

Abstract
Developmental changes in the signaling properties of NMDA receptors have been proposed to underlie the loss of plasticity that accompanies brain maturation. Calcium influx through postsynaptic NMDA receptors can stimulate neuronal gene expression via signaling pathways such as the Ras-MAP kinase (MAPK) pathway and the transcription factor cAMP response element-binding protein (CREB). We analyzed MAPK (Erk1/2) and CREB activation in response to NMDA receptor stimulation during the development of hippocampal neurons in culture. At all stages of development NMDA stimulation induced a rapid phosphorylation of CREB on Ser-133 (phospho-CREB). However, the time course of decline in phospho-CREB changed dramatically with neuronal maturation. At 7 din vitro (7 DIV) phospho-CREB remained elevated 2 hr after strong NMDA stimulation, whereas at 14 DIV phospho-CREB rose only transiently and fell back to below basal levels within 30 min. Moreover, at 14 DIV, but not at 7 DIV, NMDA receptor stimulation induced a dephosphorylation of CREB that previously had been phosphorylated by KCl depolarization or forskolin, suggesting an NMDA receptor-dependent activation of a CREB phosphatase. There was no developmental change in the time course of phospho-CREB induction that followed KCl depolarization or PKA activation, nor was there a developmental change in the time course of phospho-Erk1/2 induced by NMDA receptor activation. We suggest that, during neuronal maturation, NMDA receptor activation becomes linked specifically to protein phosphatases that act on Ser-133 of CREB. Such a developmentally regulated switch in the mode of NMDA receptor coupling to intracellular signaling pathways may contribute to the changes in neural plasticity observed during brain development.
Keywords: MAP kinase, ERK1/2, synaptic plasticity, protein phosphatase-1, gene expression, critical period
The NMDA class of ionotropic glutamate receptor is critical for activity-dependent synaptic modifications that underlie plasticity of the brain. Changes in the abundance and functional properties of NMDA receptors therefore can have important effects on synaptic plasticity. Developmental changes in the signaling properties of NMDA receptors have been proposed to underlie the loss of plasticity that accompanies brain maturation. For instance, during cortical development, changes in the gating properties of NMDA receptors correlate with maturational changes in the plasticity of visual cortex (Carmignoto and Vicini, 1992; Fox et al., 1999).
NMDA receptors are composed of NR1 subunits in combination with NR2(A–D) subunits, the latter conferring different properties on the heteromeric receptor (McBain and Mayer, 1994; Dingledine et al., 1999). The developmental change in gating properties of NMDA receptors is likely to result from a switch in subunit composition during cortical maturation, in which NR2A subunits replace or supplement NR2B-containing receptors (Monyer et al., 1994; Sheng et al., 1994;Quinlan et al., 1999; Tovar and Westbrook, 1999).
Although a change in subunit composition and hence in gating properties of NMDA receptors may contribute to the developmental regulation of NMDA receptor activity, it is probably not the only mechanism for tuning NMDA receptor signaling. Recently, it has become recognized that some aspects of NMDA receptor signal transduction depend on receptor interactions with intracellular signaling proteins mediated by the scaffold protein postsynaptic density-95 (PSD-95) or other NMDA receptor-binding proteins (Craven and Bredt, 1998; Migaud et al., 1998;Sprengel et al., 1998; Sheng and Pak, 2000). In gene targeting experiments in mice, deletion of the C-terminal tail of NR2 subunits that binds to PSD-95, or disruption of PSD-95 itself, results in defective NMDA receptor signaling and altered synaptic plasticity (Migaud et al., 1998; Mori et al., 1998; Sprengel et al., 1998).
During brain maturation the expression of many NMDA receptor-associated proteins increases and their distribution becomes localized to synapses (Cho et al., 1992; Rao et al., 1998; Wyszynski et al., 1998). We hypothesized that the mechanisms of NMDA receptor signaling can be altered during neuronal development as a result of maturational changes in the NMDA receptor-associated protein complex. We focused on NMDA receptor signaling to MAP kinase (MAPK or Erk) and to CREB, because NMDA receptor stimulation is well established to activate pathways leading to the phosphorylation and activation of these proteins in neurons (Shaywitz and Greenberg, 1999). Here we report that strong NMDA receptor stimulation of mature hippocampal neurons in culture (14 DIV or older) results in only a transient phosphorylation of CREB on Ser-133 in contrast to the prolonged CREB phosphorylation seen in immature neurons. The transience of the increase in phospho-CREB results from an NMDA receptor-dependent activation of a phosphatase that acts selectively on CREB and that is coupled to NMDA stimulation only in neurons beyond a critical stage of development. These findings suggest that synaptic plasticity can be regulated developmentally not only by modulating NMDA receptor gating kinetics but also by changing the signal output of NMDA receptors.
MATERIALS AND METHODS
Hippocampal neuron culture. Hippocampal neuron cultures were prepared from embryonic day (E) E18–E19 rat embryos dissociated with trypsin and plated on 12-well plates or 18-mm-diameter coverslips coated with poly-l-lysine (1 mg/ml in 100 mm borate buffer, pH 8.5) in MEM containing 10% fetal calf serum (FCS), 25 μg/ml insulin, 100 μg/ml transferrin, 1 mm pyruvate, and 0.6% glucose. At 4 DIV Ara-C (5 μm) was added to the medium, and at 7 DIV (and subsequently once a week) one-half of the medium was changed with fresh medium without FCS.
Cell stimulation. Cells were treated with glutamate (100 μm), NMDA (20 or 100 μm), forskolin (75 μm), or KCl (55 mm) at 3, 7, 14, or 21 DIV. Tetrodotoxin (1 μm) was added to cultures 12 hr before stimulation to reduce endogenous synaptic activity. For NMDA receptor stimulation the neurons were pretreated for 20–30 min with CNQX (40 μm) and nimodipine (5 μm). Neurons depolarized with KCl were pretreated with CNQX and aminophosphonopentanoic acid (AP-5; 100 μm). Forskolin treatment was in the presence of CNQX, AP-5, and nimodipine. In some case AP-5 or nimodipine was left out to allow multiple treatments like NMDA plus KCl or forskolin plus NMDA (see Results). All of the drugs were added directly to the medium, and during the incubation the neurons were kept in the 5% CO2 incubator.
Western blotting and quantitation. After stimulation the neurons were extracted with 80 μl of SDS sample buffer per well and boiled for 10 min. The lysates ( of the total) were separated by 10% SDS-PAGE and transferred onto nitrocellulose filters. The filters were incubated with polyclonal anti-phospho-CREB (1:1000; Upstate Biotechnology, Lake Placid, NY) or polyclonal anti-phospho-ERK1/2 (1:1000; New England Biolabs, Beverly, MA) and were visualized by chemiluminescence. Then the filters were stripped and reprobed with anti-CREB (1:250; Upstate Biotechnology) or anti-ERK1/2 (1:1000; New England Biolabs) to detect total CREB and ERK1/2. For quantitation of immunoblot signals, the band intensity was measured with a Kodak Digital Science 1D program (Rochester, NY). Because CREB antibodies and Erk1/2 antibodies (either phospho-specific or total) recognize a major doublet of bands on immunoblots, we measured the sum of both by densitometry. Each phospho-specific band intensity was equalized to the total CREB and ERK1/2 signals in the same lane. The increase in phosphorylation of CREB and ERK was normalized to the basal level and expressed as fold increase. The results were plotted with Microsoft Excel.
For Figure 7B the following antibodies were used: PP-1 rabbit polyclonal (Westphal et al., 1999) at 1:2000 and β-tubulin mouse monoclonal (Sigma, St. Louis, MO) at 1:2000.
Fig. 7.

Protein phosphatases and NMDA receptor-dependent dephosphorylation of CREB. A, NMDA receptor-dependent dephosphorylation of CREB is blocked by okadaic acid (2 μm), but not by cyclosporin A (250 μm), FK506 (1 μm), or okadaic acid (20nm). Hippocampal cultures (14 DIV) were treated with KCl or forskolin and then with NMDA +/−- phosphatase inhibitors, as indicated. Stimulation protocol, immunoblotting results, and histogram quantification are as in Figure 4. B, Immunoblot of hippocampal neuron cultures at 3, 7, 10, and 15 DIV for PP-1 levels (tubulin used as internal control).
Immunostaining. After stimulation the neurons were fixed immediately in 4% paraformaldehyde and 4% sucrose for 15 min. After permeabilization with 0.3% Triton X-100 the cells were incubated with anti-phospho-CREB (1:1000; Upstate Biotechnology) overnight at 4°C in 1× GDB buffer (30 mm phosphate buffer, pH 7.4, containing 0.2% gelatin, 0.5% Triton X-100, and 0.8 m NaCl), followed by FITC-conjugated secondary antibodies (Jackson Laboratories, Bar Harbor, ME) for 1 hr. The fluorescent images were acquired by using an interline cooled CCD camera (Princeton Instruments, Trenton, NJ) and prepared for publication with Adobe Photoshop.
RESULTS
A developmental change in the time course of NMDA receptor-induced phospho-CREB
The activation of CREB and Erk1/2 can be monitored by immunoblotting with antibodies that specifically recognize phospho-CREB (phosphorylated on Ser-133) and phospho-Erk1/2 (phosphorylated on Thr-202 and Tyr-204). As previously reported (Bading and Greenberg, 1991; Ginty et al., 1993; Xia et al., 1996), Ca2+ influx mediated by NMDA receptor activation or KCl depolarization induces phosphorylation of CREB and Erk1/2 in cultured hippocampal neurons prepared from E18–E19 rat embryos (Fig. 1). The time course of induction and maintenance of phospho-CREB and phospho-Erk1/2 was studied after various treatments of hippocampal cultures grown for 3, 7, 14, and 21 DIV. NMDA receptors were activated by 100 μm NMDA in the presence of 40 μm CNQX and 10 μm nimodipine, whereas calcium influx through voltage-gated Ca2+ channels (VGCCs) was stimulated by depolarization with 55 mm KCl in the presence of 40 μm CNQX and 100 μm APV.
Fig. 1.

Differential time courses of CREB and Erk1/2 phosphorylation and dephosphorylation in response to NMDA stimulation or KCl depolarization in cultured hippocampal neurons at 7 and 14 DIV. Representative immunoblot experiments are shown for CREB (A) and Erk1/2 (B). Phospho-CREB and phospho-Erk1/2 were immunoblotted with phospho-specific antibodies; total CREB and Erk1/2 were immunoblotted with specific antibodies that are not sensitive to a phosphorylation state. The x-axis shows time in minutes after the onset of stimulation. The y-axis shows the fold increase over basal. Results from several experiments (n = 6) are quantified in graph form for phospho-CREB (C) and phospho-Erk1/2 (D). Values are normalized to baseline. Error bars indicate ± SEM.
In hippocampal neuron cultures at 7 DIV, levels of CREB phosphorylated on Ser-133 rose rapidly after NMDA receptor stimulation, peaking within 2–5 min and remaining elevated for at least 2 hr (Fig.1A,C). Erk1/2 phosphorylation also increased rapidly in response to NMDA stimulation; it peaked by 2 min but returned close to basal levels at 2 hr (Fig. 1B,D). The same results were obtained after stimulation with glutamate (100 μm) in the presence of blockers of AMPA receptors and VGCCs and in hippocampal cultures at 3 DIV (data not shown).
In more mature hippocampal cultures (14 or 21 DIV) a markedly different time course was observed for NMDA-induced phospho-CREB levels (Fig.1A,C). The level of phospho-CREB rose during the first 2 min, but at 5 min the level decreased rapidly and by 10 min had fallen below the basal level. At 1 hr after NMDA treatment, phospho-CREB was virtually undetectable in 14 DIV cultures (Fig.1A,C). The change in time course was specific for phospho-CREB, because the phospho-Erk1/2 response to NMDA in 14 DIV cultures had the same temporal profile as at 7 DIV (Fig.1B,D). Similar results were obtained at 21 as at 14 DIV (data not shown). Thus in hippocampal cultures older than 14 DIV, there was a specific change in the duration of CREB phosphorylation induced by NMDA receptors. Instead of staying elevated (as in cultures at 7 DIV), phospho-CREB appeared to be dephosphorylated rapidly after NMDA stimulation in neurons older than 14 DIV.
We found no differences in the time course of phospho-CREB induced by KCl depolarization at 7, 14, or 21 DIV. At all developmental stages the level of phospho-CREB peaked rapidly within 2–5 min and remained elevated at 2 hr (Fig. 1A,C). KCl induced Erk1/2 phosphorylation that was more sustained than that induced by NMDA, but as with NMDA stimulation there was no difference between 7 and 14 DIV in the temporal profile of the phospho-Erk response (Fig.1B,D). Forskolin (75 μm), which activates adenylate cyclase and PKA, also induced a persistent CREB phosphorylation that was similar for both 7 and 14 DIV cultures. In both cases the phospho-CREB reached a plateau after 5 min and remained elevated for at least 2 hr (Fig.2A). Thus the transient phosphorylation followed by a rapid dephosphorylation of CREB is a specific response to NMDA receptor activation and occurs only in cultures 14 DIV or older.
Fig. 2.
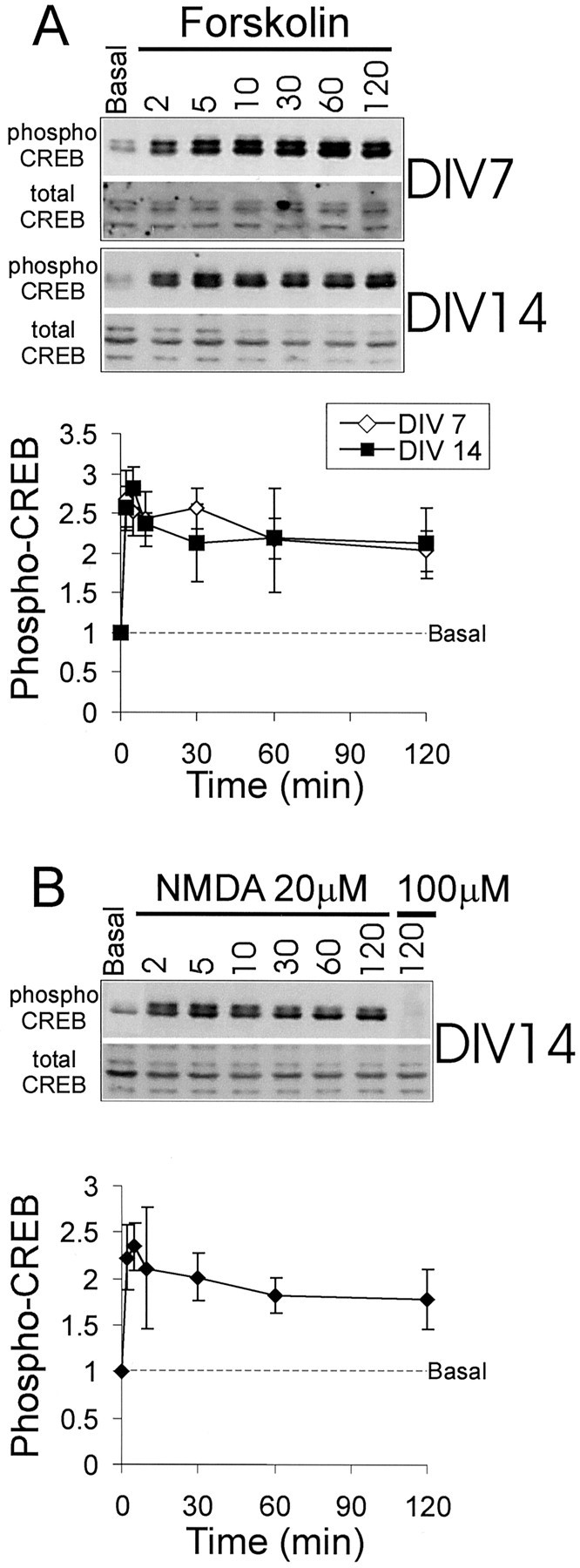
Time course of CREB phosphorylation and dephosphorylation in response to forskolin or low concentration (20 μm) NMDA in cultured hippocampal neurons at 7 and 14 DIV. Shown in A are a representative immunoblot and graphical quantification of CREB phosphorylation in response to forskolin, as shown in Figure 1. The time course of CREB phosphorylation in response to 20 μm NMDA is shown as an immunoblot and is quantified in B.
Interestingly, the transient increase followed by the persistent decrease in phospho-CREB in 14 DIV neurons was seen only in response to a strong activation of NMDA receptors. By using 20 instead of 100 μm NMDA, the time course of phospho-CREB at 14 DIV was similar to that seen at 7 DIV, remaining elevated at 2 hr (Fig.2B).
We used immunostaining in addition to immunoblotting to monitor CREB phosphorylation in hippocampal cultures and obtained similar results (Fig. 3; data not shown). Both NMDA and KCl stimulation caused a rapid increase in nuclear immunoreactivity for phospho-CREB in neurons at 14 DIV. At 90 min after NMDA stimulation, however, phospho-CREB staining in nuclei was below baseline, whereas it remained elevated 90 min after KCl depolarization (Fig. 3). Thus immunocytochemistry corroborates the Western blotting results, confirming that NMDA stimulation leads to a transient phosphorylation followed by a dephosphorylation of CREB in 14 DIV neurons.
Fig. 3.

Differential time course of CREB phosphorylation and dephosphorylation in response to NMDA and KCl as shown by immunocytochemistry for phospho-CREB in 14 DIV hippocampal neuron cultures. Cultures were stained for phospho-CREB at the indicated times (in minutes) after NMDA stimulation or KCl depolarization.
NMDA receptor stimulation blocks subsequent CREB phosphorylation by depolarization
After an initial spike the level of phospho-CREB falls to barely detectable levels by 1 hr after NMDA stimulation in 14 DIV neurons (see Fig. 1A,C). Can this dephosphorylated CREB be rephosphorylated by further stimulation with KCl or forskolin? Hippocampal neurons were treated first with 100 μm NMDA in the presence of CNQX for 1 hr, at the end of which they were stimulated with KCl or forskolin for 5 min (Fig. 4). In 14-d-old cultures the dephosphorylated CREB could not be rephosphorylated by KCl or forskolin treatments; however, Erk1/2 phosphorylation in the same cultures was stimulated further (two- to threefold) by KCl (Fig. 4). On the other hand, at 7 DIV, although the level of phospho-CREB remaining 1 hr after NMDA stimulation was still elevated above baseline, it could be increased further by KCl and forskolin. The further phosphorylation of Erk1/2 and CREB stimulated by KCl is blocked by nimodipine, confirming that it is mediated by VGCCs. Taken together, these results show that, after an initial phosphorylation of CREB, strong NMDA receptor stimulation activates a specific CREB dephosphorylation mechanism in 14 DIV neurons that cannot be overcome by stimulating classical cAMP- or Ca2+-dependent pathways of Ser-133 phosphorylation.
Fig. 4.

Strong NMDA stimulation specifically results in dephosphorylation of CREB in 14 DIV neurons, which is refractory to further rephosphorylation by forskolin or KCl. Top, The figure illustrates the protocol used for sequential stimulation by NMDA and forskolin/KCl. Middle, Shown are representative immunoblotting experiments for CREB phosphorylation (left) and Erk1/2 phosphorylation (right) for cultures at 7 and 14 DIV, as indicated. Bottom, Histograms quantify the relative levels of phospho-CREB or phospho-Erk1/2 for each condition/lane from several experiments (n = 5). Asterisk indicates significance at p < 0.01 in an unpaired, two-tailed Student's t test when compared with the value obtained in neurons stimulated additionally with KCl for 5 min or forskolin for 5 min at 7 DIV.
NMDA receptor-stimulated CREB dephosphorylation reverses cAMP or Ca2+-dependent CREB phosphorylation
We next tested whether, after KCl- or forskolin-induced phosphorylation of CREB, NMDA receptor activation can cause a dephosphorylation of CREB. Neurons first were treated with KCl in the presence of CNQX for 1 hr, at the end of which time (when the level of phospho-CREB is approximately twofold over basal) they were stimulated with 100 μm NMDA for 5 min (Fig.5). In 14 d cultures, but not in 7 d cultures, NMDA receptor activation in this protocol caused a dephosphorylation of CREB to basal levels (Fig. 5, left). An even more profound dephosphorylation of CREB was seen with 10 min treatments of NMDA or glutamate (data not shown). Significantly, NMDA did not cause dephosphorylation of Erk1/2 in the same experiments at either 7 or 14 DIV (Fig. 5, right). NMDA stimulation also reduced phospho-CREB levels after forskolin stimulation of 14 DIV cultures (Fig. 6, left), even as it caused an increase of phospho-Erk1/2 in the same protocol (Fig.6, right). After either KCl or forskolin the dephosphorylation of CREB by NMDA was blocked by AP-5, confirming that this effect is mediated by NMDA receptors (Figs. 5, 6). These data indicate that NMDA receptor stimulation in more mature neuron cultures (>14 DIV) activates a pathway that leads specifically to dephosphorylation of CREB, but not Erk1/2.
Fig. 5.

NMDA stimulation causes dephosphorylation of CREB that previously was phosphorylated in response to KCl depolarization, specifically in 14 DIV neurons. Stimulation protocol, immunoblotting results, and histogram quantification are as shown in Figure 4.Asterisk indicates significance at p< 0.01 in an unpaired, two-tailed Student's t test when compared with the values obtained in other conditions in this experiment.
Fig. 6.

NMDA stimulation causes dephosphorylation of CREB that previously was phosphorylated in response to forskolin, specifically in 14 DIV neurons. Stimulation protocol, immunoblotting results, and histogram quantification are as in Figure 4.
Two different phosphatases have been implicated in the dephosphorylation of CREB: calcineurin (PP-2B) and protein phosphatase-1 (PP-1) (Hagiwara et al., 1992; Bito et al., 1996). We tested pharmacologically whether these phosphatases play a role in NMDA-induced dephosphorylation of CREB, using 2 μmokadaic acid to block PP-1 and two specific antagonists of calcineurin, cyclosporin A (250 μm) and FK506 (1 μm). We also used a low concentration of okadaic acid (20 nm) to distinguish between PP-1 and PP-2A, because PP-2A is inhibited by okadaic acid in the 10 nm range (Cohen et al., 1990). In neurons at 14 DIV, only 2 μm okadaic acid, but not 20 nm okadaic acid or cyclosporin A or FK506, was able to prevent the NMDA receptor-mediated decrease in phospho-CREB after KCl or forskolin treatment (Fig.7A). These results are consistent with PP-1 being involved in the CREB dephosphorylation mechanism activated by NMDA receptors in >14 DIV neurons. To test whether the expression of PP-1 increases during development of hippocampal neurons in culture, we performed Western blot analysis of cultures at 3, 7, 10, and 15 DIV. Figure 7B shows that, over this 2 week period during which NMDA receptors become functionally coupled to CREB dephosphorylation, the levels of PP-1 protein stayed constant.
DISCUSSION
The major conclusions of this paper can be summarized as follows: (1) The duration of CREB Ser-133 phosphorylation induced by NMDA receptor activation depends on the developmental maturity of cultured hippocampal neurons. After 14 DIV, strong NMDA stimulation causes only a transient increase in phospho-CREB, contrasting with the prolonged increase in phospho-CREB seen at 7 DIV. (2) This developmental switch is stimulus- and pathway-specific, in that K+ depolarization (acting through VGCCs) induces the same prolonged CREB phosphorylation in young (7 DIV) and mature (>14 DIV) cultures, and the time course of NMDA receptor-stimulated phospho-Erk1/2 elevation is unaffected by culture age. (3) Strong NMDA receptor stimulation in mature neurons actually causes a robust dephosphorylation of CREB, possibly via activation of phosphatase PP-1.
The findings of this study imply a pathway-specific developmental change in the coupling of NMDA receptors to downstream signaling mechanisms. The predominant effect of NMDA receptors on CREB actually was reversed in older neurons (which showed mainly NMDA-mediated dephosphorylation of Ser-133 with strong NMDA stimuli). Significantly, this switch occurred in the absence of any apparent change in NMDA receptor-stimulated phosphorylation of Erk1/2. These features make it unlikely that the developmental change in NMDA receptor signaling to CREB is attributable simply to expression of higher levels of NMDA receptors or more active NMDA receptors in >14 DIV neurons.
Biphasic regulation of CREB phosphorylation and synaptic plasticity
The NMDA receptor-dependent CREB dephosphorylation in >14 DIV neurons depends on the concentration of NMDA. Dephosphorylation of CREB in older neurons was seen only with 100 μm NMDA, but not with 20 μm NMDA. This suggests the presence specifically in mature neurons of a biphasic Ca2+-dependent NMDA receptor-mediated mechanism, one that has a low threshold for the activation of the pathway leading to CREB phosphorylation and a higher threshold for the activation of the phosphatase pathway.
What might be the neurobiological significance of the developmental switch in the mode of NMDA receptor signaling to CREB? In addition to short-term plasticity lasting from minutes to hours, NMDA receptor activation can initiate long-term neural plasticity requiring changes in gene expression. CREB is a transcription factor widely implicated in synaptic plasticity and memory formation (Frank and Greenberg, 1994;Ghosh and Greenberg, 1995; Bito et al., 1997). Phosphorylation of CREB on Ser-133 is critical for activating the transcription of genes controlled by the CRE element (Montminy, 1997), many of which may be involved in neuronal growth and plasticity, e.g., BDNF, CaMKIV, synapsin I, somatostatin, voltage-gated potassium channels, Fos, and Jun (Sauerwald et al., 1990; Mori et al., 1993; Sassone-Corsi, 1995;Shieh et al., 1998; Tao et al., 1998).
CREB-responsive genes appear to be activated only when Ser-133 phosphorylation of CREB is prolonged, as occurs during long-lasting synaptic stimulation (Bito et al., 1996, 1997). Our study indicates that an additional factor that influences duration of CREB phosphorylation is the developmental age of the neurons. Because of the appearance of an NMDA receptor-coupled phosphatase pathway leading to CREB dephosphorylation, NMDA receptor stimulation in mature neurons is less likely to result in persistent CREB activation and hence CRE-directed gene transcription. In older neurons, NMDA receptor activation can even result in the net dephosphorylation of CREB. In immature neurons, by comparison, NMDA receptor stimulation is favored to induce CREB-dependent gene expression because of the relative lack of CREB phosphatase activity and the persistent phosphorylation of Ser-133. However, CREB phosphorylation by itself is not synonymous with gene induction (Ginty, 1997), and it remains to be confirmed whether the regulation of CREB phosphorylation by NMDA receptors correlates with gene transcriptional responses.
We speculate that the developmental increase in NMDA receptor-mediated dephosphorylation of CREB may be one of the factors that contributes to the reduction in synaptic plasticity that normally accompanies nervous system maturation. Consistent with this, a recent study showed that CRE-mediated gene expression is downregulated after the critical period in visual cortex (Pham et al., 1999). Our findings suggest that one possible mechanism for inhibiting plasticity in mature brain is the relative loss of the ability of the NMDA receptor to support persistent phosphorylation of CREB. Prolonged phosphorylation of CREB still can be achieved in mature neurons, but it may require more stringent conditions such as sustained synaptic stimulation, which appears to work in part via an activity-dependent inhibition of CREB dephosphorylation (Bito et al., 1996; Liu and Graybiel, 1996).
Using hippocampal neurons at 10–14 DIV, Hardingham et al. (1999) also found that CREB was more transiently phosphorylated in response to NMDA receptor stimulation than in response to KCl depolarization. These workers correlated the transience of this CREB phosphorylation with the ineffective induction of CRE-dependent transcription by NMDA receptors. By contrast, Hu et al. (1999), using cortical neurons at 5 DIV, reported that CREB-mediated transcription could be readily stimulated by 100 μm glutamate in a NMDA receptor-dependent manner. The apparent discrepancy between these two reports might be explained, at least in part, by our finding of a developmental switch in NMDA receptor signaling that allows CREB phosphorylation to persist in immature neurons and that abbreviates CREB phosphorylation in more developed neurons. More generally speaking, the NMDA receptor-mediated dephosphorylation of CREB in mature neurons may contribute to the observation that NMDA receptor stimulation is inefficient in activating CRE-dependent transcription.
Coupling of NMDA receptors to a phosphatase
In older neurons, NMDA receptor stimulation results in a transient phosphorylation of CREB, followed by a prolonged dephosphorylation below baseline. After KCl depolarization or PKA stimulation (which induce phosphorylation of CREB), NMDA actually causes dephosphorylation of CREB, while in the same neurons further enhancing Erk1/2 phosphorylation. These observations suggest that NMDA receptors become coupled to a specific CREB phosphatase activity as neurons mature. That the NMDA receptor-mediated dephosphorylation of CREB is blocked by 2 μm okadaic acid, but not by 20 nm okadaic acid or 250 μm cyclosporin A or 1 μm FK506, suggests that PP-1 is more critical in this pathway than PP-2A or calcineurin. This conclusion is consistent with previous reports showing that PP-1 is a major phosphatase involved in CREB dephosphorylation (Hagiwara et al., 1992; Bito et al., 1996).
Interestingly, PP-1 activity has been shown to be regulated by synaptic activity (Mulkey et al., 1994). PP-1 is concentrated in dendritic spines, perhaps by binding to spinophilin, a scaffold protein specifically localized in spines (Allen et al., 1997). PP-1 also interacts with Yotiao, an A-kinase anchoring protein (AKAP) that binds directly to the NMDA receptor (Lin et al., 1998; Westphal et al., 1999). Thus PP-1 is in the vicinity of the NMDA receptor, but it remains to be determined how it is activated after NMDA receptor stimulation. Alternatively, PP-1 might not be activated locally at the synapse but, rather, in the nucleus in response to NMDA receptor signals transduced from the synapse (Bito et al., 1997).
Developmental changes in NMDA receptor signaling
In brain the levels of many excitatory postsynaptic proteins have been shown to increase during development. For instance, there is an induction of the NR2A subunit, which is likely to be incorporated into NMDA receptors via assembly with NR1/NR2B subunits (Tovar and Westbrook, 1999). A change in the subunit composition of NMDA receptors can alter electrophysiological and pharmacological properties of the receptor–channel (Monyer et al., 1994; Dingledine et al., 1999), but it is hard to see how such biophysical changes by themselves could specifically switch the mode of NMDA receptor signal transduction to CREB. We therefore prefer the idea that the developmental switch is mediated by a quantitative or qualitative change in the protein–protein interactions that couple NMDA receptors to downstream signaling cascades.
Expression of PSD-95 and certain PSD-95-associated proteins increases during postnatal development of the brain (Cho et al., 1992; Lim et al., 1999; C. Sala and M. Sheng, unpublished observations). In mature neurons the interaction of NMDA receptors with PSD-95 may link NMDA receptors to a biochemical pathway that results in the activation of a CREB phosphatase. Whether interaction with the PSD-95 complex is required for NMDA receptor coupling to CREB dephosphorylation remains to be determined, as does the identity of the specific signaling proteins that mediate this coupling. However, it is intriguing to note in this regard that the PSD-95 knock-out mouse showed a surprising enhancement of LTP (Migaud et al., 1998), which was speculated to involve the loss of a PSD-95-dependent phosphatase (Malenka and Nicoll, 1998). It is also possible that PSD-95-independent interactions of NMDA receptors mediate the coupling of NMDA receptors to CREB dephosphorylation. A third possibility is that the NMDA receptor-dependent CREB phosphatase activity is regulated developmentally via mechanisms that do not involve protein–protein interactions, for instance, simply by elevating the expression of the CREB phosphatase. A pertinent finding here is that the levels of PP-1 do not change during the first 2 weeks of hippocampal neuron culture (Fig. 7B). Whatever the mechanism, it is clear that NMDA receptors differ significantly in their downstream effects depending on the developmental state of the neuron. Further studies on the maturation of the NMDA receptor signaling complex may shed light on the mechanisms that control postsynaptic plasticity during brain development.
Footnotes
M.S. is Assistant Investigator of the Howard Hughes Medical Institute. C.S. was supported by a fellowship from the Harvard Armenise Foundation (Department of Biological and Technical Research, San Raffaele, Italy). We thank Haruhiko Bito for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Morgan Sheng, Howard Hughes Medical Institute (Wellman 423), Massachusetts General Hospital, 50 Blossom Street, Boston, MA 02114. E-mail: sheng@helix.mgh.harvard.edu.
REFERENCES
- 1.Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase-1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 3.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 4.Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- 5.Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- 6.Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 7.Cohen P, Holmes CF, Tsukitani Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci. 1990;15:98–102. doi: 10.1016/0968-0004(90)90192-e. [DOI] [PubMed] [Google Scholar]
- 8.Craven SE, Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 9.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 10.Fox K, Henley J, Isaac J. Experience-dependent development of NMDA receptor transmission. Nat Neurosci. 1999;2:297–299. doi: 10.1038/7203. [DOI] [PubMed] [Google Scholar]
- 11.Frank DA, Greenberg ME. CREB: a mediator of long-term memory from mollusks to mammals. Cell. 1994;79:5–8. doi: 10.1016/0092-8674(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 13.Ginty DD. Calcium regulation of gene expression: isn't that spatial? Neuron. 1997;18:183–186. doi: 10.1016/s0896-6273(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 14.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 15.Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Deng T, Karin M, Shenolikar S, Montminy M. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell. 1992;10:105–113. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- 16.Hardingham GE, Chawla S, Cruzalegui FH, Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22:789–798. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- 17.Hu SC, Chrivia J, Ghosh A. Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron. 1999;22:799–808. doi: 10.1016/s0896-6273(00)80738-2. [DOI] [PubMed] [Google Scholar]
- 18.Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E. Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem. 1999;274:29510–29518. doi: 10.1074/jbc.274.41.29510. [DOI] [PubMed] [Google Scholar]
- 19.Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci. 1998;18:2017–2027. doi: 10.1523/JNEUROSCI.18-06-02017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu FC, Graybiel AM. Spatiotemporal dynamics of CREB phosphorylation: transient versus sustained phosphorylation in the developing striatum. Neuron. 1996;17:1133–1144. doi: 10.1016/s0896-6273(00)80245-7. [DOI] [PubMed] [Google Scholar]
- 21.Malenka RC, Nicoll RA. Excitatory synapses. Is bigger better? Nature. 1998;396:414–415. doi: 10.1038/24734. [DOI] [PubMed] [Google Scholar]
- 22.McBain C, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 23.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 24.Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- 25.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 26.Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluR ε2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/s0896-6273(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 27.Mori Y, Matsubara H, Folco E, Siegel A, Koren G. The transcription of a mammalian voltage-gated potassium channel is regulated by cAMP in a cell-specific manner. J Biol Chem. 1993;268:26482–26493. [PubMed] [Google Scholar]
- 28.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-2 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 29.Pham TA, Impey S, Storm DR, Stryker MP. CRE-mediated gene transcription in neocortical neuronal plasticity during the developmental critical period. Neuron. 1999;22:63–72. doi: 10.1016/s0896-6273(00)80679-0. [DOI] [PubMed] [Google Scholar]
- 30.Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- 31.Rao A, Kim E, Sheng M, Craig A. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sassone-Corsi P. Transcription factors responsive to cAMP. Annu Rev Cell Dev Biol. 1995;11:355–377. doi: 10.1146/annurev.cb.11.110195.002035. [DOI] [PubMed] [Google Scholar]
- 33.Sauerwald A, Hoesche C, Oschwald R, Kilimann MW. The 5[prime]-flanking region of the synapsin I gene. A G+C-rich, TATA- and CAAT-less, phylogenetically conserved sequence with cell type-specific promoter function. J Biol Chem. 1990;265:14932–14937. [PubMed] [Google Scholar]
- 34.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 35.Sheng M, Pak DTS. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000;62:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- 36.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 37.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 38.Sprengel R, Suchanek B, Amico C, Brusa R, Burnasheve N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SGN, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- 39.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 40.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott J. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 42.Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M. Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci. 1998;18:1383–1392. doi: 10.1523/JNEUROSCI.18-04-01383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia Z, Dudek H, Miranti C, Greenberg M. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]