

Abstract
To identify molecular targets of corticosteroid negative feedback effects on neurosecretory neurons comprising the central limb of the hypothalamo–pituitary–adrenal (HPA) axis, we monitored ether stress effects on corticotropin-releasing factor (CRF) and arginine vasopressin (AVP) heteronuclear RNA (hnRNA) expression in rats that were intact or adrenalectomized (ADX) and replaced with corticosterone (B) at constant levels ranging from nil to peak stress concentrations. Under basal conditions, relative levels of both primary transcripts varied inversely as a function of plasma B titers. In response to stress, the kinetics of CRF hnRNA responses of intact and ADX rats replaced with low B were similar, peaking at 5 min after stress. By contrast, intact rats showed a delayed AVP hnRNA response (peak at 2 hr), the timing of which was markedly advanced in ADX/low B-replaced animals (peak at 5–30 min). Transcription factors implicated in these responses responded similarly. Manipulation of B status did not affect the early (5–15 min) phosphorylation of transcription factor cAMP-response element-binding protein (CREB) but accelerated maximal Fos induction from 2 hr after stress (intact) to 1 hr (ADX). Assays of binding by proteins in hypothalamic extracts of similarly manipulated rats toward consensus CRE and AP-1 response elements supported a role for the stress-induced plasma B increment in antagonizing AP-1, but not CRE, binding. These findings suggest that glucocorticoid negative feedback at the transcriptional levels is exerted selectively on AVP gene expression through a mechanism that likely involves glucocorticoid receptor interactions with immediate-early gene products.
Keywords: arginine vasopressin, corticosterone, corticotropin-releasing factor, CREB, Fos, glucocorticoids, negative feedback, neurosecretory neurons, paraventricular nucleus
Negative feedback, or end-product inhibition, is an important regulatory mechanism in neuroendocrine systems. In one well-studied model, glucocorticoid mediators of the endocrine arm of the stress response act centrally to phasically inhibit further biosynthetic and secretory activity of the hypothalamo–pituitary–adrenal (HPA) axis (Keller-Wood and Dallman, 1984; Dallman et al., 1987). Because stimulatory drive on this axis is imparted principally by corticotropin-releasing factor (CRF) and arginine vasopressin (AVP), interacting co-secretagogues for pituitary adrenocorticotropin (ACTH) that are expressed by a common neurosecretory neuron population (Vale et al., 1981; Sawchenko et al., 1984), the molecular target(s) of corticosteroid negative feedback remain unsettled.
Threats to homeostasis posed by the internal or external environments commonly elicit coordinated neural and hormonal responses that serve to mobilize bodily resources to facilitate coping with emergency situations. Stress-related sensory information, conveyed to a population of neurosecretory neurons within the paraventricular nucleus of the hypothalamus (PVH), initiates the neuroendocrine stress cascade by provoking the release of CRF and AVP into the portal vasculature that supplies the anterior pituitary (Vale et al., 1981; Swanson et al., 1983) to stimulate the release of ACTH and, consequently, glucocorticoids from the adrenal cortex. Synaptic activation of hypophysiotropic CRF-expressing neurons commonly triggers neuropeptide gene expression to replenish depleted stores and can result in stimulus-specific alterations in cellular phenotype, including induced AVP expression (Lightman and Young, 1988; Herman et al., 1992; Herman, 1995; Makino et al., 1995). Although glucocorticoids provide the major inhibitory signal that constrains the biosynthetic and secretory activities of the HPA axis (Dallman et al., 1987), there remain uncertainties as to how basal and stress-induced corticosterone (B) secretion negatively regulates CRF and AVP expression. The recent demonstration that the transcriptional activation of the CRF and AVP genes in response to acute ether stress follow distinct time courses, with peak heteronuclear RNA (hnRNA) responses occurring at 5 min and 2 hr, respectively (Kovács and Sawchenko, 1996), provided a basis for probing the manner in which steroidal influences may participate in this differential control.
To identify molecular targets of corticosteroid negative feedback, we compared the timing and magnitude of ether stress-induced CRF and AVP hnRNA responses in the parvocellular division of the PVH of rats that were intact, adrenalectomized (ADX), or ADX and replaced with varying constant levels of corticosterone intended to approximate basal morning, basal evening, or peak stress levels of the hormone. To determine the effects of these manipulations on candidate mechanisms regulating neuropeptide gene expression, we followed in parallel the timing and steroid dependence of stress effects on the expression of transcription factors that have been implicated in the early CRF hnRNA [the phosphorylated form of the cAMP-response element-binding protein (pCREB)] and the delayed AVP hnRNA responses (Fos, the protein product of the c-fos proto-oncogene). In addition, gel shift assays were used to assess the manner in which binding affinities of proteins contained in hypothalamic extracts toward consensus DNA sequences that confer cAMP/Ca2+ and AP-1 responsiveness might vary as a function of stress and steroidal environment.
Portions of the findings have been reported previously in abstract form (Kovács and Sawchenko, 1997).
MATERIALS AND METHODS
Animals and procedures. Adult male Sprague Dawley rats were housed under controlled temperature and lighting (12 hr light/dark cycle; lights on at 6 A.M.) with food and water available ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the Salk Institute. Animals were subjected to bilateral adrenalectomy or sham operations and received subcutaneous implants of slow-release corticosterone (B) pellets (0, 35, 50, or 100 mg; Innovative Research of America) under Nembutal anesthesia 5 d before stress. For phospho-CREB immunostaining, rats were implanted with jugular catheters 2 d before challenge and anesthetized via remote intravenous injection to avoid nonspecific effects of handling and intraperitoneal injection. Rats were perfused transcardially with 4% paraformaldehyde in 0.1 m borate buffer, pH 9.5, at intervals ranging between 5 min and 4 hr after 5 min exposure to ether vapor as described (Kovács and Sawchenko, 1996). Multiple series of 30-μm-thick frozen sections were collected at 150 μm intervals and stored in cryoprotectant at −20°C until histochemical processing.
Immunocytochemistry. Fos immunoreactivity (-ir) was localized using antisera raised in rabbits against a synthetic N-terminal fragment (residues 4–17) of human Fos (sc-52, Santa Cruz Biotechnology). Specific staining was abolished by preadsorbing the antiserum overnight at 4°C with 50 μm of the synthetic peptide immunogen. Binding purified antisera raised in rabbit against both the native (unphosphorylated) and phosphorylated forms of the pCREB were provided courtesy of Dr. Marc Montminy (The Salk Institute). These were raised against synthetic peptides corresponding to residues 136–150 (native CREB), or a phosphorylated peptide (CREB128–141) spanning the protein kinase A phosphoacceptor site at Ser133. Immunoblot analyses of nuclear extracts from hypothalamus have shown that both purified antisera label a single band of the expected size and that only the pCREB antiserum discriminates unphosphorylated from protein kinase A-phosphorylated CREB (Hagiwara et al., 1993). Staining with the native CREB antiserum was eliminated by overnight preincubation at 4°C with 60 μm of the homologous synthetic peptide, whereas that yielded by the anti-pCREB serum persisted after incubation with unphosphorylated CREB128–141 in the low millimolar range. All primary antisera were applied at a 1:1000 dilution and localized using a conventional avidin–biotin immunoperoxidase protocol (Sawchenko et al., 1990) and Vectastain Elite reagents (Vector Laboratories). In addition to anesthetization through intravenous cannula to minimize the impact of handling and injection, empirically determined modifications incorporated to optimize staining for pCREB included substituting 3% BSA for 2% goat serum as a blocking agent, adding the phosphatase inhibitors (1 mm sodium vanadate and 25 mm sodium fluoride) to the perfusates and primary antiserum solutions, and performing incubations in primary antiserum in the presence of 1 mm unphosphorylated synthetic CREB128–141 to minimize nonspecific cross-reactivity.
Blood sampling and corticosterone measurement. A separate group of rats was used to measure hormonal responses to stress and manipulation of corticosteroid status. These animals were implanted 2 d before experimentation with jugular venous catheters under pentobarbital anesthesia. Cannulae were fashioned from PE50 tubing with SILASTIC tips (Dow Corning, Corning, NY), exteriorized on the neck, and extended with an additional length of PE50 tubing on the morning of the test. The animals then remained undisturbed for 3–4 hr, and blood samples were taken before and at 5, 15, 30, and 60 min after ether stress. Plasma corticosterone (B) was measured by direct radioimmunoassay without extraction, using an antiserum raised in rabbits against a corticosterone–carboxymethyloxime–BSA conjugate and an 125I-labeled corticosterone–carboxymethyloxime–tyrosine–methylester tracer. Interference with plasma transcortin was eliminated by treatment of samples at low pH. The sensitivity of the assay was 0.1 pmol per tube; intra-assay and interassay coefficients of variation were 7 and 24%, respectively.
In situ hybridization histochemistry. To monitor CRF and AVP hnRNA, riboprobes complementary to intronic sequences of the CRF and AVP genes were transcribed from plasmids provided by Dr. A. Ericsson (The Salk Institute) and Dr. T. G. Sherman (Georgetown University), respectively, in the presence of35S-UTP and35S-ATP. Hybridization and autoradiographic techniques were modified following Simmons et al. (1989). Tissue sections were mounted onto poly-l-lysine-coated slides post-fixed with 4% paraformaldehyde, then digested with Proteinase K (10 mg/ml in 50 mm Tris, pH 8, and 5 mm EDTA at 37°C, 30 min), acetylated (0.25% acetic anhydride in 0.1 mtriethanolamine, pH 8), and dehydrated. Hybridization mixture (50% formamide, 0.3 m NaCl, 10 mm Tris, pH 8, 2 mm EDTA, 1× Denhardt's, 10% dextran sulfate, 0.5 mg/ml yeast tRNA) was pipetted onto the slides (100 μl, containing probe at 107 dpm/ml) and hybridized overnight at 56°C. Sections were then rinsed in 4× SSC (1× SSC: 0.15m NaCl and 15 mm trisodium-citrate buffer, pH 7), digested with ribonuclease A (20 mg/ml in Tris-EDTA buffer with 0.5m NaCl at 37°C for 30 min), gradually desalted, and washed in 0.1× SSC at 65–75°C for 30 min. Slides were exposed to x-ray film for 24–48 hr, then dipped in NTB-2 nuclear emulsion (Kodak) and exposed to intervals ranging from 10 to 14 d (AVP hnRNA) and 4 to 6 weeks (CRF hnRNA), developed in D-19 developer, and lightly counterstained with thionin.
Analysis. Semiquantitative densitometric analysis of relative levels of hnRNAs of interest was performed on nuclear emulsion-coated slides. Relative levels of optical density were obtained by comparing a standard curve generated from brain paste standard samples containing serial dilution of35S-UTP with experimental samples using Macintosh-driven NIH Image software (versions 1.55 and 1.61). The medial parvocellular subdivision of the PVH (Swanson and Kuypers, 1980) was defined from Nissl staining patterns and aligned with corresponding dark-field images of hybridized sections by redirected sampling. Optical density readings, corrected for background, were taken at regularly spaced (150 μm) intervals, and average values were determined throughout the extent of this cell group for each animal. Scattered atopic magnocellular neurons within the parvocellular subdivision were recognized on Nissl-stained material under bright-field illumination and were excluded from the analysis of AVP hnRNA.
The number of immunopositive (Fos-ir or pCREB-ir) cell nuclei in a sampling of the dorsal medial parvocellular subdivision of the PVN as a function of treatment status were counted using NIH Image (1.55 and 1.61) software. Images were captured from immunoperoxidase-stained sections at the mid-level of the medial parvocellular subdivision using a CCD camera (Sony). The boundary of the region was outlined, and the number of positive profiles was recorded after thresholding the images to a common level. The minimum size of a profile to be considered as a c-fos/pCREB-positive cell nucleus was determined as more than five pixels. Total cell counts were taken bilaterally at regularly spaced intervals and expressed as mean ± SEM for each time point and treatment group.
Hypothalamic extracts and gel mobility shift assay. Separate groups of intact, ADX, and ADX/B-replaced rats were prepared as above and decapitated at 10 min (for assessments of CRE binding) or 2 hr (for AP-1 binding) after stress. The hypothalami were quickly dissected and frozen on dry ice. Whole-cell extracts were obtained by sonicating the tissue in cold buffer containing 20 mm HEPES, 0.4m NaCl, 20% glycerol, 5 mmMgCl2, 0.5 mm EDTA, 0.1 mm EGTA, 1% NP-40, 5 mm DTT, 1 mmPMSF, 1 mm NaF, pH 7.9. Samples were centrifuged for 10 min at 14,000 × g, and the supernatant was used for binding reaction. Double-stranded oligonucleotides, corresponding to the canonical CRE (AGAGATTGCCTGACGTCAGAGAGCTAG) or AP-1 binding sites (CGCTTGATGAGTCAGCCGGAA) were end-labeled with adenosine 5′-[γ32P]-triphosphate with T4 polynucleotide kinase (Promega). The binding reaction was performed in binding buffer (Promega) for 10 min at room temperature in the presence of poly[dIdC] to reduce nonspecific binding. Samples containing similar total amounts of protein were then run on a 4% nondenaturating polyacrylamide gel in 0.5 × Tris-borate buffer (TBE) at 200 V. Gels were dried, and the position of the DNA/protein shifted complexes were determined by autoradiography using Kodak XAR films. At least six separate repetitions of each experiment were performed. Quantitative analysis of the binding was achieved by measuring relative optical densities of specifically shifted bands using Scion image analysis software after digitizing film autoradiograms using a BioCapt system.
Data analysis and statistics. Data are expressed as mean ± SEM, and were analyzed using a one-way ANOVA, with Tukey's honestly significant difference test applied, post hoc, for individual between-group comparisons. Statistical analyses were performed using STATISTICA software for Windows (StatSoft, vers.5.1) software.
RESULTS
Plasma corticosterone levels
Hormonal response profiles confirmed ether-induced activation of B secretion in intact rats from low basal (24.6 ± 6.5 nm) to peak levels (550 ± 68 nm) at 30 min after stress. In ADX rats, plasma B levels were undetectable. Supplementation of ADX rats with constant-release subcutaneous steroid pellets was generally successful in approximating basal morning (27.5 ± 3.5 nm; ADX + 35B), circadian zenith (84 ± 28 nm; ADX + 50B), and peak stress levels (398 ± 84 nm; ADX + 100B) of the hormone, respectively. None of these groups displayed significant plasma B responses to stress.
Steroid dependence of basal hnRNA expression
Intron-specific cRNA probes were used to assess the transcriptional activities of the CRF and AVP genes in the PVH. These probes hybridize to hnRNAs, before the excision of intronic sequences to form mature mRNA, and have been validated as an index of transcriptional activation in this system (Herman et al., 1991, 1992). Although there exists a substantial steady-state pool of CRF mRNA in stress-related parvocellular neurosecretory cells in the PVH of unperturbed rats, only a few scattered cell nuclei were seen to show positive CRF hnRNA hybridization signals under resting conditions, indicating a low level of ongoing transcription (Fig.1). ADX, which provides a persistent stimulus for CRF synthesis and release, resulted in a 3.8-fold increase in relative levels of CRF hnRNA in the parvocellular division of the PVH. The lowest level of constant B replacement (35 mg pellets) completely constrained the ADX-induced hnRNA signal to values that did not differ significantly from those seen in intact controls (Fig.3). Plasma B levels achieved in ADX animals whose replacement regimens approximated the circadian- or stress-induced peak hormone concentrations also prevented the ADX-induced rise in CRF intronic expression in the parvocellular division of the PVH (Figs. 1, 3).
Fig. 1.

Time course of CRF hnRNA responses to ether stress in control (Intact) rats, adrenalectomized (ADX) rats, and ADX rats replaced with graded levels of corticosterone (B). Dark-field autoradiograms from similar rostrocaudal levels of the PVH showing nuclear hybridization signal obtained using an intron-specific cRNA probe at key time points after stress. From low resting levels, intact rats show a marked increase of CRF hnRNA signal in the dorsal aspect of medial parvocellular subdivision that peaks at 5 min after stress. ADX increases basal and peak-stress levels of the CRF primary transcript but does not alter the timing of the response. Low-level (35 mg) B replacement in ADX rats results in a situation that mirrors that seen in control animals. ADX rats supplemented with peak-stress levels of B (100 mg) do not display detectable alterations in CRF hnRNA at any post-stress time point examined. (Photomicrographs from ADX + 50B rats are comparable to those of the ADX + 100Bgroup and have been omitted for clarity.) All photomicrographs: 75× magnification. mpd, Dorsal aspect of medial parvocellular subdivision; pm, posterior magnocellular.
Fig. 3.

Time course of stress-induced changes in relative levels of CRF and AVP hnRNA as a function of corticosteroid status. Values are based on densitometric determinations over the medial parvocellular part of the PVH and are given as means ± SEM (n = 5–7/group). Note the stability of rise, peak, and decline of CRF hnRNA induction across Intact,ADX, and ADX+35B conditions, which contrasts with the marked increase and acceleration of the AVP transcriptional response. Higher levels of B replacement (50 or 100 mg) eliminate detectable basal and stress-induced expression of both AVP and CRF primary transcripts. + Differs significantly from intact control group, p < 0.05; ++ p < 0.01. * Differs significantly from basal value in rats of similar steroid treatment status,p < 0.05; **p < 0.01.
Nonmanipulated intact rats displayed AVP hnRNA signal in all acknowledged sites of AVP synthesis in the hypothalamus, including the suprachiasmatic and supraoptic nuclei, as well as in the topographically discrete magnocellular division of the PVH (Fig.2). Scattered, positively labeled nuclei detected over the parvocellular subdivision were similar in size and labeling intensity to those detected in the magnocellular compartment and were interpreted as representing ectopic magnocellular vasopressinergic cells. Densitometric analysis revealed a marked (threefold) elevation of the AVP intronic signal after ADX over the medial parvocellular compartment without any significant change detected in the magnocellular division of the nucleus (Fig.3).
Fig. 2.
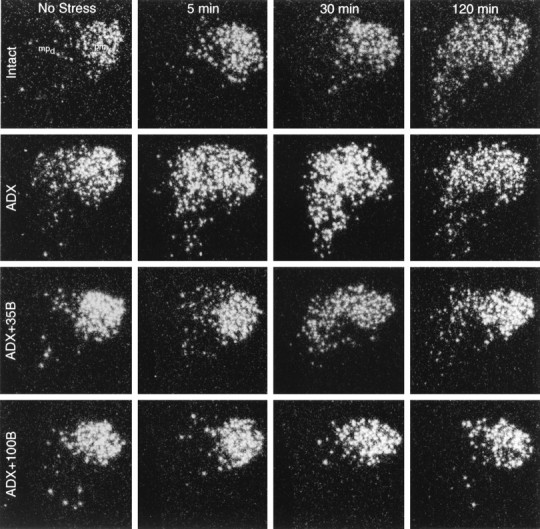
Time course of AVP hnRNA responses to acute ether stress as a function of corticosteroid status. Dark-field autoradiograms were obtained using a cRNA probe complementary to the first intron of the vasopressin gene at key time points after stress in rats that were intact, ADX, or ADX and supplemented with 35 or 100 mg constant-release B pellets. Substantial basal levels of AVP hnRNA expression are apparent in the magnocellular subdivision that do not change as a function of corticosteroid status and/or stress. Stress-induced expression of the primary transcript shows maximum at 2 hr after stress in intact animals. In ADX rats, parvocellular neurons display clear AVP hnRNA signals under basal conditions and, in addition, an accelerated AVP hnRNA response that peaks 5–30 min after stress. Low levels of B replacement (35 mg) restore the basal AVP expression in the parvocellular neurosecretory cells, but the timing of the peak stress-induced AVP transcriptional response remains accelerated, relative to that seen in intact controls. Sustained high levels of B (ADX+100B) eliminate detectable basal and stress-induced expression of AVP primary transcripts in the parvocellular neurons. (Photomicrographs from ADX + 50B rats are comparable to those of the ADX + 100B group and have been omitted for clarity.) All photomicrographs: 75× magnification.
Low levels of constant B replacement (ADX + 35B) restored the basal AVP hnRNA signal in the parvocellular division of the PVH, which was not seen to be decreased further in animals that received higher levels of B replacement (ADX + 50B and ADX + 100B groups). By contrast, no significant changes were apparent in the levels of hybridizable AVP hnRNA over the magnocellular division of the PVH and the supraoptic nucleus in any replacement group (Figs. 2, 3).
Effects of stress as a function of corticosteroid status
The magnitude and kinetics of the induction of CRF hnRNA seen in the medial parvocellular part of the PVH in intact animals after an acute ether challenge was fully compatible with previous findings (Kovács and Sawchenko, 1996), with a peak 5.8-fold elevation detected at 5 min after the termination of ether stress, diminishing to values that were not significantly different from baseline between 30 and 60 min after stress (Figs. 1, 3).
In rats submitted to ADX surgery 5 d before stress, ether exposure provoked a further twofold induction of CRF hnRNA, over and above the already elevated baseline levels of expression. However, the distribution of the signal, and the timing of the peak and decay of the hnRNA response to a similar challenge were all indistinguishable from those seen in intact animals. As noted above, ADX rats replaced with 35 mg B pellets displayed plasma hormone levels similar to intact nonstressed controls but were unable to mount a B response to stress. The maximal CRF intronic response to ether inhalation in these animals was also found to occur at the 5 min time point (6.5-fold elevation), which declined to control levels by 60 min, fully comparable to the time course exhibited by intact rats. These results suggest that stress-induced rise in circulating B is not responsible for the extinction of CRF gene expression during stress. Steroid replacement of ADX rats with 50 or 100 mg B pellets, which resulted in constant plasma B levels of 150 ± 24 and 398 ± 84 nm, respectively, were sufficient to block the stress-induced CRF hnRNA response at each time point examined.
Intact rats, which responded with a 450 nm plasma B peak 30 min after ether stress, consistently displayed a slower rise in AVP hnRNA, which was found to be maximal at 2 hr after the challenge. These results are compatible with previous findings using this model, in which AVP intronic expression showed a peak 3.2-fold elevation at 2 hr after ether inhalation (Kovács and Sawchenko, 1996). Here, ADX resulted in an elevation of basal (twofold) and maximal stress-induced (2.3-fold) AVP hnRNA levels in the medial parvocellular part of the PVH and, in addition (and in contrast to the situation with CRF), a marked shift in the timing of peak responses to 5–30 min post-stress (Figs. 2, 3). Low-level B replacement (35 mg pellet), which provided constant steroid levels corresponding to the circadian nadir, restored basal and peak levels of parvocellular AVP hnRNA expression to ones closely approximating the magnitude of those seen in adrenal-intact rats, but the timing of the response in these animals that lacked the stress-induced plasma B pulse remained accelerated. Higher levels of B replacement reduced resting parvocellular AVP hnRNA levels to near those seen in intact controls and eliminated significant stress-induced increments in this parameter (Figs. 2, 3). In contrast to the effects described above, neither stress nor manipulation of the steroid milieu resulted in any apparent or measured effects on primary AVP transcripts in magnocellular division of the PVH or in the supraoptic nucleus (data not shown).
Stress and steroidal effects on transcription factor expression
CREB has been implicated as a transcriptional regulator of many genes, including CRF, AVP, and c-fos (Sassone-Corsi et al., 1988; Seasholtz et al., 1988; Sheng et al., 1990; Verbeeck et al., 1990; Pardy et al., 1992; Guardiola-Diaz et al., 1994). CREB is a ubiquitously and constitutively expressed nuclear protein that on phosphorylation becomes transcriptionally active and recruits other cofactors at CRE sites of target genes (Gonzalez and Montminy, 1989). To determine whether steroid-dependent changes in neuropeptide gene expression may be correlated with, and potentially mediated by, alterations in CREB expression or phosphorylation, we followed the dynamics of immunoreactive CREB and pCREB expression in the PVH of rats subjected to manipulations similar to those described above.
CREB protein was found to be expressed constitutively in ostensibly all neuronal cell nuclei within the PVH. Neither exposure to ether nor any manipulation of circulating corticosterone levels resulted in consistent changes in the number, staining intensity, or the subcellular distribution of CREB-ir in the PVH (data not shown). Binding of purified antisera specific to the Ser133-phosphorylated form of CREB revealed a low level of pCREB expression within the hypothalamus under basal conditions, with immunoreactive cell nuclei seen most prominently in the suprachiasmatic and supraoptic nuclei, the lateral and anterior hypothalamic areas, and in the magnocellular, but not the parvocellular, division of the PVH. An increased number of pCREB-positive cell nuclei was detected in the stress-related neurons of ADX rats under basal (nonstressed) conditions, but low-level constant B replacement restored the number of pCREB-ir neurons to a level comparable to that seen in nonmanipulated rats (Fig.4A). In agreement with our previous results (Kovács and Sawchenko, 1996), ether exposure resulted in a marked induction of nuclear pCREB immunoreactivity within the parvocellular division of the PVH at 10 min after stress. At this time point, ADX rats showed an even more pronounced increase, which differed significantly from that seen in intact controls (Fig.4A). Groups of ADX animals that received either the lower (35 mg) or higher (100 mg) levels of B replacement described above displayed increments in stress-induced pCREB expression that were comparable to that seen in intact rats and less than that detected in unreplaced ADX animals (Fig. 4A).
Fig. 4.

Stress-induced CREB phosphorylation in parvocellular neurosecretory neurons of intact and steroid-manipulated rats. A, Mean ± SEM number of pCREB-ir cell nuclei in the parvocellular subdivision of the PVH, as a function of treatment condition, in rats killed 10 min after ether stress. All groups displayed reliable pCREB expression, with only the response of nonreplaced ADX animals differing significantly from that of the stressed controls. n = 5 per group; *p < 0.05 versus basal values, + p < 0.05 versus intact stress group.B, Comparison of time course of CREB phosphorylation in the parvocellular neurosecretory neurons of intact and adrenalectomized rats. Values are presented as mean ± SEM. Note that the timing of the induction and the decay of the response, but not the peak values, are similar in ADX versus intact rats. n = 3–4 per group.
To determine whether the lack of circulating glucocorticoids might affect the timing of CREB phosphorylation, the time course of pCREB induction in the parvocellular division of the PVH was compared in intact and ADX rats. Although ADX animals again displayed a more pronounced increment in the number of pCREB-ir neurons, the timing of the rise, peak, and decay of the response was comparable between the two groups (Fig. 4B).
The c-fos immediate-early gene has been used widely as an inducible marker of cellular activation in stress-related neural circuitry (Ceccatelli et al., 1989; Chan et al., 1993; Kovács 1998) and encodes the Fos phosphoprotein, which dimerizes with protein products of the jun family to comprise AP-1 transcription factors (Morgan and Curran, 1991; Armstrong and Montminy, 1993). To probe for corticosteroid-dependent changes in the magnitude and timing of immediate-early gene induction in our paradigm, we localized Fos protein using an N-terminally directed antiserum that does not cross-react with Fos-related antigens. The basal and stress-induced pattern of Fos-ir expression seen in intact rats exposed to acute ether inhalation was again fully compatible with our previous findings using this model (Kovács and Sawchenko 1996), with resting levels being low to undetectable, increasing to a peak at 2 hr after stress, and abating thereafter (Fig. 5). Although they exhibited comparable basal and peak counts of Fos-ir neurons, unreplaced ADX rats displayed an advance in the timing of the response, with significant elevations detectable at 30 min, and peak values achieved at 1 hr, after stress. This shift in the timing of maximal stress-induced Fos-ir was preserved in ADX rats receiving low levels of steroid replacement (35 mg pellets). The two higher levels of B replacement completely prevented significant stress-induced Fos induction in the PVH at each time point examined (Fig. 5), although, interestingly, neither the strength nor the distribution of Fos expression in extrahypothalamic areas of these animals displayed any obvious differences from those seen in intact controls (data not shown).
Fig. 5.

Ether stress-induced Fos-ir expression in the PVH as a function of corticosteroid status. Time courses of Fos-ir in the PVH of rats that were intact, ADX, or ADX and replaced with constant release B pellets. The graphs show the mean ± SEM number of Fos-ir cell nuclei in the medial parvocellular part of the PVH. Note that intact rats show peak Fos induction 2 hr after stress. ADX rats and ADX rats that received low levels of constant B substitution displayed an accelerated Fos response to acute ether stress. Higher constant levels of B prevented the stress-induced increase of Fos-ir in the hypophysiotropic zone of the PVH. n = 4–5 per group. *p < 0.05 versus baseline, nonstressed values.
Alterations in AP-1 binding by stress and steroid hormones
Gel mobility shift assays were used to analyze stress and glucocorticoid dependence of interactions between consensus DNA sequences that confer cAMP/Ca2+ and AP-1 responsiveness with proteins contained in the hypothalamic extracts from rats exposed to ether stress and/or ADX and replaced with graded levels of constant B. Although basal expression of Fos and pCREB were low or undetectable (see above), cell extracts from nonmanipulated animals displayed detectable binding toward both AP-1 and CRE oligonucleotides as demonstrated by specifically shifted bands (Fig.6). The specificity of the complexes was established by competition with unlabeled oligonucleotides and poly[dIdC]. Adrenalectomy, which failed to induce persisting Fos expression in the PVH, resulted in a significant (3.3-fold) increase in AP-1 binding in nonstressed rats that was reduced to levels not reliably different from controls by B replacement. Extracts from all groups exposed to ether stress displayed significantly increased (1.9- to 2.2-fold) AP-1 binding compared with nonstressed controls, but these were comparable across levels of steroid replacement in ADX animals. Affinities of hypothalamic extracts toward consensus CRE oligonucleotides (data not shown) displayed no consistent variation as a function of steroid status and did not differ in any marked or reliable manner among the stressed groups investigated.
Fig. 6.

Stress and corticosteroid dependence of AP-1 binding by hypothalamic extracts. Top, Specifically shifted bands (arrow) indicative of specific binding to consensus AP-1 oligonucleotides were detected in hypothalamic whole-cell extracts obtained from rats under each treatment condition. ADX resulted in a marked increase of AP-1 binding, which was reduced in a dose-related manner by B replacement. No such steroid-dependent decrease of AP-1 binding is seen in stressed, steroid-supplemented rats, although a clear effect of stress alone is evident across treatment conditions. Bottom, Quantitative densitometric analysis of AP-1 binding by hypothalamic extracts as a function of treatment condition. Values provided are mean ± SEM (n = 6–10), expressed as a percentage of nonstressed control values. *p < 0.05 versus nonstressed control condition.
DISCUSSION
It is generally held that corticosteroid negative feedback confers a means by which to limit depletion of corticotropin-releasing peptides in the event that any acute stress may be prolonged or repeated and to minimize exposure to adverse catabolic and immunosuppressive effects of sustained elevations in circulating glucocorticoids. Despite the fact that CRF is acknowledged as the principal corticotropin-releasing peptide of the mammalian hypothalamus on the basis of potency and its obligate requirement for stress-induced ACTH release (Antoni, 1986), the present results support the view that it is the AVP, and not the CRF, gene that is the principal target of glucocorticoid-mediated transcriptional suppression during stress (Fig.7). Because glucocorticoid inhibition of peptide release occurs over multiple time domains that may involve distinct mechanisms (Keller-Wood and Dallman, 1984), the extent to which the present findings may generalize to this level of analysis is unclear. It is nonetheless of interest to point out that the results are consistent with the emerging consensus that AVP is the principal regulated variable that imparts situation-specific drive on the axis, whereas CRF serves mainly to impose stimulatory tone (for review, seeAntoni, 1993). In identifying the expression, as well as the release, of AVP as a principal regulated variable governing HPA function during stress, the results sharpen the focus of efforts to explore the basis for the gamut of systemic and affective disorders in which dysfunction of the HPA axis in general, and of feedback control in particular, has been postulated to play a causal or exacerbating role.
Fig. 7.

Summary of acute stress-induced changes in CRF and AVP transcription as a function of corticosteroid status. Graphs, based on data presented in Figure 3, plot optical densities corresponding to relative levels of CRF (dashed line) and AVP (solid line) primary transcripts in the parvocellular division of PVH and corticosteroid (dotted line) concentrations measured during 5 min of ether stress in different groups. In intact rats, the early induction of CRF hnRNA occurs before the stress-induced rise in plasma B, which peaks at 30 min, and precedes the peak AVP transcriptional response that occurs at 2 hr after stress. ADX rats, which do not display detectable plasma B secretion, show elevated basal levels of both transcripts and, in addition, an accelerated AVP response to stress with no significant changes seen in the dynamics of the CRF intronic response. ADX rats supplemented with constant low B (ADX+35B) to maintain initial resting hormone titers displayed a restoration of basal transcriptional activities of both genes, but the AVP response remained accelerated, relative to intact controls. Rats with high constant B replacement (ADX+100B) do not display any significant elevation of either transcript at any time point examined.
Differential steroid effects on the stress-induced transcriptional activation of CRF and AVP expression
ADX-induced increases in CRF and AVP mRNA and peptide expression in parvocellular neurosecretory neurons are well documented (Kiss et al., 1984; Sawchenko et al., 1984; Jingami et al., 1985; Wolfson et al., 1985; Kovács et al., 1986; Young et al., 1986a,b;Kovács and Mezey, 1987; Swanson and Simmons, 1989). These effects clearly extend to the hnRNA level, indicating that increased biosynthetic activity is at least partly attributable to enhanced transcriptional activity. In this, our observations are consistent with those of Herman and colleagues (1992, 1995) who demonstrated a rapid upregulation of primary CRF and AVP transcripts after steroid withdrawal, but are in conflict with others (Ma et al., 1997b) who reported no significant rise in CRF hnRNA levels 6 d after ADX.
These effects are clearly steroid dependent. In nonstressed ADX rats, replacement regimens designed to mimic basal plasma B levels completely constrain the transcriptional activity of CRF and AVP genes in parvocellular neurons to levels comparable to those seen in intact controls. This level of B supplementation is sufficient to saturate type I corticosteroid receptors (Reul and de Kloet, 1985) and has been reported to restore baseline secretory activity in the HPA axis (Akana et al., 1985, 1988) and parvocellular AVP hnRNA expression (Herman, 1995) but is reportedly insufficient to normalize ADX-induced AVP (Herman, 1995) or CRF (Swanson and Simmons, 1989) mRNA expression. Higher levels of constant B replacement did not result in any further detectable decrease from the already low basal levels of CRF and AVP hnRNA expression, indirectly supporting a role for type I corticosteroid receptors in determining basal- but not stress-induced activity of the HPA axis (Bradbury et al., 1994).
We reported previously stark differences in timing of peak CRF (5 min) and AVP (120 min) hnRNA responses to acute ether stress in otherwise nonmanipulated rats, supporting an involvement of distinct mechanisms governing the expression of genes encoding the two major ACTH secretagogues, in vivo. Here we report for the first time that fast and delayed glucocorticoid feedback affects not only the secretory behavior of the HPA axis but also the transcriptional activity of genes that encode the two main corticotropin secretagogues, with the AVP gene being the principal target of glucocorticoid-mediated transcriptional suppression during stress.
ADX rats with and without low level constant B replacement, which are incapable of generating a stress-induced increment in plasma B, displayed a robust and consistent advance in the timing of the AVP hnRNA response to acute ether challenge. By contrast, the absence of this capacity affected neither the timing nor the magnitude of stress-induced activation of CRF primary transcript levels in this same cell group. These data are in agreement with the findings of Ma et al. (1997b), demonstrating a rapid decrease in AVP, but not CRF, hnRNA to acute corticosterone injections in ADX animals, and again support the view that AVP is the primary regulated variable governing HPA function during stress and plays a pivotal role in the maintenance of axis activity, particularly under conditions of prolonged or repeated stimulation (Ma et al., 1997a).
Changes in CRF hnRNA in response to acute ether exposure precede the stress-induced peak in plasma B, and this disparity essentially rules out the possibility that the bolus B secreted during stress is a major determinant of the magnitude of the CRF transcriptional response. The possibility remains that it might play a role in its extinction, although we did not detect any sustained elevation of CRF (or AVP) primary transcripts after stress in ADX rats with or without low B supplementation. This result is in contrast with those ofHerman (1995), who reported a prolongation of AVP transcriptional activation in ADX/B-replaced rats in a restraint stress paradigm. Although the basis for this disparity is unclear, one intriguing possible explanation is that it may be attributable to differences in circuitries that drive and/or modulate HPA responsiveness in these two distinct stress models.
In assessing the potential involvement of transcription factors in the activation of CRF and AVP gene expression, correlative evidence was provided to implicate CREB phosphorylation in the rapid CRF hnRNA response and to indicate a requirement for additional factors, such as the products of inducible immediate-early genes that require de novo protein synthesis in the delayed AVP intronic response (Kovács and Sawchenko, 1996; Kovács et al., 1998). With respect to possible involvement of CREB, we failed to adduce evidence for reliable changes in the CREB phosphorylation in parvocellular neurons as a function of the corticosteroid status. CREB phosphorylation was slightly elevated in ADX rats, but in contrast with other findings (Legradi et al., 1997), steroid replacement did not affect stress-induced pCREB-ir in the cell group of interest. Adrenal status did, however, markedly affect ether-induced Fos expression, with a significant advancement of the peak seen in ADX rats and a complete suppression of ether-induced Fos-ir observed in ADX rats replaced with constant high levels of B. This shift in timing of Fos protein induction might be involved in the accelerated AVP hnRNA response seen in ADX rats replaced with constant low levels of B.
Possible mechanisms of glucocorticoid effects on CRF and AVP expression
Results from each phase of the present analysis support distinct mechanisms of glucocorticoid involvement in the transcriptional control of CRF and AVP expression under basal and challenged conditions. Although parvocellular neurosecretory neurons express type II glucocorticoid receptors (Uht et al., 1988), providing a potential basis for the feedback inhibition of CRF and AVP transcription, the mechanisms that underlie this repression have remained elusive. We suggest that what has been classically termed “slow feedback” determines the basal transcriptional activity of both genes and depends on glucocorticoid levels before stress. Three non-mutually exclusive possible mechanisms of action may be surmised from the existing literature. One may involve direct corticosteroid receptor interactions with their respective consensus DNA recognition sequence (Roberts et al., 1979; Drouin et al., 1989; Cairns et al., 1993). Both the CRF and AVP genes contain cis-acting elements that could confer glucocorticoid repression. Three to five distinct glucocorticoid response elements (GREs) have been identified in the promoter region of the CRF gene (Roche et al., 1988; Guardiola-Diaz et al., 1996), and the presence of a functional glucocorticoid regulatory element within the proximal 5′ flanking region of the AVP gene has been reported recently (Burke et al., 1997). Alternatively, glucocorticoid-mediated repression of both basal and stress-induced CRF and/or AVP expression might result from interference with DNA binding of other transcription factors (Akerblom et al., 1988, Pearce and Yamamoto 1993). Glucocorticoid repression of forskolin-stimulated CRH-reporter expression in AtT-20 cells has been shown to occur via direct or indirect interference with a CRE, rather than GRE, site (Rosen et al., 1992; Guardiola-Diaz et al., 1996). Finally, glucocorticoid influences may be mediated in a manner not directly dependent on their DNA binding capabilities. A wealth of evidence is available to document the ability of glucocorticoid receptor complex to bind directly to Jun protein and thereby repress or decrease AP-1 activity (Diamond et al., 1990; Schule et al., 1990; Stauber et al., 1990; Yang-Yen et al., 1990; Unlap and Jope, 1994). Our results are coarsely compatible with such a mechanism, in showing an increase of AP-1 binding in ADX rats, which varies inversely as a function of steady-state B levels. Although the strength of the conclusions that may be drawn from gel shift analyses that made use of whole hypothalamic extracts and consensus DNA binding sequences are limited, it is clear from the present experiments that the steroid dependence of AP-1 binding under basal and stressed conditions is different. Further studies using more precise tissue sampling techniques and promoter-specific nucleotides, along with supershift analysis of binding complexes, are needed to probe the nature of the interactions between glucocorticoids, inducible transcription factors, and DNA regulatory elements under stress conditions.
The so-called “fast feedback” effects of the stress-induced plasma B pulse, which have been viewed as being exerted specifically on peptide release, also clearly affect neuropeptide gene expression, targeting selectively the timing of AVP transcriptional activation. The rapidity of ether-induced nuclear expression of nascent CRF transcripts (Kovács and Sawchenko, 1996; our present observations) essentially eliminates the possibility that the stress-induced B peak is a significant determinant of either the initiation or the extinction of the response. By contrast, fast feedback effects on stress-induced AVP expression might involve multiple mechanisms, including cross-interference with the ability of inducible transcription factors to bind their cognate DNA response elements. Although our results highlight AP-1 binding moieties as potential participants in such interactions, any specific role they may play in this regard remains to be determined.
Footnotes
This work was supported by a grant from the US-Hungarian Science and Technology Joint Fund (JF328), an International Research Scholars Program award from the Howard Hughes Medical Institute, the Hungarian Science Research Foundation, OTKA (K.J.K.), and National Institutes of Health Grant NS-21182 (P.E.S.). We thank Drs. A. Ericsson and T. G. Sherman for generously providing plasmids, Dr. M. Montminy for pCREB antisera, and Dr. R. L. Cole for helpful discussions on gel-shift assays, and we gratefully acknowledge the assistance of Carlos Arias and Orsolya Szalay (technical), Kris Trulock (graphic/photographic), and Belle Wamsley (editorial).
Correspondence should be addressed to Dr. Krisztina J. Kovács, Laboratory of Molecular Neuroendocrinology, Institute of Experimental Medicine, Szigony u. 43, Budapest, H-1083 Hungary. E-mail:kovacs@koki.hu.
REFERENCES
- 1.Akana SF, Cascio CS, Shinsako J, Dallman MF. Corticosterone: narrow range required for normal body and thymus weight and ACTH. Am J Physiol. 1985;249:R527–R532. doi: 10.1152/ajpregu.1985.249.5.R527. [DOI] [PubMed] [Google Scholar]
- 2.Akana SF, Jacobson L, Cascio CS, Shinsako J, Dallman MF. Constant corticosterone replacement normalizes basal adrenocorticotropin (ACTH) but permits sustained ACTH hypersecretion after stress in adrenalectomized rats. Endocrinology. 1988;122:1337–1342. doi: 10.1210/endo-122-4-1337. [DOI] [PubMed] [Google Scholar]
- 3.Akerblom IE, Slater EP, Beato M, Baxter JD, Mellon PE. Negative regulation by glucocorticoids through interference with cAMP responsive enhancer. Science. 1988;241:350–353. doi: 10.1126/science.2838908. [DOI] [PubMed] [Google Scholar]
- 4.Antoni FA. Hypothalamic control of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin-releasing factor. Endocr Rev. 1986;7:351–378. doi: 10.1210/edrv-7-4-351. [DOI] [PubMed] [Google Scholar]
- 5.Antoni FA. Vasopressinergic control of pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol. 1993;14:76–122. doi: 10.1006/frne.1993.1004. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong RC, Montminy MM. Transsynaptic control of gene expression. Annu Rev Neurosci. 1993;16:17–29. doi: 10.1146/annurev.ne.16.030193.000313. [DOI] [PubMed] [Google Scholar]
- 7.Bradbury MJ, Akana SF, Dallman MF. Roles of type I and II corticosteroid receptors in regulation of basal activity in the hypothalamo-pituitary-adrenal axis during the diurnal trough and the peak: evidence for a nonadditive effect of combined receptor occupation. Endocrinology. 1994;134:1286–1296. doi: 10.1210/endo.134.3.8119168. [DOI] [PubMed] [Google Scholar]
- 8.Burke ZD, Ho MY, Morgan H, Smith M, Murphy D, Carter D. Repression of vasopressin gene expression by glucocorticoids in transgenic mice: evidence of a direct mechanism mediated by proximal 5′ flanking sequence. Neuroscience. 1997;78:1177–1185. doi: 10.1016/s0306-4522(96)00603-3. [DOI] [PubMed] [Google Scholar]
- 9.Cairns C, Cairns W, Okret S. Inhibition of gene expression by steroid hormone receptors via negative glucocorticoid response element: evidence for the involvement of DNA-binding and agonistic effects of the antiglucocorticoid/antiprogestin RU486. DNA Cell Biol. 1993;12:695–702. doi: 10.1089/dna.1993.12.695. [DOI] [PubMed] [Google Scholar]
- 10.Ceccatelli S, Villar MJ, Goldstein M, Hökfelt T. Expression of c-fos immunoreactivity in transmitter-characterized neurons after stress. Proc Natl Acad Sci USA. 1989;86:9569–9573. doi: 10.1073/pnas.86.23.9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan RKW, Brown ER, Ericsson A, Kovács KJ, Sawchenko PE. A comparison of two immediate-early genes, c-fos and NGFI-B, as markers for functional activation in stress-related neuroendocrine circuitry. J Neurosci. 1993;13:5126–5138. doi: 10.1523/JNEUROSCI.13-12-05126.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dallman MF, Akana SF, Cascio CS, Darlington DN, Jacobson L, Levine N. Regulation of ACTH secretion: variation on a theme B. Recent Prog Horm Res. 1987;43:113–167. doi: 10.1016/b978-0-12-571143-2.50010-1. [DOI] [PubMed] [Google Scholar]
- 13.Diamond MI, Miner JN, Yashinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249:1266–1272. doi: 10.1126/science.2119054. [DOI] [PubMed] [Google Scholar]
- 14.Drouin J, Trifiro MA, Plante RK, Nemer M, Eriksson P, Wrange O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene expression. Mol Cell Biol. 1989;9:5305–5314. doi: 10.1128/mcb.9.12.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 16.Guardiola-Diaz HM, Boswell C, Seasholtz AF. The cAMP-responsive element in the corticotropin-releasing hormone gene mediates transcriptional regulation by depolarization. J Biol Chem. 1994;269:14784–14791. [PubMed] [Google Scholar]
- 17.Guardiola-Diaz HM, Kolinske JS, Gates LH, Seasholtz AF. Negative glucocorticoid regulation of cyclic adenosine 3′5′-monophosphate-stimulated corticotropin-releasing hormone-reporter expression in AtT-20 cells. Mol Endocrinol. 1996;10:317–329. doi: 10.1210/mend.10.3.8833660. [DOI] [PubMed] [Google Scholar]
- 18.Hagiwara M, Brindle P, Harootunian A, Armstrong R, Rivier J, Vale W, Tsien R, Montminy MR. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herman JP. In situ hybridization analysis of vasopressin gene transcription in the paraventricular and supraoptic nuclei of the rat: regulation by stress and glucocorticoids. J Comp Neurol. 1995;363:15–27. doi: 10.1002/cne.903630103. [DOI] [PubMed] [Google Scholar]
- 20.Herman JP, Schafer MK-H, Watson SJ, Sherman TG. In situ hybridization analysis of arginine vasopressin gene transcription using intron-specific probes. Mol Endocrinol. 1991;5:1447–1456. doi: 10.1210/mend-5-10-1447. [DOI] [PubMed] [Google Scholar]
- 21.Herman JP, Schafer MK-H, Thompson RC, Watson SJ. Rapid regulation of corticotropin-releasing hormone gene transcription in vivo. Mol Endocrinol. 1992;6:1061–1069. doi: 10.1210/mend.6.7.1324419. [DOI] [PubMed] [Google Scholar]
- 22.Herman JP, Cullinan WE, Morano MI, Akil H, Watson SJ. Contribution of the ventral subiculum to inhibitory regulation of the hypothalamo-pituitary-adrenocortical axis. J Neuroendocrinol. 1995;7:475–482. doi: 10.1111/j.1365-2826.1995.tb00784.x. [DOI] [PubMed] [Google Scholar]
- 23.Jingami H, Matsukura S, Numa S, Imura H. Effects of adrenalectomy and dexamethasone administration on the level of preprocorticotropin-releasing factor messenger ribonucleic acid (mRNA) in the hypothalamus and adrenocorticotropin/β-lipotropin precursor mRNA in the pituitary in rats. Endocrinology. 1985;117:1314–1320. doi: 10.1210/endo-117-4-1314. [DOI] [PubMed] [Google Scholar]
- 24.Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- 25.Kiss JZ, Mezey É, Skirboll L. Corticotropin-releasing factor-immunoreactive neurons of the paraventricular nucleus become vasopressin positive after adrenalectomy. Proc Natl Acad Sci USA. 1984;81:1854–1858. doi: 10.1073/pnas.81.6.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kovács KJ. c-Fos as a transcription factor: a stressful review from a functional map. Neurochem Int. 1998;33:287–297. doi: 10.1016/s0197-0186(98)00023-0. [DOI] [PubMed] [Google Scholar]
- 27.Kovács K, Mezey É. Dexamethasone inhibits corticotropin-releasing factor gene expression in the rat paraventricular nucleus. Neuroendocrinology. 1987;46:365–368. doi: 10.1159/000124846. [DOI] [PubMed] [Google Scholar]
- 28.Kovács KJ, Sawchenko PE. Sequence of stress-induced alterations in indices of synaptic and transcriptional activation in parvocellular neurosecretory neurons. J Neurosci. 1996;16:262–273. doi: 10.1523/JNEUROSCI.16-01-00262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovács KJ, Sawchenko PE. Glucocorticoid negative feedback is exerted selectively on vasopressin gene transcription in parvocellular neurosecretory neurons. Soc Neurosci Abstr. 1997;23:797.8. doi: 10.1523/JNEUROSCI.20-10-03843.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovács K, Kiss JZ, Makara GB. Glucocorticoid implants around the hypothalamic paraventricular nucleus prevent the increase of corticotropin-releasing factor and arginine vasopressin immunostaining induced by adrenalectomy. Neuroendocrinology. 1986;44:229–234. doi: 10.1159/000124650. [DOI] [PubMed] [Google Scholar]
- 31.Kovács KJ, Arias C, Sawchenko PE. Protein synthesis blockade differentially affects the stress-induced transcriptional activation of neuropeptide genes in parvocellular neurosecretory neurons. Mol Brain Res. 1998;54:85–91. doi: 10.1016/s0169-328x(97)00324-0. [DOI] [PubMed] [Google Scholar]
- 32.Legradi G, Holzer D, Kapcala LP, Lechan RM. Glucocorticoids inhibit stress-induced phosphorylation of CREB in corticotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Neuroendocrinology. 1997;66:86–97. doi: 10.1159/000127224. [DOI] [PubMed] [Google Scholar]
- 33.Lightman SL, Young WS., III Corticotropin-releasing factor, vasopressin and pro-opiomelanocortin mRNA responses to stress and opiates in the rats. J Physiol (Lond) 1988;403:511–523. doi: 10.1113/jphysiol.1988.sp017261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma X-M, Levy A, Lightman SL. Emergence of an isolated arginine vasopressin (AVP) response to stress after repeated restraint: a study of both AVP and corticotropin-releasing hormone messenger ribonucleic acid (RNA) and heteronuclear RNA. Endocrinology. 1997a;138:4351–4357. doi: 10.1210/endo.138.10.5446. [DOI] [PubMed] [Google Scholar]
- 35.Ma X-M, Levy A, Lightman SL. Rapid changes of heteronuclear RNA for arginine vasopressin but not for corticotropin releasing hormone in response to acute corticosterone administration. J Neuroendocrinol. 1997b;9:723–728. doi: 10.1046/j.1365-2826.1997.00646.x. [DOI] [PubMed] [Google Scholar]
- 36.Makino S, Smith MA, Gold PW. Increased expression of corticotropin-releasing hormone and vasopressin messenger ribonucleic acid (mRNA) in the hypothalamic paraventricular nucleus during repeated stress: association with reduction in glucocorticoid receptor mRNA levels. Endocrinology. 1995;136:3299–3309. doi: 10.1210/endo.136.8.7628364. [DOI] [PubMed] [Google Scholar]
- 37.Morgan JI, Curran T. Stimulus-transcription coupling in the nervous system: involvement of inducible proto-oncogenes fos and jun. Annu Rev Neurosci. 1991;14:421–451. doi: 10.1146/annurev.ne.14.030191.002225. [DOI] [PubMed] [Google Scholar]
- 38.Pardy K, Adan RAH, Carter DA, Seah V, Burbach JPH, Murphy D. The identification of a cis-acting element involved in cyclic 3′,5′-adenosine monophosphate regulation of bovine vasopressin gene expression. J Biol Chem. 1992;267:21746–21752. [PubMed] [Google Scholar]
- 39.Pearce D, Yamamoto KR. Mineralocorticoid and glucocorticoid receptor activities distinguished by nonreceptor factors at a composite response element. Science. 1993;259:1161–1165. doi: 10.1126/science.8382376. [DOI] [PubMed] [Google Scholar]
- 40.Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 41.Roberts JL, Budarf ML, Baxter JD, Herbert E. Selective reduction of proadenocorticotropin/endorphin proteins and mRNA activity in mouse pituitary tumor cells by glucocorticoids. Biochemistry. 1979;18:4907–4915. doi: 10.1021/bi00589a019. [DOI] [PubMed] [Google Scholar]
- 42.Roche PJ, Crawford RJ, Fernley RT, Tregear GW, Coghlan JP. Nucleotide sequence of the gene coding for ovine corticotropin-releasing factor and regulation its mRNA level by glucocorticoids. Gene. 1988;71:421–431. doi: 10.1016/0378-1119(88)90059-5. [DOI] [PubMed] [Google Scholar]
- 43.Rosen LB, Majzoub JA, Adler GK. Effects of glucocorticoid on corticotropin-releasing hormone gene regulation by second messenger pathways in NPLC and AtT-20 cells. Endocrinology. 1992;130:2237–2244. doi: 10.1210/endo.130.4.1547737. [DOI] [PubMed] [Google Scholar]
- 44.Sassone-Corsi P, Visvader J, Ferland L, Mellon PL, Verma IM. Induction of proto-oncogene fos transcription through the adenylate-cyclase pathway: characterization of a cAMP-responsive element. Genes Dev. 1988;2:1529–1538. doi: 10.1101/gad.2.12a.1529. [DOI] [PubMed] [Google Scholar]
- 45.Sawchenko PE, Swanson LW, Vale WW. Co-expression of corticotropin-releasing factor and vasopressin immunoreactivity in parvocellular neurosecretory neurons of the adrenalectomized rat. Proc Natl Acad Sci USA. 1984;81:1883–1887. doi: 10.1073/pnas.81.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sawchenko PE, Cunningham ET, Jr, Mortrud MT, Pfeiffer SW, Gerfen CR. Phaseolus vulgaris-leucoagglutinin (PHA-L) anterograde axonal transport technique. Methods Neurosci. 1990;3:247–260. [Google Scholar]
- 47.Schule R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM. Functional antagonism between oncoprotein c-jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 48.Seasholtz AF, Thompson RC, Douglas JO. Identification of a cyclic adenosine monophosphate-responsive element in the rat corticotropin-releasing hormone gene. Mol Endocrinol. 1988;2:1311–1319. doi: 10.1210/mend-2-12-1311. [DOI] [PubMed] [Google Scholar]
- 49.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 50.Simmons DM, Arriza JL, Swanson LW. A complete protocol for in situ hybridization of messenger RNAs in brain and other tissues with radiolabeled single-stranded RNA probes. J Histotechnol. 1989;12:169–181. [Google Scholar]
- 51.Stauber C, Altschmeid J, Akerblom IE, Marron JL, Mellon PL. Mutual cross-interference between glucocorticoid receptor and CREB inhibits transactivation in placental cells. New Biol. 1990;4:527–540. [PubMed] [Google Scholar]
- 52.Swanson LW, Kuypers HGJM. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and the organization of projections to the pituitary, dorsal vagal complex and spinal cord as demonstrated by retrograde fluorescence double labeling methods. J Comp Neurol. 1980;194:555–570. doi: 10.1002/cne.901940306. [DOI] [PubMed] [Google Scholar]
- 53.Swanson LW, Simmons DM. Differential steroid hormone and neural influences on peptide mRNA levels in CRH cells of the paraventricular nucleus: hybridization histochemical study in the rat. J Comp Neurol. 1989;285:413–435. doi: 10.1002/cne.902850402. [DOI] [PubMed] [Google Scholar]
- 54.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 55.Uht RM, McKelvy JF, Harrison RW, Bohn MC. Demonstration of glucocorticoid receptor-like immunoreactivity in glucocorticoid-sensitive vasopressin and corticotropin-releasing factor neurons in the hypothalamic paraventricular nucleus. J Neurosci Res. 1988;19:405–411. doi: 10.1002/jnr.490190404. [DOI] [PubMed] [Google Scholar]
- 56.Unlap T, Jope RS. Dexamethasone attenuates kainate-induced AP-1 activation in rat brain. Mol Brain Res. 1994;24:275–282. doi: 10.1016/0169-328x(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 57.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and β-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 58.Verbeeck MAE, Adan RAH, Burbach JPH. Vasopressin gene expression is stimulated by cyclic AMP in homologous and heterologous expression systems. FEBS Lett. 1990;272:89–93. doi: 10.1016/0014-5793(90)80455-r. [DOI] [PubMed] [Google Scholar]
- 59.Wolfson B, Manning RW, Davis LG, Arentzen R, Baldino F., Jr Co-localization of corticotropin-releasing factor and vasopressin mRNA in neurons after adrenalectomy. Nature. 1985;315:59–61. doi: 10.1038/315059a0. [DOI] [PubMed] [Google Scholar]
- 60.Yang-Yen H-F, Chambard J-C, Sun Y-L, Smeal T, Schmidt TJ, Drouin J, Karin M. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 61.Young WS, III, Mezey É, Siegel RE. Vasopressin and oxytocin mRNAs in adrenalectomized and Brattleboro rats: analysis by quantitative in situ hybridization histochemistry. Mol Brain Res. 1986a;1:231–241. doi: 10.1016/0169-328x(86)90029-x. [DOI] [PubMed] [Google Scholar]
- 62.Young WS, III, Mezey É, Siegel RE. Quantitative in situ hybridization histochemistry reveals increased levels of corticotropin-releasing factor mRNA after adrenalectomy in rats. Neurosci Lett. 1986b;70:198–203. doi: 10.1016/0304-3940(86)90463-5. [DOI] [PubMed] [Google Scholar]