

Abstract
The proteolipid protein (PLP) gene encodes two myelin-specific protein isoforms, DM-20 and PLP, which are members of the highly conserved lipophilin family of transmembrane proteins. While the functions of this family are poorly understood, the fact that null mutations of the PLP gene cause leukodystrophy in man is testament to the importance of DM-20 and PLP in normal CNS function. PLP differs from DM-20 by the presence of a 35 amino acid domain exposed to the cytoplasm, which is not encoded by other lipophilin genes and appears to have arisen in amphibians ∼300 million years before present. However, the lipophilin gene family can be traced back at least 550 million years and is represented inDrosophila and silkworms. Thus, from an evolutionary perspective PLP can reasonably be anticipated to perform functions in CNS myelin that cannot be accomplished by other lipophilins. Herein we use a novel knock-in strategy to generate mice expressing wild-type levels of a Plp gene that has been modified to encode only DM-20. Although DM-20 is incorporated into functional compact myelin sheaths in young animals, our data show that the 35 amino acid PLP-specific peptide is required to engender the normal myelin period and to confer long-term stability on this multilamellar membrane.
Keywords: CNS, Drosophila melanogaster, embryonic stem cells, cre recombinase, DM-20, homologous recombination, invertebrate, knock-out mice, lipophilin family, M6a, M6b, myelin, neurodegeneration, Pelizaeus-Merzbacher disease, PLP, proteolipid protein, silkworm
The lipophilin family comprises at least three genes in terrestrial vertebrates: proteolipid protein (PLP), M6a, and M6b (Kitagawa et al., 1993; Yan et al., 1993; Gow, 1997) that encode a larger number of gene products through alternative splicing. Lipophilins are small integral membrane proteins, and several are known to beN-glycosylated (Waehneldt et al., 1987). The correspondence between molecular weights predicted from cDNAs and those determined empirically indicates that lipophilins generally lack cleavable signal peptides, thereby orienting the amino termini toward the cell cytoplasm. Analyses of several family members has revealed four transmembrane domains (Popot et al., 1991; Weimbs and Stoffel, 1992;Gow et al., 1997), and the degree of primary structure conservation suggests this is likely to be a common feature of all family members. The lipophilins have been studied most extensively in mice and humans and are expressed in myelinating cells, neurons, heart, kidney, and thymus (Baumrind et al., 1992; Campagnoni et al., 1992; Lagenaur et al., 1992; Pribyl et al., 1996; Bongarzone et al., 1999).
From an evolutionary perspective, anecdotal evidence suggests that PLP may be functionally unique among lipophilin family members (Kitagawa et al., 1993). PLP differs from its smaller splice isoform, DM-20, by the presence of 35 amino acids in the cytoplasmic domain near the middle of the protein. The 105 nucleotides encoding this PLP-specific peptide comprise exon 3b (DM-20 mRNA splices from exon 3a to 4) and are not found in lower vertebrates. Presumably, the appearance of exon 3b in amphibians ∼300 million years before present imparted new functions on the protein or proteins encoded by the ancestral PLP gene that proved to be advantageous to myelinating cells. Indeed, the importance of this evolutionary event is emphasized by the replacement of P0 with PLP as the dominant integral membrane protein in the CNS of higher vertebrates (Yoshida and Colman, 1996).
Despite extensive efforts, the functions of most lipophilins remain undefined. DM-20 and PLP have been examined in greatest detail, particularly regarding myelin stability. However, most studies have focused on PLP, and few have attempted or succeeded in distinguishing this protein from DM-20 at the level of function. Previously, we demonstrated that although both isoforms share a common topology (Gow et al., 1997), the impact of missense mutations on their intracellular trafficking is often dissimilar (Gow et al., 1994; Gow and Lazzarini, 1996) despite the fact that structural changes in both proteins are detectable using conformationally sensitive antibodies (Gow et al., 1997). Importantly, data from in vitro experiments recapitulate aspects of the DM-20/PLP trafficking defect in the dysmyelinating rumpshaker mouse where mutant DM-20 is incorporated into myelin, but PLP remains largely in the cell body (Mitchell et al., 1992; Gow et al., 1998). Thus, the PLP-specific peptide appears to confer extreme sensitivity to changes in higher-ordered protein structure.
Herein we dissect the functions of DM-20 and PLP by asking a simple question: can DM-20 functionally replace PLP in compact myelin? To answer this question we generated two mutations in the Plpgene using a novel homologous recombination strategy in embryonic stem cells. In the first mutant (the neo mouse) we ablated expression of the Plp gene. The second mutant (therec mouse) was derived from the first but expresses wild-type levels of a Plp gene that is modified to encode only DM-20. We show that these manipulations yield the intended results. Furthermore, neo mice develop a slowly progressive neurodegenerative disease throughout the CNS that is manifested in the second year of life as diminishing motor coordination. Most importantly, this phenotype is not appreciably altered by incorporating DM-20 into compact myelin at levels comparable to that of DM-20 plus PLP in controls. These data convincingly demonstrate that DM-20 cannot functionally replace PLP in compact myelin, and we conclude that the PLP-specific peptide confers properties on PLP that are critical to the long-term stability and normal function of this membrane.
MATERIALS AND METHODS
Constructing the Plp targeting plasmid for homologous recombination. The human PLP cDNA was used to screen 300,000 colonies from a 129 Sv/Ev genomic library in lambda Dash II. Two genomic clones were purified and found to contain exons 2 through 7 of the mouse Plp gene (Fig.1). An 8 kb EcoRI fragment containing exons 2–7 was cloned downstream of a herpes simplex virus-thymidine kinase cassette, and site-directed mutagenesis was used to inactivate the splice-donor site at the 3′ end of exon 3 from AAG↓gtt to AAG↓ctt, thereby creating anHindIII restriction site that can be used to identify this mutant allele; uppercase letters represent the nucleotide sequence of the final codon in exon 3, and lowercase letters reflect the sequence of intron 3. Finally, a single loxP site was also introduced into the 5′ end of intron 3 at position +49 by PCR-mediated, site-directed mutagenesis, and a floxed PGKneo cassette was introduced into this site by cre-mediated recombination inEscherichia coli using double selection on ampicillin/kanamycin plates. The coding strand for thePGKneo cassette selected is the noncoding strand of thePlp gene.
Fig. 1.

Genetic characterization of mice derived from embryonic stem cells. A, Schematic of the wild-typePlp gene and the targeting construct electroporated into ES cells (neo). Exons are numbered 1–7.Arrows show the binding sites of oligonucleotide primers used to identify different Plp alleles.Asterisk, A HindIII site introduced to destroy the splice-donor site of exon 3b; triangles,loxP sites; P, PstI;E, EcoRI. B, Schematic of the targeted Plp gene after removal of thePGKneo cassette with cre recombinase. C,Southern blots distinguish wild-type and rec mice fromneo mice. D, PCR products from tail DNA of male mice distinguish wild-type, neo, andrec alleles. The DNA fragment sizes of the products are indicated at the left. H3, HindIII digestion.
Generation of Plp-null embryonic stem cells,neo, and rec mice. Three homologous recombinant embryonic stem cell (ES) clones were obtained from a Southern blot screen of 300 G418-resistant colonies. Chimeric male mice derived from clone 17 or clone 34 transmitted the Plp-nullallele (neo allele) through the germline, and the knock-out mice generated were phenotypically indistinguishable. Subsequent characterization and genetic manipulations were performed on mice derived from ES cell clone 34. Southern blots of tail DNA digested withPstI and electrophoresed on 0.8% agarose gels yielded 8.5 kb (wild-type allele) or 4.3 kb (knock-out allele) bands when hybridized with a 1.2 kb PstI-EcoRI fragment of intron 1 flanking the 5′ end of the targeting construct (Fig. 1). The presence of the PGKneo cassette was revealed as a 1.8 kb band using a 0.6kb PstI neomycin coding region to probe Southern blots of HindIII/XhoI-digested tail DNA. To generate rec (DM-20 only) mice, male neo(DM-20/PLP absent) mice were mated with superovulated female C57Bl/6tac mice, and the 0.5 d embryos were harvested for pronuclear injection with a supercoiled plasmid, pOG231 (O'Gorman et al., 1991), encoding cre-recombinase driven by the hCMV promoter. Injected embryos were reimplanted in pseudopregnant hosts. This method of effecting recombination is very efficient; PCR analyses showed that thePGKneo cassette was not detected in 14 of 18 female mice (78%) born from surrogate hosts.
Southern blots, Northern blots, and PCR. Blots were hybridized as follows: a prehybridization for 2 hr at 42°C in 50% formamide, 6× SSPE, 5× Denhardt's solution, 25 mmphosphate, pH 6.9, 0.3% SDS, 0.4 mg/ml sonicated salmon sperm DNA, and a 24 hr hybridization at 42°C with 105(Southern blots) or 107 (Northern blots) cpm probe/ml of prehybridization solution. Blots were washed for 2 × 15 min at room temperature in 2× SSPE/ and 1% SDS then 2 × 15 min in 0.2× SSPE and 0.1% SDS at 55°C for all probes. The wild-type neo (DM-20/PLP absent) and rec (DM-20 only) alleles of the Plp gene are routinely distinguished by PCR of tail DNA using 30 cycles of amplification at 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. The primers used are: AG25, 5′-AAC ACC ACC GGA GCA GTC AGG CAG A-3′; AG263, 5′-CTC ACT CAA AAG CTA GGC CTC AGG AG-3′, and; AG264, 5′-AGA ACG AGA TCA GCA GCC TCT GTT CC-3′. In male mice, AG25/263 primers generate a 300 bp product from the wild-type allele, and AG25/264 primers generate a 360 bp fragment from the Plp-knockout allele. The PGKneo cassette is deleted in rec (DM-20 only) mice; thus, the AG25/264 primers do not yield a product. However, the AG25/263 primers generate a 300 bp wild-type product and a 335 bp product from the rec(DM-20 only) allele. Syntheses of cDNAs were primed with oligo-dT using Moloney murine leukemia virus reverse transcriptase (Life Technologies, Gaithersburg, MD). Two primers, AG25 and AG11 (5′-AAT GTA CAC AGG CAC AGC AGA GCA G-3′) are used to amplify cDNAs encoding PLP (252 bp) and DM-20 (146 bp). FokI digestion of the PLP product yields two DNA fragments of 155 and 97 bp.
Western blots. Mouse brains were homogenized at 4°C in 0.32 m sucrose centrifuged over sucrose step gradients to float the myelin membrane fractions (Norton, 1974). Purified water-shocked myelin or whole-brain homogenates were dissolved in modified sample buffer (Laemmli, 1970) containing 5% SDS and 0.5% β-mercaptoethanol, heated to 50°C for 10 min and electrophoresed overnight at 40 V on 12.5% SDS-polyacrylamide slab gels (20 μg protein/lane). Proteins were transferred to nitrocellulose at 60 V for 5 hr in 0.03% SDS, 25 mm Tris buffer, pH 8.3, 0.192 m glycine, and 20% methanol. The blots were blocked in 5% milk powder dissolved in TBS for 1 hr. Primary antibodies were diluted into 0.5% milk and TBS and incubated at room temperature overnight, and secondary antibody-alkaline phosphatase conjugates were incubated with the blots for 1 hr then developed using nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate.
Immunocytochemistry. Mice were perfused intracardially with fresh 4% paraformaldehyde/0.1 m sodium phosphate buffer, pH 7.4, for 45–60 min before the brains were dissected and embedded in OCT. Cryostat sections were stained as previously described (Gow et al., 1998) with antibodies against several myelin proteins: Rt αPLP diluted 1:1 (clone AA3); Ms αMOG at 1:1 (clone 8–18C5); Ms αMBP and Ms αCNPase at 1:1000 (Sternberger).
Electron microscopy and immunogold labeling. Six-month-old mice were perfused through the left ventricle briefly with saline, followed by 2% paraformaldehyde, 1% glutaraldehyde, and 0.1m sodium phosphate buffer, pH7.2, and finally with 4% glutaraldehyde and 0.1 m sodium phosphate buffer at 4°C. Cerebellum, optic nerve, and cervical spinal cord were post-fixed in osmium, block-stained with uranyl acetate, and embedded in Epon (Friedrich and Mugnaini, 1981). Average compact myelin period was measured in cerebellum, optic nerve, and cervical spinal cord from electron micrographs at 125,000× using a micrometer lens. For immunogold labeling, thin sections from cerebellar white matter were etched with saturated sodium metaperiodate for 10 min, quenched for 30 min with 50 mm NH4Cl in 0.1m phosphate buffer, pH7.4, treated for 30 min with 0.2% Triton X-100 in PBS, and blocked for 30 min in 1% BSA–fish gelatin. Sections were then labeled with Rt anti-PLP antibodies (undiluted hybridoma; clone AA3) for 1 or 19 hr and 10 nm gold-conjugated to Gt-anti-rat antibodies (EY Labs, San Mateo, CA) for 2 hr then contrasted for 5 min with 2% uranyl acetate.
Rotarod analyses. A 2 d training and testing protocol was used to measure motor coordination using an Ugo Basile (Varese, Italy) rotarod according to the procedure by Gow et al. (1999). Day 1 of the protocol was used to train the mice, and on day 2, the average of three time intervals for which the mice remained on an accelerating horizontal rod was recorded. A cohort of mice, comprising six wild-type, six neo (DM-20/PLP absent; triangles), and fiverec (DM-20 only; circles) males, were subjected to the rotarod analyses at 6, 12, 14, 15, and 16 months of age.
RESULTS
Homologous recombination in mouse ES cells is a powerful technique with which to uncover the functions of proteins in vivo. In the vast majority of cases, genes are inactivated by deletion of the proximal promoter or coding region, and function is assessed in the absence of the gene product or products. Strategies have also been developed to introduce subtle mutations, such as single amino acid changes (Wu et al., 1994); however, a difficulty with these strategies is that mutations that cause lethality in young hemizygotes, or severely reduce fertility, preclude the establishment of breeding colonies. We have developed a strategy to introduce potentially lethal changes into genes, which enables the generation of breeding colonies provided that infertility or early death does not ensue in the absence of expression of the gene. This methodology is particularly advantageous for sex-linked genes because most ES cells in common use are derived from the blastocysts of male embryos, which harbor a single copy of the X chromosome.
Generation of knock-out (neo) and DM-20 only (rec) mice
In the current study, we introduced a potentially lethal exon 3 splice-donor mutation into the Plp gene on theX-chromosome using a two-stage procedure. Previous studies have shown that ablating Plp gene expression is tolerated in the first year of life, after which these animals develop an overt neurological phenotype characterized by demyelination and neuronal degeneration (Boison and Stoffel, 1994; Klugmann et al., 1997). Thus, in the first stage we prepared a targeting construct and inserted aPGKneo gene into the noncoding strand of the Plpgene in intron 3 (Fig. 1A, neo allele). Black rectangles in the diagram represent coding exons 2–7 of thePlp gene. Exon 3 is divided into 3a and 3b at the cryptic splice-donor site, which is used to generate mRNA encoding DM-20. White boxes show the noncoding regions of exons 1 and 7, respectively. ThePGKneo gene is inserted 49 bp downstream of the canonical exon 3b splice-donor site AG↓gt, which is inactivated in the targeting construct with a single nucleotide change to AG↓ct (Fig.1A, asterisk). This mutation generates aHindIII restriction enzyme site, which is used to detect the mutated splice site.
Although antisense suppression has previously been used to generatePlp-null mice (Boison and Stoffel, 1994), we augmented this design with 34 bp loxP sites that flank thePGKneo cassette (Fig. 1A, triangles). This important modification makes possible the removal of thePGKneo cassette by cre-recombinase-mediated recombination in the second stage of this study, a manipulation that reactivates expression of the mutant Plp locus and leaves behind a single loxP site in intron 3 (Fig. 1B, triangle). Importantly, this recombination event does not remove the disrupted splice-donor site of exon 3b (Fig. 1B, asterisk), the function of which is to preclude the generation of mRNA encoding PLP (white rectangle in exon 3). Thus, the resultantrec (DM-20 only) mice can only synthesize DM-20 from the mutant Plp locus. Southern blots distinguish wild-type,neo (DM-20/PLP absent), and rec mice (Fig.1C); however, we routinely use three oligonucleotide primers (Fig. 1A) to genotype wild-type (+/Y), neo (neo/Y) andrec (rec/Y) male mice by PCR (Fig.1D), and the mutated exon 3 splice-donor site is revealed by HindIII (Fig. 1D, H3) digestion of PCR products.
Genetic manipulation of the Plp gene appropriately modifies gene expression
To determine if our genetic manipulations in ES cells produced the desired results in mice we examined expression of the mutatedPlp locus in neo and rec male mice at the levels of steady-state RNA and protein (Fig.2). Maximal expression of thePlp gene in mouse brain occurs at ∼18 d after birth, and Northern blots probed with a PLP cDNA show strong expression of this gene in wild-type mice (Fig. 2A). In contrast,Plp expression is not detectable in neo mice, indicating that antisense-mediated suppression by the PGKneocassette is operating as expected. Furthermore, from long exposures of these Northern blots we are unable to detect a hybridization signal from neo mice, and we estimate that expression of thePlp gene is suppressed by at least 1000-fold. On the other hand, after cre recombinase-mediated excision of the PGKneocassette, the level of Plp expression in rec mice is indistinguishable from wild-type littermates. These data demonstrate that neither the mutated exon 3 splice site nor the presence of theloxP site in intron 3 alter the stability of mRNA derived from the mutant Plp gene. These data are verified by Western blotting of purified myelin (Fig. 2B) or total homogenate from the brains of mutant mice at p18 using a monoclonal antibody that recognizes the C-termini of both PLP and DM-20. WhilePlp gene products are undetectable in neo mice, only DM-20 is present in myelin from rec mice. Importantly, the amount of DM-20 from these animals approximates the additive amounts of PLP and DM-20 in wild-type myelin, suggesting that the level of Plp gene expression in rec mice is normal but that all processed transcripts encode DM-20.
Fig. 2.

Analyses of Plp gene expression from wild-type, neo, and rec male mice.A, Northern blots showing equal steady-state levels of mRNA in wild-type and rec mice. B,Western blots showing equal levels of Plp gene products in wild-type and rec mice. C, RT-PCR analyses of Plp gene expression in wild-type,neo, and rec mice at postnatal day 18 and embryonic day 13.5. Molecular sizing markers are shown at theleft.
In addition to Northern and Western blotting, we used RT-PCR to detect very low levels of Plp expression in neo andrec mice. Besides the maximal expression of thePlp gene in mice at p18, a small peak of expression also occurs in the brain during midgestation (Ikenaka et al., 1992; Timsit et al., 1992). The primers used for RT-PCR hybridize to exons 3 and 4 (Fig. 1A, AG25, AG11) and distinguish PLP and DM-20 encoding mRNAs by size (Fig. 2C). At p18, most of the mRNA in wild-type brain encodes PLP, which is reflected in the relative band intensities for PLP and DM-20. In neo mice,Plp gene products are not detected even after using the PCR sample as the template in a second round of amplification for 20 cycles. Only mRNA encoding DM-20 is detected in rec mice, which demonstrates that the cryptic splice donor site at the 3′ end of exon 3a is efficiently used. At e13.5 we detect mRNAs encoding both PLP and DM-20 and the relative band intensities indicate that DM-20 mRNA is the dominant species. The identity of the PLP band was further verified by digestion of the PCR products with the FokI restriction enzyme to yield the anticipated 155 and 97 bp fragments (data not shown). The expression of mRNAs encoding PLP and DM-20 has previously been reported in human fetuses (Grever et al., 1997) but not in mouse embryos (Ikenaka et al., 1992; Timsit et al., 1992); thus, the current data provide a satisfying demonstration of the evolutionary conservation ofPlp gene expression. In rec embryos, only mRNA encoding DM-20 is detected, which again shows that the exon 3a splice-donor site is efficiently used.
Myelinogenesis appears normal in neo andrec mice
To determine if myelinogenesis proceeds normally in the mutant mice we stained parasagittal cryostat sections of the neocortex from p18 mice with antibodies against DM-20/PLP and myelin-oligodendrocyte glycoprotein (MOG). MOG is generally viewed as a late marker of myelinogenesis, and its expression appears to coincide with the final stages of myelin sheath formation. DM-20/PLP-positive myelin sheaths are visible throughout the cortex of wild-type mice (Fig.3a), which are also strongly stained with MOG antibodies (Fig. 3d). Several oligodendrocyte cell bodies are present in this field (arrows) that are stained by both antibodies. Although lacking PLP, oligodendrocytes and myelin sheaths in the recmice are indistinguishable from wild-type littermates in size and shape as well as in antibody staining (Fig. 3c,f). Immunogold labeling (Fig. 3i) of compact myelin in the deep cerebellar white matter of rec mice demonstrates that DM-20 is incorporated into compact myelin similarly to wild-type animals (Fig. 3g). Myelin sheaths in neo mice are not labeled with anti-DM-20/PLP antibodies (Fig. 3b,h); however, morphologically normal oligodendrocytes and myelin sheaths are stained with anti-MOG antibodies (Fig. 3e).
Fig. 3.
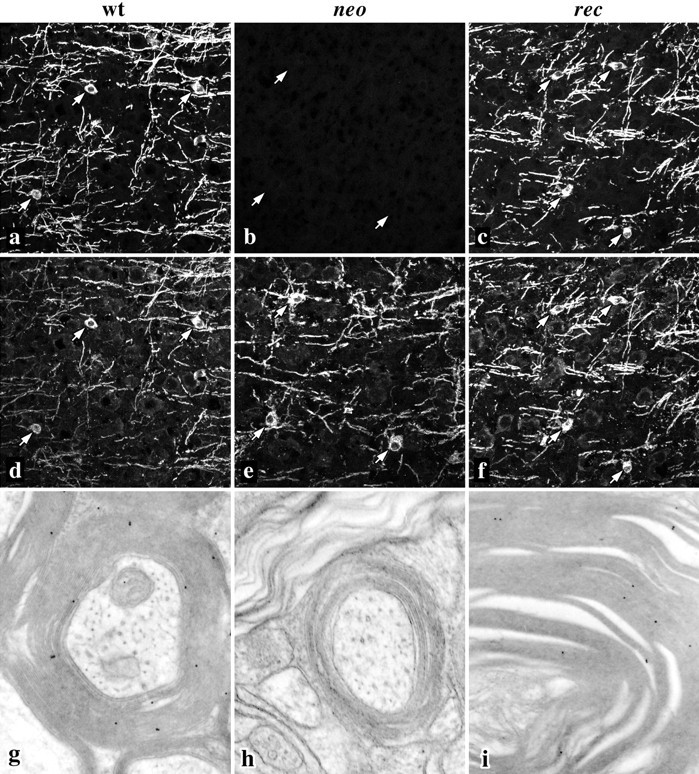
Myelinogenesis appears normal in the cortex of wild-type, neo, and rec mice. Confocal micrographs show myelin sheaths and oligodendrocyte cell bodies stained with antibodies against DM-20/PLP (a–c) and MOG (d–f). DM-20/PLP staining in neomice is absent, but arrows mark the positions of oligodendrocyte cell bodies that stain normally with anti-MOG antibodies. g–i, Immunogold labeling (10 nm) of thin sections from cerebellar white matter tracts. Antibodies recognizing both DM-20 and PLP label compact myelin sheaths in wild-type and rec mice but do not label sheaths fromneo mice. Magnifications: a–f, 160×;g–i, 27,000×.
CNS compact myelin period is perturbed in neo andrec mice
A further demonstration of the nonequivalent functions of PLP and DM-20 is apparent at high magnification in electron micrographs of cerebellar white matter. Transverse sections of compact myelin from wild-type, neo, and rec mice are shown in Figure4 where closed circles mark the major dense lines of the myelin sheaths from neo andrec mice, and open circles highlight these structures in wild-type myelin. The most prominent pathological features are statistically significant changes in the period of neo andrec myelin compared to controls: the period of wild-type myelin is 9.4 nm, whereas the periods for neo andrec myelin are 8.3 and 11.3 nm, respectively (Table1).
Fig. 4.

Compact myelin period is altered inneo and rec mice. Electron micrographs of regions of compact myelin in the cerebellum of 6-month-old mice.Filled circles highlight the major dense lines inneo and rec myelin, and open circles highlight wild-type major dense lines.
Table 1.
The period of compact myelin is perturbed in the CNS ofneo and rec mice
| Genotype | Myelin spacing (nm) (mean ± SEM) | n |
|---|---|---|
| Wild type | 9.4 ± 0.10 | 10 |
| neo | 8.3 ± 0.10* | 10 |
| rec | 11.3 ± 0.12* | 10 |
*p < 0.0001 (ANOVA).
Aging neo and rec mice develop motor coordination abnormalities
Previous studies show that the behavioral phenotype of youngPlp-null mice is indistinguishable from wild-type littermates; however, as these mutants age they develop widespread demyelination in the CNS that is manifested by reduced proficiency in a rotarod motor performance test. Accordingly, we used this analysis to determine if DM-20 can mimic the function of PLP and avert the development of neurological abnormalities in aging rec mice. Wild-type mice (Fig. 5,squares) perform consistently over the duration of the experiment and remain on the rod for ∼240 sec. Althoughrec mice (Fig. 5, circles) perform well in this analysis up to 1 year of age, their proficiency decreases shortly thereafter and is similar in time course and severity to the slowly progressive disease that stems from the absence of PLP inneo mice. The performance of neo mice (Fig. 5,triangles) in the current study is similar to findings from other laboratories (Boison and Stoffel, 1994; Klugmann et al., 1997). Thus, the rotarod data show that Plp gene products are critical for normal function in the CNS. Moreover, the current study provides strong evidence that replacement of PLP with roughly equal amounts of DM-20 in compact myelin causes disease in aging mice and is a clear demonstration that the cytoplasmic PLP-specific peptide encoded by exon 3b imparts unique functions on PLP that cannot be recapitulated by DM-20.
Fig. 5.

Rotarod analyses from a cohort of mice between 6 and 16 months. Wild-type mice, Squares;neo mice, triangles; recmice, circles. Data represents the means from five (rec) or six (wild-type or neo) mice at different ages ± SEM.
Neurodegeneration is similar in neo andrec mice
To further characterize the phenotype of rec mice identified in the rotarod analyses, plastic-embedded sections from optic nerve, cerebellum (Fig. 6), and cervical spinal cord were examined by light and electron microscopy. Six-month-old mice were chosen for these experiments to determine if pathology is apparent at a significantly earlier age than that suggested from the rotarod performance analyses. Semithin sections from wild-type mice (Fig. 6a) show a region of white matter (WM) subjacent to the granular (IGL) and molecular layers (ML) in the cerebellum. Within the white matter, longitudinal and transverse myelinated fibers are evenly stained with toluidine blue. In contrast, white matter regions from neo and rec mice appear disorganized, and darkly stained fibers scattered throughout the sections (Fig. 6b,c, arrows) are indicative of degenerative disease. Occasional redundant myelin profiles are also apparent in the mutants that seem to be more common in neo mice than inrec mice (arrowheads). The granular and molecular layers in neo mice appear normal, although there is some evidence of pathology in the granular layer of rec mice.
Fig. 6.
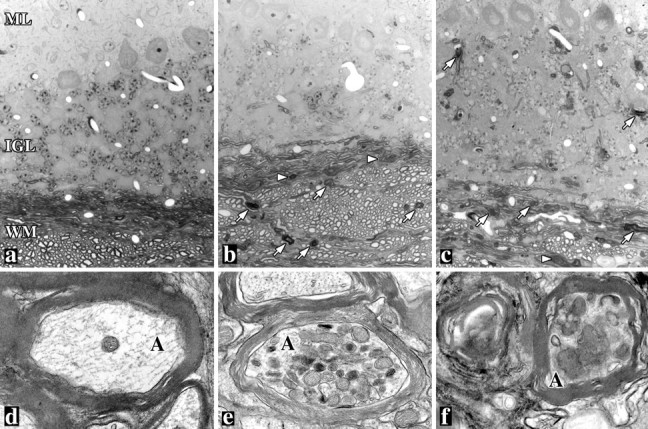
White matter tract degeneration in the cerebella of 6-month-old mutant mice. Light (a–c) and electron (d–f) micrographs from plastic sections of wild-type (a, d), neo (b, e), and rec (c, f) mice show widespread degenerative changes in the mutants, including degenerating myelin sheaths (arrows) and redundant myelin (arrowheads). Axonal spheroids filled with mitochondria are evident in electron micrographs fromneo and rec mice. ML, Molecular layer; IGL, internal granular layer;WM, white matter; A, axon. Magnifications: a–c, 350×; d–f, 18,000×.
Ultrastructural examination of white matter from neo andrec mice reveals widespread neurodegenerative changes, including axonal spheroids filled with mitochondria and other organelles (Fig. 6e,f), degenerating axons, redundant myelin sheaths, myelin vacuolization, and diffuse gliosis (data not shown). We did not observe this pathology in wild-type littermates. Thus, the prominent pathology in different brain regions fromneo and rec mice at least 6 months before the manifestation of a behavioral phenotype indicates that the absence of PLP induces a slowly progressive disease beginning at an early age and only becomes apparent after reaching a clinical threshold in the second year of life. Furthermore, the similarities in pathology betweenneo and rec mice is further evidence that DM-20 cannot substitute for PLP in the CNS. From the current analysis we cannot formally exclude the possibility that disease in therec mice arises from an inherent toxicity of DM-20; in absolute terms, this protein is present in the myelin of recmice at approximately fivefold higher levels than in wild-type sheaths. Nonetheless, we consider this explanation to be unlikely to account for pathogenesis in rec mice because of the similarity of disease to that in neo mice in terms of both time course and severity.
The lipophilin family is represented in invertebrates
Kitagawa et al. (1993) have presented convincing evidence that three lower vertebrate genes DMα, DMβ, andDMγ are the antecedents of PLP, M6a, andM6b in terrestrial vertebrates, respectively, and have estimated that the lipophilin gene family arose ∼440 million years before present. However, recent sequence data indicates that this gene family is also represented in invertebrates. An expressed sequence tag (EST) clone encoding the N-terminal 220 amino acids of an invertebrate gene from the silkworm, Bombyx mori, with homology to vertebrate lipophilins (GenBank accession number AU000676) shares 83% similarity at the amino acid level with three ESTs from the fruit fly, Drosophila melanogaster (GenBank accession numbers AA141336, AA696516, and AA816816). We have used these ESTs to delineate a Drosophila gene on chromosome 3L/78D4 (GenBank accession numbers AC017581 and AC010049) comprising five exons with canonical splice sites and spanning 4 kb of genomic sequence. We have verified the Flybase sequences by PCR cloning and sequencing this gene from wild-type Drosophila. Interestingly, exon B is alternatively spliced in a manner analogous to the M6a andM6b genes in mammals (Olinsky et al., 1996) (A. Gow, unpublished data). The open reading frame of the Drosophilagene encodes a protein with 39–48% similarity to vertebrate lipophilins (Fig. 7). Moreover, inspection of the exon boundaries reveals a surprising level of conservation with mammalian lipophilins (Fig. 7,arrowheads). In the Drosophila gene, the exon boundaries divide codons between two exons (i.e., the codons are split after the first or second nucleotide), and the splice site positions within these codons are conserved in the hM6b (GenBank accession numbers AC003035 and AC003037), hDM-20/PLP(GenBank accession number Z73964) and hM6a genes (Olinsky et al., 1996). Such a precise maintenance of gene structure is consistent with a high degree of conservation through evolution and lends strong support for inclusion of this invertebrate gene in the lipophilin family. Of particular interest is the penultimate exon in theDrosophila lipophilin gene, which is homologous to exons 3a and 4 of the PLP gene. With the sequencing of theDrosophila genome now complete, it is likely that this organism harbors a single lipophilin gene in which a peptide with homology to the PLP-specific peptide is absent (i.e., exon 3b ofPLP).
Fig. 7.

Amino acid alignments of vertebrate and invertebrate lipophilins. ClustalW alignment of human (h), Drosophila(d), and silkworm (s) lipophilins. Amino acid identities between different proteins appear on a darkly shaded background, whereas similarities are shown on a light background. Downturned arrowheads mark the exon boundaries for the human PLP and M6bgenes; upturned arrowheads mark the exon boundaries forDrosophila M6a.
DISCUSSION
We have generated two Plp mutant mice using homologous recombination in ES cells to determine if PLP and DM-20 are functionally equivalent in CNS compact myelin. In the first mutant, we have knocked-out the Plp gene (neo mice) and have demonstrated that these animals exhibit a slowly progressive neurodegenerative disease in the CNS that begins before 6 months of age and is detectable using a motor coordination test in the second year of life. These data accord with the findings from two other groups who have ablated Plp gene expression in mice (Boison et al., 1995; Klugmann et al., 1997). In the second mutant, we have knocked-in a single base pair change into the Plp gene, which forces all mRNA derived from this modified gene to encode DM-20. Although this protein is incorporated into compact myelin, rec mice exhibit a slowly progressive neurodegenerative disease in the CNS that is similar in time course and severity to disease in the knock-out mice (neo mice). Thus, the central feature of the current study is a clear demonstration that DM-20 cannot functionally replace PLP in compact CNS myelin. The simplest interpretation of these data is that the PLP-specific peptide confers on PLP unique properties that are crucial for normal CNS myelin compaction and long-term stability.
Several aspects of pathology observed in the neo andrec mice have been documented in naturally occurring hypomyelinating mutants such as jimpy, shiverer, andquaking mice (Rosenfeld and Friedrich, 1983; Hogan and Greenfield, 1984). These animals harbor deletions or mutations in genes that are expressed in oligodendrocytes and neurons and encode proteins with disparate functions, from serving as a structural component in compact myelin to modulating alternative splicing of myelin gene transcripts. Together with the current data, these observations suggest that pathogenesis is likely to reflect the perturbation of a crucial aspect of the axoglial interaction that must be maintained throughout the life of an animal for normal function, rather than to reveal specific properties of the particular gene products under investigation. In this light, the age at which a behavioral phenotype becomes apparent may be a useful indicator of the extent of disruption to the axoglial unit and highlights the need for standardized test paradigms to detect such abnormalities.
A number of groups have generated transgenic mice harboring supernumerary copies of the Plp gene to overexpress DM-20 and PLP or have introduced into the germline transgenes encoding one or the other of these proteins (Mastronardi et al., 1993; Kagawa et al., 1994; Nadon et al., 1994; Readhead et al., 1994; Johnson et al., 1995). Although CNS pathology in these animals bears some resemblance to that of the mutant mice examined in the current study, the age at which behavioral symptoms become apparent in the other studies are positively correlated with the extent of transgene overexpression and often appear a few months after birth (Ikenaka and Kagawa, 1995). In recmice, we demonstrate at the levels of steady-state RNA and protein that the expression of the “Dm-20 gene“ is comparable to the level of wild-type Plp gene expression in littermate controls. Furthermore, pathogenesis in rec mice (normal levels of Plp gene product) is unchanged from that inneo mice (no Plp gene products). These data indicate that the phenotype of the rec mice is unlikely to be related to changes in gene expression stemming from our genetic manipulation of intron 3 of the Plp gene.
Although there are no significant differences observed in the time course and severity of neurodegeneration between neo andrec mice, we have consistently observed subtle alterations of compact myelin period throughout the CNS of these mutants. On the one hand, the absence of DM-20 and PLP in the myelin membrane causes a 12% reduction in period, whereas on the other hand, replacement of PLP with comparable levels of DM-20 causes the myelin period to increase by 20% above normal. The mechanism underlying these changes is unknown, but similar shifts in period have previously been documented in the dysmyelinating mutants. For example, Duncan (1990) has shown that the reduced period observed in the md rat mutant stems from a collapse of the spacing between the extracellular surfaces of adjacent myelin lamellae at the intraperiod line. The Plp gene in these animals harbors a missense mutation that disrupts the trafficking of DM-20 and PLP through the secretory pathway (Gow, unpublished data); thus md myelin is largely devoid of these proteins and may be akin to myelin in the neo mice. These data identify regulation of the spacing at the apposition of the extracellular membrane surfaces in compact myelin as a major function of DM-20 and PLP.
We are unable to offer a substantiated explanation for the expanded myelin period in rec mice; however, the absence of the PLP-specific peptide in myelin from these animals does not account for the abnormality because this peptide domain is also absent inneo myelin where membrane period is decreased. An alternative explanation is that the intraperiod line may be increased in rec mice compared to controls. In considering possible mechanisms that may mediate this increase, we note that DM-20 and PLP share a common topology (Gow et al., 1997) and that the amino acid sequences of the extracellular domains of these proteins are identical. Nevertheless, the topography of the extracellular domains may be distinct and governed by the relative positions of charged residues in the cytoplasmic regions of DM-20 and PLP (Fig.8). Previous studies show that the boundaries between extramembranous and transmembrane domains are often punctuated with charged amino acids (Hartmann et al., 1989). This rule of thumb reasonably describes the C-terminal residues of the PLP-specific peptide, His-Pro-Asp-Lys, which abut the third transmembrane domain beginning, Phe-Val-Gly-Ile. In the absence of the PLP-specific peptide, the cytoplasmic domain of DM-20 contains charged residues, but the closest is five residues removed from the third transmembrane domain, i.e.,Lys-Gly-Leu-Ser-Ala-Thr abuts Phe-Val-Gly-Ile. Placement of Lys at the cytoplasmic surface of the lipid bilayer would cause the insertion of Gly-Leu-Ser-Ala-Thr into the membrane and could reasonably be expected to dislocate several amino acids at the other side of the bilayer, thereby increasing the size of the second extracellular domain and forcing apart the intraperiod line.
Fig. 8.

Topographical models of PLP and DM-20 in a membrane bilayer. Four transmembrane domains, a–d, are depicted for both proteins (Gow et al., 1997) in which numbers represent the amino acids in the vicinity of the membrane surfaces. In this model, the amino acids K150 of PLP and K110 of DM-20 define the beginning of the third transmembrane domain for each protein (Hartmann et al., 1989). Note that F151 in PLP is at the bilayer surface but that F116 in DM-20 is displaced toward the center of the membrane by the five amino acids, Gly-Leu-Ser-Ala-Thr. This displacement could increase the size of the second extracellular domain.
In addition to studies in mammals, the lipophilin gene family has been extensively characterized in marine vertebrates. Three genes have been identified in multiple species: DMα, DMβ, andDMγ, none of which have been found to encode a peptide homologous to the terrestrial vertebrate PLP-specific peptide. Herein we show that the lipophilin family arose substantially earlier than previously thought and is represented in invertebrates that diverged from chordates at least 550 million years before present. Hydrophobicity profiles of the invertebrate lipophilins identify four domains of sufficient length to span the bilayer, which suggests that the overall topology of these proteins may be similar to other family members. Amino acid alignments with the second extracellular domains of the vertebrate lipophilins are of particular interest (Table2). The four cystine residues which delineate this domain are conserved in all lipophilins and have been shown for DM-20/PLP to form disulfide bridges in vivo(Weimbs and Stoffel, 1992). We have previously suggested that the lipophilin family may be divided into two subgroups on the basis of a short motif in this domain, with unknown function, between the second and third conserved cystine residues in group A lipophilins (Gow, 1997). The invertebrate lipophilin proteins exhibit little homology with this motif and greatest similarity to the DMβ/M6agenes in group B.
Table 2.
Amino acid consensus sequences for two subgroups in the lipophilin family
| Lipophilin group | Protein | Amino acid sequences of second extracellular domains | |||||
|---|---|---|---|---|---|---|---|
| A | hDM-20/hM6b | W | C | CΦD | PWNAFPGK Φ CG | C | |
| DMα/DMγ | W | C | CVD | PWNASPGK Φ CG | C | ||
| Consensus* | W X2 | C X16–17 | CΦD X7 | PWNA PGK Φ CG | X6 | C | |
| B | hM6a | W | C | CLD | TIGEEKKIC | C | |
| DMβ | W | C | CFD | PIHEQKTVC | C | ||
| sM6a | W | C | CID | VKQEDMKIC | C | ||
| dM6a | W | C | CID | TKLEDMKVC | C | ||
| Consensus* | W X2 | C X8–11 | CΦD X7–11 | E. ΦC | X8–9 | C | |
*Single-letter codes are used to specify amino acids; X denotes any amino acid; Φ denotes an hydrophobic amino acid. DM-20/PLP, M6a, and M6b sequences are from human; DMα, DMβ, and DMγ sequences are from Squalus acanthias; dM6a sequence is fromDrosophila melanogaster; sM6a sequence is from the silkworm,Bombyx mori.
In summary, we have identified functional differences in the proteins encoded by the PLP gene using a novel knock-in strategy, and we conclude that DM-20 cannot functionally replace PLP in compact CNS myelin. Mice that do not express the Plp gene develop a slowly progressive neurodegenerative disease in the CNS in the second year of life, and we demonstrate that DM-20 fails to rescue this phenotype, even when present in myelin at levels that are comparable to the endogenous levels of PLP and DM-20 in wild-type littermates. Finally, these results accord with arguments made on evolutionary grounds that the presence of the PLP-specific peptide confers on PLP unique properties that are absent in DM-20.
Footnotes
This work was supported by grants to A.G. from the National Multiple Sclerosis Society of America (RG2891A1) and the Human Frontiers Science Program (RG318/97), and a doctorate fellowship to B.S. from the University of Modena. We thank Valerie Williams (Department of Cell Biology, Mount Sinai School of Medicine) who immunogold-labeled sections for electron microscopy.
Correspondence should be addressed to Alexander Gow, Brookdale Center for Developmental and Molecular Biology, Box 1020, Mount Sinai School of Medicine, One Gustave L. Levy Place, New York, NY 10029. E-mail:ag4@doc.mssm.edu.
REFERENCES
- 1.Baumrind NL, Parkinson D, Wayne DB, Heuser JE, Pearlman AL. EMA: a developmentally regulated cell-surface glycoprotein of CNS neurons that is concentrated at the leading edge of growth cones. Dev Dynamics. 1992;194:311–325. doi: 10.1002/aja.1001940407. [DOI] [PubMed] [Google Scholar]
- 2.Boison D, Stoffel W. Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc Natl Acad Sci USA. 1994;91:11709–11713. doi: 10.1073/pnas.91.24.11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boison D, Bussow H, D'Urso D, Muller H-W, Stoffel W. Adhesive properties of proteolipid protein are responsible for the compaction of CNS myelin sheaths. J Neurosci. 1995;15:5502–5513. doi: 10.1523/JNEUROSCI.15-08-05502.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bongarzone ER, Campagnoni CW, Kampf K, Jacobs EC, Handley VW, Schonmann W, Campagnoni AT. Identification of a new exon in the myelin proteolipid protein gene encoding novel protein isoforms that are restricted to the somata of oligodendrocytes and neurons. J Neurosci. 1999;19:8349–8357. doi: 10.1523/JNEUROSCI.19-19-08349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campagnoni CW, Garbay B, Micevych P, Pribyl T, Kampf K, Handley VW, Campagnoni AT. DM20 mRNA splice product of the myelin proteolipid protein gene is expressed in the murine heart. J Neurosci Res. 1992;33:148–155. doi: 10.1002/jnr.490330119. [DOI] [PubMed] [Google Scholar]
- 6.Duncan ID. Dissection of the phenotype and genotype of the X-linked myelin mutants. In: Skoff RP, Colman DR, editors. Myelination and dysmyelination, Vol 605. New York Academy of Sciences; New York: 1990. pp. 110–121. [DOI] [PubMed] [Google Scholar]
- 7.Friedrich VL, Mugnaini E. Preparation of neural tissues for electron microscopy. In: Heimer L, Robards M, editors. A handbook of neuroanatomical tract tracing techniques. Plenum; New York: 1981. pp. 345–375. [Google Scholar]
- 8.Gow A. Redefining the lipophilin family of proteolipid proteins. J Neurosci Res. 1997;50:659–664. doi: 10.1002/(SICI)1097-4547(19971201)50:5<659::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 9.Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- 10.Gow A, Friedrich VL, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- 11.Gow A, Gragerov A, Gard A, Colman DR, Lazzarini RA. Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J Neurosci. 1997;17:181–189. doi: 10.1523/JNEUROSCI.17-01-00181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA. CNS myelin and sertoli cell tight junction strands are absent in Osp/Claudin 11-null mice. Cell. 1999;99:649–659. doi: 10.1016/s0092-8674(00)81553-6. [DOI] [PubMed] [Google Scholar]
- 14.Grever WE, Weidenheim KM, Tricoche M, Rashbaum WK, Lyman WD. Oligodendrocyte gene expression in the human fetal spinal cord during the second trimester of gestation. J Neurosci Res. 1997;47:332–340. doi: 10.1002/(sici)1097-4547(19970201)47:3<332::aid-jnr11>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann E, Rapoport TA, Lodish HF. Predicting the orientation of eukaryotic membrane-spanning proteins. Proc Natl Acad Sci USA. 1989;86:5786–5790. doi: 10.1073/pnas.86.15.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan EL, Greenfield S. Animal models of genetic disorders of myelin. In: Morell P, editor. Myelin. Plenum; New York: 1984. pp. 489–534. [Google Scholar]
- 17.Ikenaka K, Kagawa T. Transgenic systems in studying myelin gene expression. Dev Neurosci. 1995;17:127–136. doi: 10.1159/000111281. [DOI] [PubMed] [Google Scholar]
- 18.Ikenaka K, Kagawa T, Mikoshiba K. Selective expression of DM-20, an alternatively spliced myelin proteolipid protein gene product, in developing nervous system and in nonglial cells. J Neurochem. 1992;58:2248–2253. doi: 10.1111/j.1471-4159.1992.tb10970.x. [DOI] [PubMed] [Google Scholar]
- 19.Johnson RS, Roder JC, Riordan JR. Over-expression of the DM-20 myelin proteolipid causes central nervous system demyelination in transgenic mice. J Neurochem. 1995;64:967–976. doi: 10.1046/j.1471-4159.1995.64030967.x. [DOI] [PubMed] [Google Scholar]
- 20.Kagawa T, Ikenaka K, Inoue Y, Kuriyama S, Tsujii T, Nakao J, Nakajima K, Aruga J, Okano H, Mikoshiba K. Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron. 1994;13:427–442. doi: 10.1016/0896-6273(94)90358-1. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa K, Sinoway MP, Yang C, Gould RM, Colman DR. A proteolipid protein gene family: expression in sharks and rays and possible evolution from an ancestral gene encoding a pore-forming polypeptide. Neuron. 1993;11:433–448. doi: 10.1016/0896-6273(93)90148-k. [DOI] [PubMed] [Google Scholar]
- 22.Klugmann M, Schwab MH, Puhlhofer A, Schneider A, Zimmermann F, Griffiths IR, Nave K-A. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18:59–70. doi: 10.1016/s0896-6273(01)80046-5. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Lagenaur C, Kunemund V, Fischer G, Fushiki S, Schachner M. Monoclonal M6 antibody interferes with neurite extension of cultured neurons. J Neurobiol. 1992;23:71–88. doi: 10.1002/neu.480230108. [DOI] [PubMed] [Google Scholar]
- 25.Mastronardi FG, Ackerley CA, Arsenault L, Roots BI, Moscarello MA. Demyelination in a transgenic mouse: a model for multiple sclerosis. J Neurosci Res. 1993;36:315–324. doi: 10.1002/jnr.490360309. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell LS, Gillespie SC, McAllister F, Fanarraga ML, Kirkham D, Kelly B, Brophy PJ, Griffiths IR, Montague P, Kennedy PG. Developmental expression of major myelin protein genes in the CNS of X-linked hypomyelinating mutant rumpshaker. J Neurosci Res. 1992;33:205–217. doi: 10.1002/jnr.490330204. [DOI] [PubMed] [Google Scholar]
- 27.Nadon NL, Arnheiter H, Hudson LD. A combination of PLP and DM20 transgenes promotes partial myelination in the jimpy mouse. J Neurochem. 1994;63:822–833. doi: 10.1046/j.1471-4159.1994.63030822.x. [DOI] [PubMed] [Google Scholar]
- 28.Norton WT. Isolation of myelin from nerve tissue. Methods Enzymol. 1974;31:435–444. doi: 10.1016/0076-6879(74)31049-x. [DOI] [PubMed] [Google Scholar]
- 29.O'Gorman S, Fox DT, Wahl GM. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science. 1991;251:1351–1355. doi: 10.1126/science.1900642. [DOI] [PubMed] [Google Scholar]
- 30.Olinsky S, Loop BT, DeKosky A, Ripepi B, Weng W, Cummins J, Wenger SL, Yan Y, Lagenaur C, Narayanan V. Chromosomal mapping of the human M6 genes. Genomics. 1996;33:532–536. doi: 10.1006/geno.1996.0231. [DOI] [PubMed] [Google Scholar]
- 31.Popot J-L, Pham-Dinh D, Dautigny A. Major myelin proteolipid: the 4-alpha-helix topology. J Membr Biol. 1991;120:233–246. doi: 10.1007/BF01868534. [DOI] [PubMed] [Google Scholar]
- 32.Pribyl TM, Campagnoni C, Kampf K, Handley VW, Campagnoni AT. The major myelin protein genes are expressed in the human thymus. J Neurosci Res. 1996;45:812–819. doi: 10.1002/(SICI)1097-4547(19960915)45:6<812::AID-JNR18>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 33.Readhead C, Schneider A, Griffiths IR, Nave K-A. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 34.Rosenfeld J, Friedrich VL. Axonal swellings in jimpy mice: does lack of myelin cause neuronal abnormalities? Neuroscience. 1983;10:959–966. doi: 10.1016/0306-4522(83)90233-6. [DOI] [PubMed] [Google Scholar]
- 35.Timsit SG, Bally-Cuif L, Colman DR, Zalc B. DM-20 mRNA is expressed during the embryonic development of the nervous system of the mouse. J Neurochem. 1992;58:1172–1175. doi: 10.1111/j.1471-4159.1992.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 36.Waehneldt TV, Matthieu J-M, Malotka J, Joss J. A glycosylated proteolipid protein is common to CNS myelin of recent lungfish (Ceratodidae, Lepidosirenidae). Comp Biochem Physiol. 1987;88B:1209–1212. doi: 10.1016/0305-0491(87)90025-3. [DOI] [PubMed] [Google Scholar]
- 37.Weimbs T, Stoffel W. Proteolipid protein (PLP) of CNS myelin: positions of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implications for the membrane topology of PLP. Biochemistry. 1992;31:12289–12296. doi: 10.1021/bi00164a002. [DOI] [PubMed] [Google Scholar]
- 38.Wu H, Liu X, Jaenisch R. Double replacement: strategy for efficient introduction of subtle mutations into the murine Col1a-1 gene by homologous recombination in embryonic stem cells. Proc Natl Acad Sci USA. 1994;91:2819–2823. doi: 10.1073/pnas.91.7.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan Y, Lagenaur C, Narayanan V. Molecular cloning of M6: identification of a PLP/DM20 gene family. Neuron. 1993;11:423–431. doi: 10.1016/0896-6273(93)90147-j. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida M, Colman DR. Parallel evolution and coexpression of the proteolipid proteins and protein zero in vertebrate myelin. Neuron. 1996;16:1115–1126. doi: 10.1016/s0896-6273(00)80138-5. [DOI] [PubMed] [Google Scholar]