

Abstract
Hypoxia-induced suppression of NMDA receptors (NMDARs) in western painted turtle (Chrysemys picta) cortical neurons may be critical for surviving months of anoxic dormancy. We report that NMDARs are silenced by at least three different mechanisms operating at different times during anoxia. In pyramidal neurons from cerebrocortex, 1–8 min anoxia suppressed NMDAR activity (Ca2+ influx and open probability) by 50–60%. This rapid decrease in receptor activity was controlled by activation of phosphatase 1 or 2A but was not associated with an increase in [Ca2+]i. However, during 2 hr of anoxia, [Ca2+]i in cerebrocortical neurons increased by 35%, and suppression of NMDARs was predicted by the increase of [Ca2+]i and controlled by calmodulin. An additional mechanism of NMDAR silencing, reversible removal of receptors from the cell membrane, was found in cerebrocortex of turtles remaining anoxic at 3°C for 3–21 d. When suppression of NMDARs was prevented with phosphatase inhibitors, tolerance of anoxia was lost. Silencing of NMDARs is thus critical to the remarkable ability of C. picta to tolerate life without oxygen.
Keywords: anoxia, turtles, NMDA receptor, intracellular calcium, phosphatase, receptor downregulation
Typical mammalian neurons are exquisitely sensitive to oxygen deprivation, suffering irreversible injury after brief periods of anoxia. In contrast, western painted turtles (Chrysemys picta) are unusually tolerant of anoxia, surviving 24–48 hr of anoxia at 25°C and 4–5 months at 2–3°C during winter dormancy (Ultsch and Jackson, 1982). Survival of neurons in these remarkable animals may involve an “arrest” of ion channels that decreases excitability, reduces ion translocation, and preserves [ATP] during the energetic stress imposed by anaerobic conditions (Hochachka, 1986; Lutz, 1992). Indeed, sodium channel abundance and NMDA receptor (NMDAR) activity decrease in C. picta neurons during anoxia (Perez-Pinzon et al., 1992; Buck and Bickler, 1995; Bickler, 1998).
One of the more striking features of the adaptation of turtle neurons to anoxia is their ability to maintain [Ca2+]i at near normal levels. Excessive Ca2+ influx and Ca2+ toxicity that occurs in anoxic mammalian neurons does not occur in turtle neurons (Bickler, 1998). This is a critical adaptation because ionized [Ca2+] in C. picta CSF increases more than sixfold after just 10 d of anoxia and must reach levels far greater after several months of dormancy (Cserr et al., 1988). NMDAR suppression appears to be critical for avoiding excessive Ca2+ influx under these conditions (Bickler and Buck, 1998), but the mechanisms by which NMDARs are controlled to accomplish this are largely unknown. Adenosine participates in the regulation of NMDARs in turtle cerebrocortex during anoxia. However, adenosine does not appear to explain all of the receptor downregulation because adenosine A1receptor antagonists fail to fully prevent NMDAR suppression (Buck and Bickler, 1995, 1998). In addition, adenosine increases and decreases in a cyclic manner during anoxia (Lutz and Kabler, 1997), whereas NMDAR suppression is maintained more or less constant over hours to weeks (Bickler, 1998).
The regulation of NMDAR activity by phosphorylation of one or more subunits is an important mechanism in the plasticity of glutamatergic synapses (Swope et al., 1999). We hypothesized that suppression of NMDAR function during anoxia might be controlled by mechanisms similar to those involved in the long-term depression (LTD) of mammalian glutamatergic synapses. Suppression of NMDARs in LTD is exerted by the activation of phosphatase 1/2A or the calcium–calmodulin-dependent phosphatase calcineurin (Mulkey et al., 1993, 1994; Tokuda and Hatase, 1998). The latter is a possible mechanism in turtle neurons because [Ca2+]i increases 70–100 nm during anoxia (Bickler, 1998). Finally, because sodium channel abundance decreases during anoxia (Perez-Pinzon et al., 1992), it is possible that NMDARs are similarly downregulated as a mechanism of suppressing receptor function. In this paper, we report that NMDARs are silenced by at least three different mechanisms operating at different times during anoxia: dephosphorylation requiring minutes, Ca2+-dependent control operating over several hours, and removal of receptors from the cell membrane over days to weeks.
MATERIALS AND METHODS
These studies were sanctioned by the University of California at San Francisco Committee on Animal Research and conform to relevant National Institutes of Health guidelines for the care of experimental animals. C. picta collected in spring, summer, and autumn were obtained from Lemberger (Oshkosh, WI). The animals were mainly females and weighed 250–650 gm.
All tissue used in these studies was obtained from the cerebrocortex, which is a 1-mm-thick sheet of tissue in this species. After decapitation, the entire brain was removed and placed in oxygenated (95% O2–5% CO2) turtle artificial CSF (aCSF) at 3–5°C (aCSF in mm: 97 NaCl, 26.5 NaHCO3, 2.0 NaH2PO4, 2.6 KCl, 2.5 CaCl2, 2.0 MgCl2, 20 glucose, and 10 HEPES, pH 7.4 at 20°C). Six to eight 3 × 4 mm pieces of cerebrocortex was obtained from each cortex by cutting with fine scissors (Blanton et al., 1989). Hippocampal slices from Sprague Dawley rats were obtained by standard methods (Dingledine, 1984).
NMDA receptor function in turtle neurons was assessed with cell-attached patch-clamp recordings and by measuring NMDAR-mediated Ca2+ fluxes (NMDA Δ Ca2+) with fura-2. Pyramidal neurons used for both patch-clamp recording and [Ca2+]imeasurements are located within 50 μm of the ventral surface of the cortical sheets.
Patch-clamp analysis of turtle NMDA receptor open probability. Cell-attached patch-clamp recordings of NMDAR currents and open probability were measured and analyzed as described by Buck and Bickler (1998). Cortical sheets were supported by nylon mesh in a recording chamber and held in place by a coil of platinum wire. Perfusate was gravity-fed (flow of 2–3 ml/min) from glass bottles gassed with either 95% O2–5% CO2 or 95% N2–5% CO2. During anoxic experiments, the head space above the recording chamber was continuously flushed with 95% N2–5% CO2 gas. Less than 8 min was required to decrease the PO2 (Clark oxygen electrode) in the chamber to <1 mmHg. Studies were done at 25°C.
Single-channel NMDAR recordings were made with fire-polished 6–10 MΩ electrodes containing (in mm): NaCl 115, CsCl 5, CaCl2 2.5, EGTA 10, HEPES acid 10, glycine 0.001, and NMDA 0.01, pH 7.4. Cell-attached 5–20 GΩ seals were obtained using a blind technique. Four diagnostic criteria were used to identify single-channel NMDAR currents (Buck and Bickler, 1998).
NMDAR activity measured by Ca2+influx. We also assessed the activity of cortical NMDARs by measuring the increase in [Ca2+]i (NMDA Δ Ca2+) during application of NMDA to cortical sheets or acutely dissociated neurons. Increase in [Ca2+]i was measured with fura-2. The methods for dissection, loading cortical sheets with fura-2, and measuring [Ca2+]i changes are described by Buck and Bickler (1995). During fura-2 loading, slices were continuously bubbled with 95% O2–5%CO2 or 95% N2–5% CO2, depending on planned experiments. NMDA Δ Ca2+ was measured during application of NMDA (final concentration of 100 or 200 μm) to cortical sheets mounted on a specially designed holder in a fluorometer cuvette. Action potentials and neurotransmitter release that might be triggered by NMDA under these conditions was prevented with 1 μmtetrodotoxin, 0.1 μm ω-conotoxin GIVa, and 0.5 μm agatoxin IVa. In pilot experiments, we found that blocking L-type voltage-gated Ca2+ channels with 1 μm nimodipine or 100 μmBa2+ did not significantly change measured NMDA Δ Ca2+. Therefore, we assume that application of NMDA causes an increase in [Ca2+]i that is essentially Ca2+ influx through the NMDAR. In studies with dissociated neurons, 1 μmglycine and 0.2 mmMg2+ was used to increase Ca2+ influx via the NMDAR.
Dissociated neurons from the cerebral cortex were obtained after digestion of cortical sheets with 0.05% trypsin and 0.01% pronase. The tissue was triturated onto coverslips coated with Cell-Tak (Collaborative Research, Bedford MA). Cells were loaded with 2 μm fura-2 AM for 15 min, and then [Ca2+]i changes during NMDA application was measured with a Photon Technology International (Brunswick, NJ) fluorometer system and an inverted microscope (Bickler and Hansen, 1998). Coverslips were mounted in a gas tight recording chamber perfused with aCSF. NMDA was delivered to the chamber via an Automate Inc. (Berkeley, CA) perfusion system. In experiments involving anoxia, 1 mm sodium dithionite was added to the perfusate solutions to keep PO2<0.5 mmHg.
Cell viability measurements. Cell viability was assessed by histological appearance of cortical pyramidal neurons in sections of cortical sheets. Sheets were fixed in paraformaldehyde, dehydrated, and embedded in paraffin. They were sectioned (7 μm) and stained with hematoxlyn–eosin and examined with light microscopy. A blinded observer then recorded the percentage of surviving cells in 50 × 100 μm areas. Two of these areas were examined in each slice, and the results were averaged. Dead cells were identified by holes left in the sections, or by cells with grossly altered nuclei or cytoplasm. For comparison, measurements of viability were also made in rat CA1 neurons in hippocampal slices as described previously (Bickler and Hansen, 1998).
Trypan blue exclusion was also used as an index of viability in cortical sheets. Tissue was incubated in 0.2% trypan blue for 10 min after an insult and/or recovery. After rinsing in aCSF for 10 min, the presence of blue staining was assessed visually with a dissecting microscope.
ATP concentration was measured with a luciferin–luceriferase assay as described by Buck and Bickler (1995).
NMDAR abundance in membrane fractions from brain cortex homogenates was measured with SDS-PAGE–Western blots. Total protein in the membrane fractions was measured with a standard spectrophotometric assay (Enhanced Protein assay; Pierce, St. Louis, MO), and equal amounts of protein were added to each lane. Immunoblotting to detect turtle NMDARs was done with rat NMDA NR1 antibodies, which reacted strongly with the turtle analog. Primary and secondary antibodies and blocking protein were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Quantitation of gel staining was done with NIH Image software.
Statistics. Statistical tests were performed using JMP (SAS Institute, Cary, NC). Results in the text and figures are reported as mean ± SE.
RESULTS
Comparative survival of mammalian and turtle neurons during anoxia
To investigate the basis for differences in anoxia sensitivity of turtle and mammalian neurons, we measured membrane potential, ATP levels, trypan blue exclusion, and neuron histology in turtle cortical neurons and rat hippocampal CA1 neurons. In turtle neurons during a 3 hr period of in vitro anoxia (medium bubbled with 95% N2–5% CO2 and containing 1 mm dithionite, PO2 <0.1 mmHg), membrane potential remained stable between −65 and −80 mV, [ATP] remained within 80% of control, trypan blue was excluded, and histological appearance was retained. In contrast, >85% of rat CA1 neurons were killed after 10 min of anoxia. The noncompetitive NMDAR antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) prevented death of rat neurons (Fig. 1A). Thus, at least one difference between turtle and mammalian neurons is an absence of NMDAR-mediated cell death in turtle neurons during anoxia.
Fig. 1.

Comparison of Chrysemyscerebrocortical neurons and rat CA1 neurons. A, Differences in viability of turtle and rat neurons 5 hr after bouts of anoxia. *p < 0.05 denotes significant difference between turtle and rat groups (ANOVA). B, Anoxia protects turtle neurons from NMDA neurotoxicity, but not when NMDA receptor activity is stimulated with 0.1 nm forskolin or 0.5 μm okadaic acid. *p < 0.05 denotes significant difference from control (Dunnett's test).Numbers above bars indicate number of slices studied, and error bars show 1 SEM.
Is NMDAR silencing critical for cell survival during anoxia?
To examine the coupling between NMDAR function and cell survival or death in turtle neurons, cell survival after exposure to NMDA under oxic (95%O2–5%CO2) and anoxic conditions (95% N2–5% CO2) was measured. In the presence of oxygen, >80% of pyramidal neurons in cortical sheets were killed by exposure to NMDA (100 μm NMDA for 1 hr, wash for 5 hr). With exposure to 200 μm NMDA for 2–4 hr or with 100 μm NMDA and an overnight recovery period, 90–100% of neurons were killed. However, NMDA toxicity was prevented by a 1 hr pretreatment with anoxia, suggesting that anoxia attenuates NMDAR-dependent cell death (Fig. 1B). When the activity of NMDARs was increased by treatment with 0.1 nm forskolin or 0.5 μmokadaic acid (see Figs. 4B, 6C), cell death during anoxia increased significantly. Cell death in anoxic forskolin-treated slices was prevented by 10 μmMK-801. Thus, survival of turtle neurons during anoxia is dependent specifically on reduction in NMDAR-mediated events.
Fig. 4.

Role of phosphatases in the regulation of NMDA receptors during anoxia. A, Example showing that the nonspecific phosphatase inhibitor okadaic acid prevents NMDA receptor inactivation in a dissociated pyramidal neuron during anoxia.B, Phosphatase inhibitors prevent receptor silencing in cortical sheets during anoxia. Bars show mean NMDA Δ Ca2+ responses during normoxia, anoxia, and anoxia with 1 μm calyculin or 120 nm okadaic acid.Numbers above bars indicate n values. *p < 0.05 compared with oxic control (Dunnett's test). C, Decreases in NMDA receptor open probability during anoxia are prevented with calyculin A (1 μm). *p < 0.05 denotes significant difference from control (Dunnett's test); n = 9, both groups.D, Calyculin does not affect NMDA receptor open probability with oxygen available; n = 5.
Fig. 6.
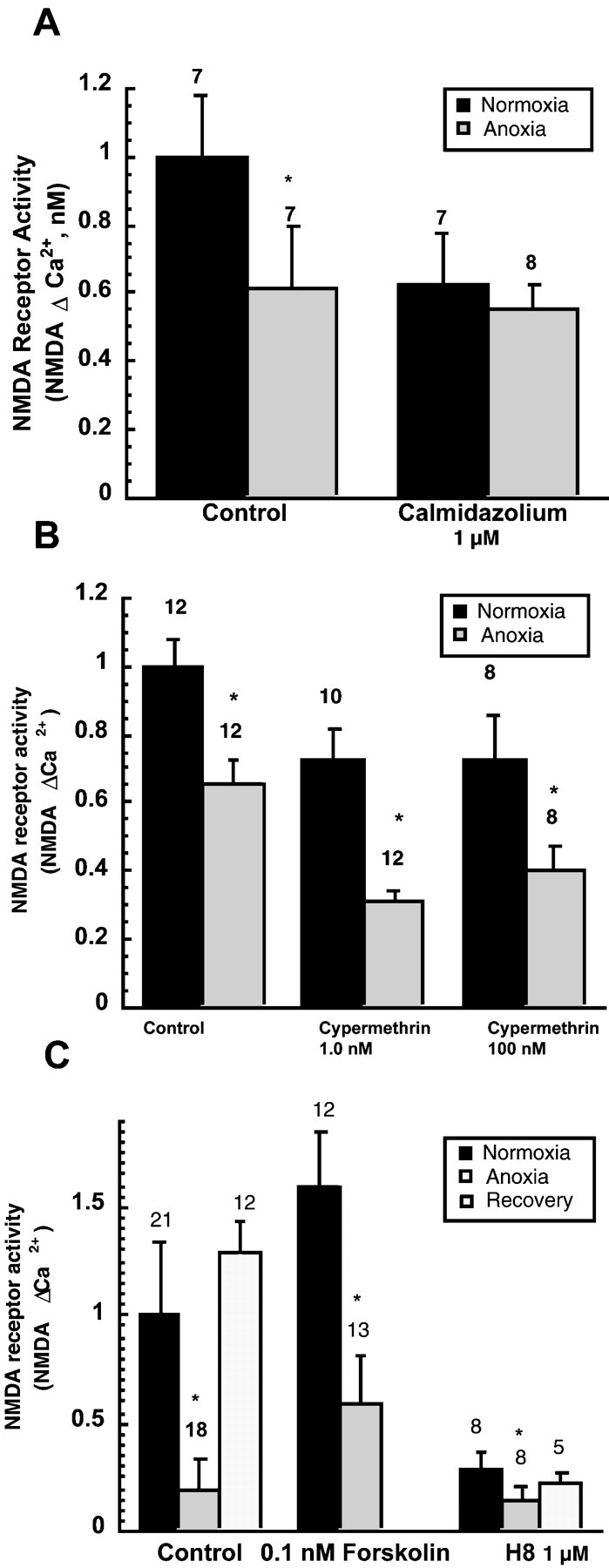
Regulation of NMDA receptor activity during anoxia by Ca2+-dependent processes. A, Inhibition of the Ca2+ binding protein calmodulin reduced NMDA receptor silencing during anoxia. Cortical sheets were treated with 10 nm calmidazolium 30 min before and during 2 hr anoxia or with O2 present. NMDA Δ Ca2+ was then measured. B, The calcineurin inhibitor cypermethrin, at two concentrations, did not prevent NMDA receptor silencing during anoxia. C, Effects of increasing PKA activity with forskolin or inhibiting it with H8 on NMDA receptor activity during normoxia or anoxia. Test compounds were present during the 2 hr period before assay of receptor activity. For all graphs, bars show means ± SEM, and numbersare number of observations; and *p < 0.05 denotes significant difference compared with control condition within each group (ANOVA).
Changes in NMDAR activity during anoxia
The apparent lack of NMDAR-dependent cell death in anoxic turtle neurons led us to test whether NMDARs are suppressed during anoxia and by what mechanisms. To do this, we measured NMDAR activity with patch-clamp electrodes and with fura-2 measurements of NMDA Δ Ca2+. In cell attached patches, 15–90 min of anoxia reduced NMDAR open probability by 50–59% (n= 9, p < 0.01) (Fig.2A). The first significant decrease in receptor open probability was seen 15 min after the beginning the anoxic perfusate. Because 7–8 min is required before the PO2 in the recording chamber falls to <1 mmHg (Clark O2 electrode), this decrease in receptor activity probably requires <7–8 min to occur. No change in single-channel receptor current amplitude during anoxia was observed.
Fig. 2.

Decrease in NMDA receptor activity during anoxia.A, Decrease in mean NMDA receptor open probability in cell-attached patches during 90 min of anoxia. Patch pipettes contained 10 μm NMDA. *p < 0.05 denotes significant decrease compared with normoxic control (Dunnett's test). Error bars show 1 SEM; n = 6–8 for each data point. B, Examples of [Ca2+]i changes produced by 100 μm NMDA (arrows) in cortical sheets in normoxic and anoxic conditions and after recovery from anoxia.C, Mean NMDA receptor activity (NMDA Δ Ca2+) in cortical sheets during anoxia and recovery.D, Example of the decrease in NMDA receptor activity observed during anoxia in dissociated cortical neurons. NMDA (100 μm) was applied for 10 sec periods.
During 2 hr of anoxia, the amplitude of NMDA Δ Ca2+ observed in neurons in cortical sheets decreased by 53% (n = 24, p < 0.001) (Fig. 2B,C). Measurements of NMDA Δ Ca2+ in dissociated pyramidal neurons obtained from cortical sheets showed that suppression of NMDAR activity occurs within 1–2 min of anoxia (Fig. 2D). In dissociated neurons, the mean decrease in the amplitude of the NMDA Δ Ca2+ was 67 ± 14% (n = 5, p < 0.01). Anoxia did not increase [Ca2+]iin any of the dissociated neurons.
Changes in NMDAR activity and abundance during anoxia
We next investigated whether a decrease in the number of receptors present in the membrane of neurons might account for part of the decrease in NMDA Δ Ca2+ observed in intact cortex. Antibody staining revealed no detectable decrease in receptor abundance after 3 hr of anoxia either at room temperature (22–25°C) or during submergence in 3°C anoxic water (Fig.3). Because the brain PO2 probably does not reach zero for 1 hr after submergence (Jackson, 1968), the duration of the actual period of anoxia in these animals brains was probably ∼2 hr. Over 3–21 d of anoxia, however, a significant decrease in NMDAR abundance was seen. Within 5–7 hr of reoxygenation, receptor abundance returned to baseline in the 21 d anoxic animals (Fig. 3, bottom panel). In room air breathing turtles, no change in receptor abundance occurred with short-term (3 hr) or long-term (3 weeks) reduction in body temperature to 3°C.
Fig. 3.

Changing abundance of NMDA receptors during anoxia and recovery. Top portion shows representative Western immunoblots of NR1 NMDA subunits. Bottom graph shows changes in receptor abundance during 3 hr to 3 weeks of anoxia (submergence in 3°C anoxic water) and recovery.
Role of receptor phosphorylation control in NMDAR silencing
To test whether phosphorylation control participates in NMDAR silencing during anoxia, we exposed isolated neurons to the nonspecific phosphatase inhibitor okadaic acid. We found that low concentrations of okadaic acid (120 nm) prevented the anoxia-induced suppression of NMDA Δ Ca2+(n = 5, p < 0.01) (Fig.4A). To further investigate the role of phosphatases, we measured NMDA Δ Ca2+ and NMDAR open probability in neurons in cortical sheets. In cortical sheets, the nonspecific phosphatase inhibitor okadaic acid, as well as calyculin A (a specific inhibitor of serine/threonine phosphatases 1and 2A), prevented suppression of NMDA Δ Ca2+ responses in pyramidal neurons during anoxia (Fig. 4B). Furthermore, calyculin A prevented decreases in cell-attached receptor open probability during anoxia but had no effect on receptor activity with oxygen present (Fig.4C,D).
Role of Ca2+ in NMDAR regulation during anoxia
During 2 hr of anoxia, [Ca2+]i in neurons in cortical sheets increased from 135 ± 6 to 202 ± 5 nm (n = 6, p < 0.001) (Fig. 5A). This increase in [Ca2+]i predicted the depression NMDAR activity during anoxia (regression significant,p < 0.01). NMDAR activity was predicted by [Ca2+]i during oxic conditions as well (Fig. 5B).
Fig. 5.
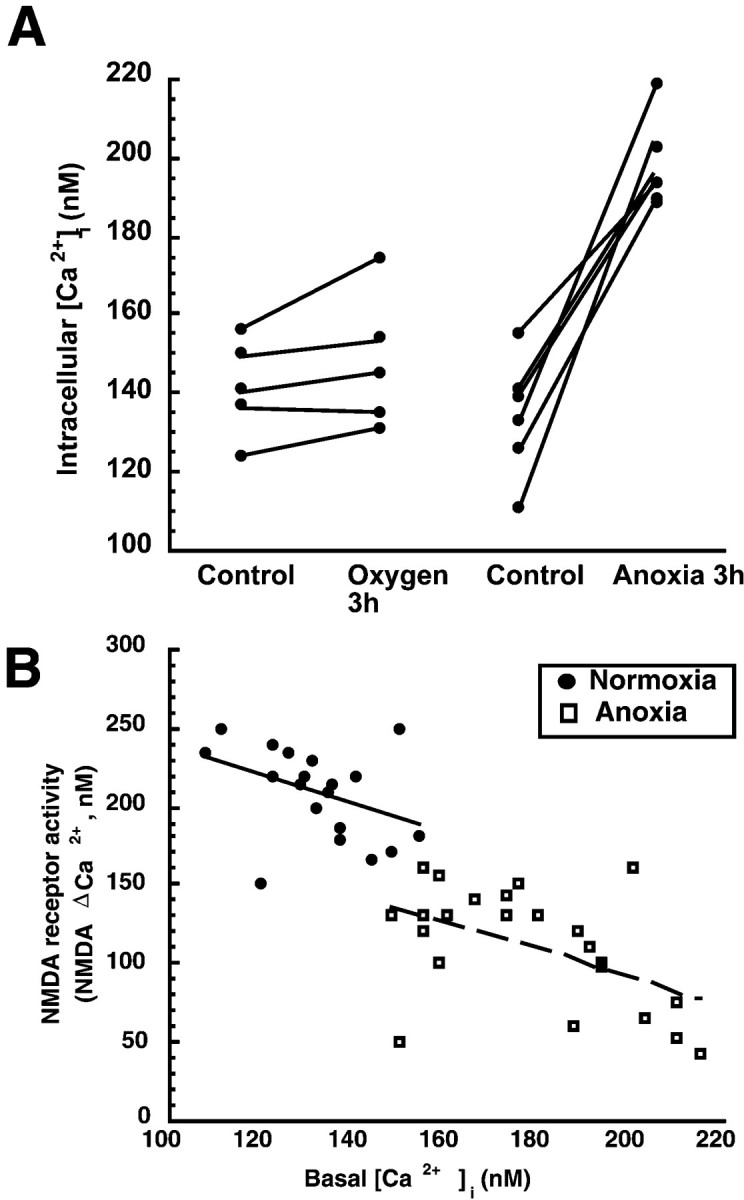
[Ca2+]i predicts NMDA receptor activity in both oxic and anoxic neurons in cortical sheets. A, Two hours of in vitro anoxia increased [Ca2+]i by 32% (70 nm). B, Significant correlation of [Ca2+]i to NMDA receptor activity in both oxic or anoxic neurons. Lines show least-squares linear regression for each group of neurons.
We examined whether the calcium-dependent signaling protein calmodulin participates in NMDAR regulation during anoxia. Calmidazolium, an inhibitor of calmodulin, decreased NMDAR activity ∼38% in the presence of oxygen, suggesting that calmodulin normally acts as a positive modulator of NMDAR activity. During anoxia, calmidazolium prevented a further decrease in NMDAR activity, suggesting that calmodulin, or another signaling molecule associated with it, normally plays some role in the inactivation of receptor function (Fig.6A). We also investigated whether the calcium–calmodulin-dependent phosphatase calcineurin (phosphatase 2B) plays a role in suppressing NMDAR function. Cypermethrin, a specific calcineurin inhibitor, was used. We found that calcineurin inhibition produced a slight, nonsignificant decrease in NMDAR activity in both aerobic and anaerobic conditions. However, cypermethrin did not prevent anoxia from suppressing NMDAR activity, suggesting that activation of calcineurin does not contribute to the inactivation of NMDARs during anaerobic conditions (Fig.6B).
Protein kinase A (PKA) is an important regulator of NMDARs under aerobic conditions (Swope et al., 1999), and because PKA can be activated by increases in [Ca2+]i (via calmodulin, adenylate cyclase, and cAMP), we determined whether signaling via this pathway contributes to receptor suppression during anoxia. We found that NMDARs are controlled by PKA activity during both aerobic and anaerobic conditions but that silencing of NMDARs during anoxia occurred regardless of whether PKA activity was stimulated or inhibited (Fig. 6C). Pretreatment of cortical sheets with low concentrations of forskolin (0.1–1.0 nm), which stimulates cAMP production and activates PKA, increased NMDA Δ Ca2+ by ∼50% (Fig. 6C). Anoxia still resulted in a decrease in NMDAR function during anoxia, although the response was larger than in untreated anoxic cerebrocortex. H8, an inhibitor of PKA, decreased NMDAR, with a further suppression during anoxia still present. Together, the results with PKA stimulation and inhibition show that, although PKA–cAMP strongly influences the activity of the NMDAR under both aerobic and anaerobic conditions, receptor silencing during anoxia is not significantly controlled by these actions.
DISCUSSION
Importance of NMDA receptor silencing to surviving anoxia
We have shown that the silencing of NMDARs in anoxic turtle neurons occurs by at least three mechanisms of different time courses. Suppression of receptors within minutes of anoxia is controlled by protein phosphorylation. During several hours of anoxia, receptors are silenced by processes related to elevated [Ca2+]i, whereas over days to weeks of oxygen deprivation, receptors are removed from the cell membrane. These different controls achieve depression of NMDAR function that may enable turtle neurons to survive the energetic challenge of prolonged oxygen deprivation that accompanies diving or winter dormancy. Energy expenditure in anoxic turtle tissues is reduced by 90–99% (Buck and Hochachka, 1993), an adaptation termed “metabolic arrest” (Hochachka, 1986). Metabolic arrest enables brain [ATP] to remain within 80% of normal during several hours of anoxia (Buck et al., 1998). Reduction in ion leakage and ion pumping (“ion channel arrest”) enables continued ion homeostasis despite dramatically reduced energy production (Lutz, 1992; Buck and Hochachka, 1993). Suppression of NMDAR function may play a significant role in both saving energy and preserving critical ion gradients. A decrease in Ca2+ influx through the NMDAR is particularly critical because large increases in [Ca2+]e [possibly up to 25 mm (Ultsch and Jackson, 1982; Cserr et al., 1988)] occur during weeks to months of anoxic dormancy. The potential for injury caused by elevated [Ca2+]i is therefore reduced by silencing of NMDA receptors. Ca2+ overload in mammalian neurons is a key factor causing cell death in ischemia or hypoxia (Kristian and Siesjo, 1998; Lee et al., 1999), and this danger is thus avoided by turtle neurons. The pattern of NMDAR regulation we have observed seems suited to the biology of diving and dormancy in this animal.
Mechanisms of NMDAR regulation during anoxia
During the first few minutes of anoxia, the turtle NMDAR is inactivated by a mechanism involving phosphatase 1 or 2A. These phosphatases decrease the activity of mammalian NMDARs and may play important roles in the long-term control of glutamatergic synapses (Mulkey et al., 1993). In mammalian neurons, these phosphatases control NMDAR function by altering the open probability of the channel rather than by changing receptor current amplitude (Wang et al., 1994), the same effect exerted by anoxia in turtle neurons. In this respect, the rapid reduction in turtle NMDAR activity during anoxia is similar to long-term depression in mammalian glutamatergic synapses. In contrast to mammalian long-term depression, however, rapid alterations in NMDAR activity during anoxia apparently do not involve calcium-dependent phosphatase activation (Mulkey et al., 1994), because receptor inactivation was observed in the absence of increases in [Ca2+]i.
The activity of NMDARs in turtle neurons was influenced by cAMP–protein kinase A, similar to mammalian receptors (Swope et al., 1999). However, we did not find that alterations in control based on protein kinase A played a role in suppressing NMDAR function during anoxia. Protein kinase C, like protein kinase A, acts to increase NMDAR activity (Swope et al., 1999) in mammalian receptors. It is possible that receptor phosphorylation by one of these kinases is important in the reactivation of NMDARs during recovery from anoxia, but we have not studied this.
Dephosphorylation of NMDARs by phosphatases early in anoxia may be related to later changes in NMDAR expression or distribution. We speculate that dephosphorylation tags inactive NMDARs for internalization at a later time. Receptor internalization–redistribution may be a common phenomenon controlling receptor function. It has been described for metabotropic glutamate receptors (Doherty et al., 1999), nicotinic acetylcholine receptors (Akaaboune et al., 1999), and GABAC receptors (Filippova et al., 1999) and may contribute to modulating synaptic plasticity (Carroll et al., 1999).
Role of Ca2+ in NMDAR regulation
In turtle neurons, [Ca2+]i increases ∼30% above basal during 2 hr of anoxia. This increase, which remains stable through 6 weeks of anoxia (Bickler, 1998), is small compared with the very large anoxia-induced increase that occurs in mammalian neurons (Lipton and Loebner, 1990). In turtle neurons, the moderate increase in [Ca2+]i apparently serves an important signaling function during anoxia. In these neurons, elevated [Ca2+]iis associated with cell survival rather than death and with suppression of NMDAR activity. This increase is related to a suppression of NMDAR activity in a complex way. First, [Ca2+]i predicts NMDA receptor function, both during oxic and anoxic conditions, indicating that elevated [Ca2+]i exerts a feedback influence on this pathway of Ca2+influx, perhaps serving to limit excessive Ca2+ accumulation. The effect of [Ca2+]i on NMDAR activity is greater in anoxic neurons. Second, Ca2+-dependent control is exerted on NMDARs via calmodulin, but the effector molecule(s) remains to be defined. Although we found that calmodulin was an important control over NMDAR activity during both aerobic and anaerobic conditions, we were unable to identify whether calmodulin directly influences NMDAR activity by binding to the receptor (Wyszynski et al., 1997; Zhang et al., 1998) or whether it controls the receptor via calmodulin-dependent protein kinases or phosphatases. The Ca2+-dependent phosphatase calcineurin plays a role in depressing NMDAR function in long-term depression (Mulkey et al., 1994) in mammalian neurons. However, we found that inhibiting calcineurin did not change NMDAR suppression during anoxia. Although we studied several concentrations of the calcineurin inhibitor cypermethrin, it is possible that this antagonist is not effective against reptilian phosphatases.
Calcium-dependent depolymerization of the cytoskeleton is responsible for NMDAR desensitization, which occurs when [Ca2+]i reaches high levels (Rosenmund and Westbrook, 1993). Recent work suggests that Ca2+-bound calmodulin separates the NR1 subunit from actin, thereby decreasing receptor currents (Zhang et al., 1998). Thus, our experiments that involved inhibition of calmodulin may have interrupted Ca2+ signaling by both inhibiting the action of a Ca2+–calmodulin-dependent phosphatase and preventing receptor–cytoskeleton disaggregation. We cannot evaluate these possibilities because we did not determine whether anoxia produced the type of receptor desensitization described by Rosenmund and Westbrook (1993).
We have not ruled out the contribution of other possible controls of NMDAR function during anoxia. Adenosine, which accumulates in the extracellular space during anoxia, has been shown to contribute to NMDAR suppression (Buck and Bickler, 1998). Elevated [H+] is also likely to contribute to NMDAR suppression in the turtle brain during anoxia. Brain extracellular pH in turtles falls to 6.4 during 10 d of anoxia, and by the end of winter, dormancy is undoubtedly much lower (Ultsch and Jackson, 1982; Cserr et al., 1988). Intracellular pH in cortical sheets drops to 6.6 within 1 hr of anoxia (Buck et al., 1998), so reduced pH has the potential of exerting an important influence on NMDA receptor function (Traynelis and Cull-Candy, 1990). Ionized Ca2+ and Mg2+levels increase many fold in blood and CSF during long-term anoxia (Jackson et al., 1984; Cserr et al., 1988). The net effect of these changes is a stable depression of NMDAR activity to ∼50% of normal during 6 weeks of anoxia (Bickler, 1998).
Turtle neurons as models for identifying new neuroprotection strategies
The present study offers several insights into the role of glutamate receptors and [Ca2+]i in determining neuron survival during anoxia. Turtle neurons survive anoxia by suppressing NMDAR function, which is not surprising in view of the protective qualities of NMDAR antagonists in mammalian brain ischemia (Lee et al., 1999). However, NMDAR activity is only suppressed, not eliminated. This may suggest that optimal survival during hypoxic or ischemic stress is achievable by a controlled decrease NMDAR activity and that strategies aimed at high-degree blockade of NMDARs may not be viable. Knowledge of how turtle neurons regulate an appropriate level of NMDAR activity may lead to new approaches to neuroprotection compatible with this concept. Another finding of potential importance is that turtle neurons survive prolonged anoxia while [Ca2+]i rises ∼70–100 nm, a change that may be protective because it suppresses NMDAR activity. Strategies aimed at allowing a modest increase in [Ca2+]i in ischemic mammalian neurons may be worth considering.
Footnotes
This work was supported by National Institutes of Health Grants R29 GMS 55212 (P.E.B.) and PO1 NS 35902 (P.H.D.). We thank Bonnie Hansen, Melanie Vose, Rick Liniger, and Breandan Sullivan for technical support.
Correspondence should be addressed to Dr. Philip Bickler, Department of Anesthesia, Sciences 261, University of California at San Francisco, 513 Parnassus Avenue, San Francisco, CA 94143-0542. Email:bicklerp@anesthesia.ucsf.edu.
Dr. Donohoe's present address: National Institute of Medical Research, London, UK NW7 1AA.
Dr. Buck's present address: Department of Zoology, University of Toronto, Toronto, Ontario, Canada M5S 3G5.
REFERENCES
- 1.Akaaboune M, Culican SM, Turney SG, Lichtman JW. Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science. 1999;286:503–507. doi: 10.1126/science.286.5439.503. [DOI] [PubMed] [Google Scholar]
- 2.Bickler PE. Reduction of NMDA receptor activity in cerebrocortex of turtles (Chrysemys picta) during 6 wk of anoxia. Am J Physiol. 1998;275:R86–R91. doi: 10.1152/ajpregu.1998.275.1.R86. [DOI] [PubMed] [Google Scholar]
- 3.Bickler PE, Buck LT. Adaptations of vertebrate neurons to hypoxia and anoxia: maintaining critical calcium concentrations. J Exp Biol. 1998;201:1141–1152. doi: 10.1242/jeb.201.8.1141. [DOI] [PubMed] [Google Scholar]
- 4.Bickler PE, Hansen BM. Hypoxia-tolerant neonatal CA1 neurons: relationship of survival to evoked glutamate release and glutamate receptor-mediated calcium changes in hippocampal slices. Dev Brain Res. 1998;106:57–69. doi: 10.1016/s0165-3806(97)00189-2. [DOI] [PubMed] [Google Scholar]
- 5.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 6.Buck L, Bickler P. Role of adenosine in NMDA receptor modulation in the cerebral cortex of an anoxia-tolerant turtle (Chyrsemys picta belli). J Exp Biol. 1995;198:1621–1628. doi: 10.1242/jeb.198.7.1621. [DOI] [PubMed] [Google Scholar]
- 7.Buck LT, Bickler PE. Adenosine and anoxia reduce N-methyl-d-aspartate receptor open probability in turtle cerebrocortex. J Exp Biol. 1998;210:289–297. doi: 10.1242/jeb.201.2.289. [DOI] [PubMed] [Google Scholar]
- 8.Buck LT, Hochachka PW. Anoxic suppression of Na+-K+-ATPase and constant membrane potential in hepatocytes: support for channel arrest. Am J Physiol. 1993;265:R1020–R1025. doi: 10.1152/ajpregu.1993.265.5.R1020. [DOI] [PubMed] [Google Scholar]
- 9.Buck LT, Espanol M, Litt L, Bickler P. Reversible decreases in ATP and PCr concentrations in anoxic turtle brain. Comp Biochem Physiol [A] 1998;120:633–639. doi: 10.1016/s1095-6433(98)10079-x. [DOI] [PubMed] [Google Scholar]
- 10.Carroll RC, Lissin D V, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- 11.Cserr H, DePasquale M, Jackson D. Brain and cerebrospinal fluid composition after long-term anoxia in diving turtles. Am J Physiol. 1988;255:R338–R343. doi: 10.1152/ajpregu.1988.255.2.R338. [DOI] [PubMed] [Google Scholar]
- 12.Dingledine R. Brain slices. Plenum; New York: 1984. [Google Scholar]
- 13.Doherty AJ, Coutinho V, Collingridge GL, Henley JM. Rapid internalization and surface expression of a functional, fluorescently tagged G-protein-coupled glutamate receptor. Biochem J. 1999;341:415–422. [PMC free article] [PubMed] [Google Scholar]
- 14.Filippova N, Dudley R, Weiss DS. Evidence for phosphorylation-dependent internalization of recombinant human rho1 GABAC receptors. J Physiol (Lond) 1999;518:385–399. doi: 10.1111/j.1469-7793.1999.0385p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hochachka P. Defense strategies against hypoxia and hypothermia. Science. 1986;231:234–241. doi: 10.1126/science.2417316. [DOI] [PubMed] [Google Scholar]
- 16.Jackson DC. Metabolic depression and oxygen depletion in the diving turtle. J Appl Physiol. 1968;24:503–509. doi: 10.1152/jappl.1968.24.4.503. [DOI] [PubMed] [Google Scholar]
- 17.Jackson DC, Herbert CV, Ultsch GR. The comparative physiology of diving in North American freshwater turtles. II. Plasma ion balance during prolonged anoxia. Physiol Zool. 1984;57:632–640. [Google Scholar]
- 18.Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- 19.Lee J-M, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 20.Lipton P, Loebner D. Mechanisms of intracellular calcium accumulation in the CA1 region of rat hippocampus during anoxia in vivo. Stroke. 1990;21:60–64. [PubMed] [Google Scholar]
- 21.Lutz P. Mechanisms for anoxic survival in the vertebrate brain. Annu Rev Physiol. 1992;54:601–618. doi: 10.1146/annurev.ph.54.030192.003125. [DOI] [PubMed] [Google Scholar]
- 22.Lutz P, Kabler S. Release of adenosine and ATP in the brain of the freshwater turtle (Trachemys scripta) during long-term anoxia. Brain Res. 1997;769:281–286. doi: 10.1016/s0006-8993(97)00719-1. [DOI] [PubMed] [Google Scholar]
- 23.Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 24.Mulkey RM, Endo S, Shenolikar S, Malanka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 25.Perez-Pinzon M, Rosenthal M, Sick T, Lutz P, Pablo J, Mash D. Downregulation of sodium channels during anoxia: a putative survival strategy of turtle brain. Am J Physiol. 1992;262:R712–R715. doi: 10.1152/ajpregu.1992.262.4.R712. [DOI] [PubMed] [Google Scholar]
- 26.Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduced NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- 27.Swope SL, Moss SI, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phophoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- 28.Tokuda M, Hatase O. Regulation of neuronal plasticity in the central nervous system by phosphorylation and dephosphorylation. Mol Neurobiol. 1998;17:137–156. doi: 10.1007/BF02802028. [DOI] [PubMed] [Google Scholar]
- 29.Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-d-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–349. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- 30.Ultsch G, Jackson D. Long-term submergence at 3°C of the turtle Chrysemys picta belli, in normoxic and severely hypoxic water. I. Survival, gas exchange, and acid-base status. J Exp Biol. 1982;96:11–28. doi: 10.1242/jeb.97.1.87. [DOI] [PubMed] [Google Scholar]
- 31.Wang L-Y, Orser BA, Brautigan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- 32.Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]