rydel for critical reading of this manuscript; M. Gordon for preliminary ELISA results; J. Carroll and S. Gonzales for preparation of graphics; G. Howard and S. Ordway for editorial assistance; and D. McPherson for administrative assistance.
Drs. Mucke and Masliah contributed equally to this study.
Correspondence should be addressed to Dr. Lennart Mucke, Gladstone Institute of Neurological Disease, P.O. Box 419100, San Francisco, CA 94141-9100. E-mail: lmucke@gladstone.ucsf.edu.
REFERENCES
- 1.Alford MF, Masliah E, Hansen LA, Terry RD. A simple dot-immunobinding assay for quantification of synaptophysin-like immunoreactivity in human brain. J Histochem Cytochem. 1994;42:283–287. doi: 10.1177/42.2.8288869. [DOI] [PubMed] [Google Scholar]
- 2.Alloul K, Sauriol L, Kennedy W, Laurier C, Tessier G, Novosel S, Contandriopoulos A. Alzheimer's disease: a review of the disease, its epidemiology and economic impact. Arch Gerontol Geriatr. 1998;27:189–221. doi: 10.1016/s0167-4943(98)00116-2. [DOI] [PubMed] [Google Scholar]
- 3.Bartoo GT, Nochlin D, Chang D, Kim Y, Sumi SM. The mean Aβ load in the hippocampus correlates with duration and severity of dementia in subgroups of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:531–540. doi: 10.1097/00005072-199705000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Evolution of neuronal changes in the course of Alzheimer's disease. J Neural Transm [Suppl 105] 1998;53:127–140. doi: 10.1007/978-3-7091-6467-9_11. [DOI] [PubMed] [Google Scholar]
- 6.Brown DF, Risser RC, Bigio EH, Tripp P, Stiegler A, Welch E, Eagan KP, Hladik CL, White CLI. Neocortical synapse density and Braak stage in the Lewy body variant of Alzheimer disease: a comparison with classic Alzheimer disease and normal aging. J Neuropathol Exp Neurol. 1998;57:955–960. doi: 10.1097/00005072-199810000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai X-D, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 9.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 10.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the β-amyloid precursor protein in familial Alzheimer's disease causes increased β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 11.Citron M, Teplow DB, Selkoe DJ. Generation of amyloid β protein from its precursor is sequence specific. Neuron. 1995;14:661–670. doi: 10.1016/0896-6273(95)90323-2. [DOI] [PubMed] [Google Scholar]
- 12.Cummings BJ, Pike CJ, Shankle R, Cotman CW. β-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging. 1996;17:921–933. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 13.D'Hooge R, Nagels G, Westland CE, Mucke L, De Deyn PP. Spatial learning deficit in mice expressing human 751-amino acid β-amyloid precursor protein. NeuroReport. 1996;7:2807–2811. doi: 10.1097/00001756-199611040-00080. [DOI] [PubMed] [Google Scholar]
- 14.Davis JN, Chisholm JC. The “amyloid cascade hypothesis” of AD: decoy or real McCoy? Trends Neurosci. 1997;20:558–559. doi: 10.1016/s0166-2236(97)85989-9. [DOI] [PubMed] [Google Scholar]
- 15.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–304. doi: 10.1016/0197-4580(95)00013-5. [DOI] [PubMed] [Google Scholar]
- 16.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 17.Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E, HNRC Group Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 19.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan J, Yankner BA. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 20.Goate A, Chartier-Harlin M-C, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 21.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 22.Gouras GK, Xu HX, Jovanovic JN, Buxbaum JD, Wang R, Greengard P, Relkin NR, Gandy S. Generation and regulation of beta-amyloid peptide variants by neurons. J Neurochem. 1998;71:1920–1925. doi: 10.1046/j.1471-4159.1998.71051920.x. [DOI] [PubMed] [Google Scholar]
- 23.Greenberg SM, Koo EH, Selkoe DJ, Qiu WQ, Kosik KS. Secreted β-amyloid precursor protein stimulates mitogen-activated protein kinase and enhances τ phosphorylation. Proc Natl Acad Sci USA. 1994;91:7104–7108. doi: 10.1073/pnas.91.15.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-Protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 26.Honer WG, Dickson DW, Glesson J, Davies P. Regional synaptic pathology in Alzheimer's disease. Neurobiol Aging. 1992;13:375–382. doi: 10.1016/0197-4580(92)90111-a. [DOI] [PubMed] [Google Scholar]
- 27.Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer‘s disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang FS, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 29.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knowles RB, Gomez-Isla T, Hyman BT. Aβ associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol. 1998;57:1122–1130. doi: 10.1097/00005072-199812000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SJ, Liyanage U, Bickel PE, Xia WM, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 33.Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masliah E, Fagan A, Terry R, DeTeresa R, Mallory M, Gage R. Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP43 in the dentate gyrus of the adult rat. Exp Neurol. 1991a;113:131–142. doi: 10.1016/0014-4886(91)90169-d. [DOI] [PubMed] [Google Scholar]
- 35.Masliah E, Terry RD, Alford M, DeTeresa RM, Hansen LA. Cortical and subcortical patterns of synaptophysin-like immunoreactivity in Alzheimer's disease. Am J Pathol. 1991b;138:235–246. [PMC free article] [PubMed] [Google Scholar]
- 36.Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA. Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol. 1992a;32:321–329. doi: 10.1002/ana.410320304. [DOI] [PubMed] [Google Scholar]
- 37.Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, DeTeresa R, Terry RD. Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J Neuropathol Exp Neurol. 1992b;51:404–414. doi: 10.1097/00005072-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 39.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett. 1994;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 40.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masliah E, Raber J, Alford M, Mallory M, Mattson MP, Yang D, Wong D, Mucke L. Amyloid protein precursor stimulates excitatory amino acid transport: implications for roles in neuroprotection and pathogenesis. J Biol Chem. 1998;273:12548–12554. doi: 10.1074/jbc.273.20.12548. [DOI] [PubMed] [Google Scholar]
- 42.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 43.Milward EA, Papadopoulos R, Fuller SJ, Moir RD, Small D, Beyreuther K, Masters CL. The amyloid protein precursor of Alzheimer's disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 44.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Van Den Haute C, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 45.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human β-amyloid precursor protein. Proc Natl Acad Sci USA. 1995;92:5341–5345. doi: 10.1073/pnas.92.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid β protein precursor in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 47.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of β-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 48.Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K. The amyloid precursor protein of Alzheimer's disease in the reduction of copper(II) to copper(I). Science. 1996;271:1406–1409. doi: 10.1126/science.271.5254.1406. [DOI] [PubMed] [Google Scholar]
- 49.Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 50.Okamoto T, Takeda S, Giambarella U, Murayama Y, Matsui T, Katada T, Matsuura Y, Nishimoto I. Intrinsic signaling function of APP as a novel target of three V642 mutations linked to familial Alzheimer's disease. EMBO J. 1996;15:3769–3777. [PMC free article] [PubMed] [Google Scholar]
- 51.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 53.Quon D, Wang Y, Catalano R, Scardina JM, Murakami K, Cordell B. Formation of β-amyloid protein deposits in brains of transgenic mice. Nature. 1991;352:239–241. doi: 10.1038/352239a0. [DOI] [PubMed] [Google Scholar]
- 54.Rockenstein EM, McConlogue L, Tan H, Gordon M, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of transgenic mice and humans with Alzheimer's disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 55.Routtenberg A. Measuring memory in a mouse model of Alzheimer's disease. Science. 1997;277:839–841. doi: 10.1126/science.277.5327.839. [DOI] [PubMed] [Google Scholar]
- 56.Snyder SW, Ladror US, Wade WS, Wang GT, Barrett LW, Matayoshi ED, Huffaker HJ, Kraft GA, Holzman TF. Amyloid-β aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys J. 1994;67:1216–1228. doi: 10.1016/S0006-3495(94)80591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Storey E, Cappai R. The amyloid precursor protein of Alzheimer's disease and the Aβ peptide. Neuropathol Appl Neurobiol. 1999;25:81–97. doi: 10.1046/j.1365-2990.1999.00164.x. [DOI] [PubMed] [Google Scholar]
- 58.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 60.Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- 61.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer‘s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 62.Terry RD, Masliah E, Hansen LA. The neuropathology of Alzheimer disease and the structural basis of its cognitive alterations. In: Terry RD, Katzman R, Bick KL, Sisochia SS, editors. Alzheimer disease. Lippincott Williams & Wilkins; Philadelphia: 1999. pp. 187–206. [Google Scholar]
- 63.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 64.Turner RS, Suzuki N, Chyung ASC, Younkin SG, Lee VMY. Amyloids β40 and β42 are generated intracellularly in cultured human neurons and their secretion increases with maturation. J Biol Chem. 1996;271:8966–8970. doi: 10.1074/jbc.271.15.8966. [DOI] [PubMed] [Google Scholar]
- 65.Wilson CA, Doms RW, Lee VM-Y. Intracellular APP processing and Aβ production in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:787–794. doi: 10.1097/00005072-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Amyloidogenic role of cytokine TGF-β1 in transgenic mice and Alzheimer’s disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- 67.Yankner BA. New clues to Alzheimer's disease: unraveling the roles of amyloid and tau. Nat Med. 1996;2:850–852. doi: 10.1038/nm0896-850. [DOI] [PubMed] [Google Scholar]
- 68.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 69.Younkin SG. Evidence that Aβ42 is the real culprit in Alzheimer's disease. Ann Neurol. 1995;37:287–288. doi: 10.1002/ana.410370303. [DOI] [PubMed] [Google Scholar]