

Abstract
ADAM proteases, defined by extracellular disintegrin and metalloprotease domains, are involved in protein processing and cell–cell interactions. Using wobbler (WR) mutant mice, we investigated the role of ADAMs in neurodegeneration and reactive glia activation in the CNS. We found that ADAM8 (CD 156), a suspected leukocyte adhesion molecule, is expressed in the CNS and highly induced in affected CNS areas of WR mice, in brainstem and spinal cord. ADAM8 mRNA and protein are found at low levels throughout the normal mouse CNS, in neurons and oligodendrocytes. In the WR CNS regions in which neurodegeneration occurs, ADAM8 is induced in neurons, reactive astrocytes, and activated microglia. Similarly, the proinflammatory cytokine tumor necrosis factor α (TNF-α) is upregulated and shows the same cellular distribution. In primary astrocytes from wild-type and WR mice, in primary cerebellar neurons, and in mouse motoneuron-like NSC19 cells, ADAM8 expression was induced up to 15-fold by mouse TNF-α, in a dose-dependent manner. In both cell types, ADAM8 was also induced by human TNF-α, indicating that TNF receptor type I (p55) is involved. Induction of ADAM8 mRNA was suppressed by treatment with an interferon-regulating factor 1 (IRF-1) antisense oligonucleotide. We conclude that IRF-1-mediated induction of ADAM8 by TNF-α is a signaling pathway relevant for neurodegenerative disorders with glia activation, proposing a role for ADAM8 in cell adhesion during neurodegeneration.
Keywords: metalloprotease-disintegrins, neurodegeneration, reactive gliosis, cell adhesion, ADAM8 (CD 156), tumor necrosis factor α, TNF-α, IRF-1
In neurodegenerative diseases, neuronal cell death is usually accompanied by activation of glial cells, astrocytes, and microglia (Kreutzberg, 1996; Ridet et al., 1997;Stoll and Jander, 1999; Streit et al., 1999). Glia activation involves remodeling of the extracellular matrix (ECM) (Chen and Strickland, 1997), cell adhesion (Ridet et al., 1997) and signaling through the ECM by shedding of cytokines, cytokine receptors, and growth factors (Rose-John and Heinrich, 1994). Extracellular proteinases capable of these functions are metalloprotease-disintegrins [(ADAMs) a disintegrin and metalloprotease domain], forming a large family of proteins with structural homology to snake venom metalloprotease (Wolfsberg et al., 1995; Blobel, 1997). Approximately 30 ADAM family members have been identified in several animal species (Schlöndorff and Blobel, 1999). Proteolysis of membrane-bound surface molecules, ectodomain shedding (Peschon et al., 1997), was recently demonstrated for tumor necrosis factor α (TNF-α) (Black et al., 1997; Moss et al., 1997), heparin-binding epidermal growth factor (Izumi et al., 1998), and amyloid precursor protein (Buxbaum et al., 1998; Lammich et al., 1999), which are cleaved by ADAMs 17, 9 or 17, and 10, respectively. A role in cell adhesion has been demonstrated for ADAM12 and ADAM15. ADAM12 is involved in fusion of myoblasts (Yagami-Hiromasa et al., 1995; Zolkiewska, 1999). ADAM (MDC) 15 is the only metalloprotease-disintegrin containing, within the integrin-binding loop of the disintegrin domain, an RGD sequence (Krätzschmar et al., 1996; Herren et al., 1997), which binds to integrin avβ3(Zhang et al., 1998).
The proinflammatory cytokine TNF-α has been implicated in a variety of neurodegenerative diseases, including multiple sclerosis (for review, see Sharief, 1998) and Alzheimer's disease (for review, seeMattson et al., 1997) in which a dual role can be ascribed to TNF-α. On the one hand, TNF-α is able to trigger neuronal death by either promoting apoptotic pathways (Knoblach et al., 1999) or suppressing survival signals (Venters et al., 1999); on the other hand, TNF-α production in neurons after ischemic or excitotoxic brain injury has neuroprotective effects, as demonstrated with mice lacking TNF receptors (Bruce et al., 1996). This view was supported by work on Alzheimer's disease in which TNF-α has local neuroprotective effects by inducing antiapoptotic pathways (Tarkowski et al., 1999). Therefore, analysis of neuronal target genes of TNF-α under pathological conditions may provide clues for its functions in the CNS.
As a model for neurodegeneration and glia activation, we used wobbler mutant mice (genotype, wr/wr; phenotype, WR) (Duchen and Strich, 1968). Neuropathological changes in WR mice are characterized by a general atrophy of forelimb muscles (Duchen and Strich, 1968;Sedehizade et al., 1997) attributable to neurodegeneration in brainstem (BS), spinal cord (SC) and, to a lesser extent, the cerebellum (Cbl) (Duchen and Strich, 1968). In response to neurodegeneration, a strong astrogliosis (Laage et al., 1988) and hyper-ramified microglia were observed in affected CNS regions (Rathke-Hartlieb et al., 1999), a common feature in a number of CNS diseases.
We analyzed the expression of ADAM proteinases in normal mice intending to define ADAMs regulated by cytokines responding to neurodegeneration in the mouse CNS.
MATERIALS AND METHODS
Animals. Breeding and experimental use of mice was performed in agreement with the German law on the protection of animals, with a permit by the local authorities. The wrmutation was maintained on a C57BL/6 background. Usually, mutant mice with a manifest WR phenotype (25–72 d old) and their normal littermates were used for biochemical analysis.
Materials. Recombinant human and mouse TNF-α were obtained from Boehringer Mannheim (Mannheim, Germany) in sterile PBS at a concentration of 5 μg/ml. Cycloheximide was obtained from Sigma (Dreieich, Germany).
Oligonucleotides. For reverse transcription (RT)-PCR analysis, we used the following primers: ADAM8 [X13335, nucleotides (nt) 27–47, nt 486–465; 459bp]; TNF-α (M38296, nt 208–229, nt 669–648; 461bp); interleukin1β (IL-1β) (M15131, nt 58–81, nt 665–641; 607bp); IL-6 (J03783, nt 214–239, nt 686–664; 424bp); interferon-γ (IFN-γ) (K00083, nt 22–43, nt 295–274; 273bp); IL-18 (D49949, nt 122–144, nt 554–532); interferon-regulating factor 1 (IRF-1) (M21065, nt 229–250, 559–580); and L7 (M29015, nt 254–274, nt 610–590; 356bp). Antisense phosphorothioate IRF-1 oligonucleotides were used as described previously (Horiuchi et al., 1997). Oligonucleotides were obtained from TIB MolBiol (Berlin, Germany).
Antibodies. A polyclonal anti-TNF-α antibody was obtained from Endogen (Woburn, MA). Rabbit anti-mouse ADAM8 antibody (pc-anti-ADAM8) was raised against a synthetic peptide corresponding to amino acids (aa) 766–780 of mouse ADAM8. The polyclonal serum was further purified by affinity chromatography using IgG-Sepharose columns as described. For a monoclonal antibody (mc-anti-ADAM8), recombinant ADAM8 protein was obtained in Escherichia coli using the pET11d vector carrying the ADAM8 cDNA sequence encoding signal peptide, prodomain, metalloprotease domain, and most of the disintegrin domain, and was immunized into Lou rats. Spleen cells prepared from the rat were fused with mouse myeloma line SP2/0-Agl4 cells.
Tissue preparation. For protein and RNA preparation and histological analysis, mice were killed by decapitation. The CNS was dissected into cerebrum (Cbr), containing telencephalon and diencephalon, Cbl, BS, and cervical SC with cervical segments C1 to C8. As control tissues, testes and lung tissue were removed. For cryosections, the respective parts of the CNS were mounted in freezing medium and rapidly shock frozen in propane–liquid nitrogen.
Immunocytochemistry. For immunofluorescence, cryosections (10 μm) of CNS tissues were fixed for 5 min in 3% paraformaldehyde at room temperature or with methanol at −20°C for 6 min. For monoclonal antibody, goat anti-mouse IgG (Dianova, Hamburg, Germany) conjugated with Cy3 (red) served as a secondary antibody. For double staining, a monoclonal Cy-3-conjugated anti-glial fibrillary acidic protein (GFAP) (1:800; Sigma), a monoclonal anti-CNPase (1:200; Sigma), or rat anti-CD45 (PharMingen, Ruesselsheim, Germany) was used together with polyclonal ADAM8 (1:100) or anti-TNF (1:100) antibody. As secondary antibodies, we used goat anti-rat IgG (1:200; Dianova) or goat-anti-rabbit IgG (1:200; Dianova), either conjugated with Cy2 or Cy3.
In situ hybridization. Riboprobes specific for mouse ADAM8 were prepared from a cDNA fragment of the ADAM8 prodomain (nt 27–486) cloned in pBluescript (Stratagene, La Jolla, CA). A BLAST search with this sequence revealed that this probe has no significant homology to prodomains of other known ADAMs. The riboprobes were generated byin vitro transcription with SP6 (antisense) and T7 (sense) polymerase with digoxigenin-conjugated 11-UTP. CNS tissues were fixed in 3% paraformaldehyde in buffered saline and embedded in paraffin according to standard procedures. Deparaffinized sections (5 μm) were hybridized with riboprobes (400 ng/ml) overnight at 55°C and subsequently washed with 0.2× SSC–0.1% SDS at 55°C for 1 min., 0.2× SSC–0.1% SDS at 55°C for 10 min, and 0.2× SSC–50% formamide for 30 min at 55°C, followed by 0.2× SSC–25% formamide for 15 min. After washing, samples were treated with RNase A (10 μg/ml in 2× SSC) for 15 min at 37°C. Bound riboprobe was detected with an anti-digoxigenin antibody (Boehringer Mannheim) coupled to alkaline phosphatase and subsequent color reaction using nitro blue tetrazolium–5-bromo-4-chloro-3-indolyl phosphate as substrate.
RT-PCR. Total RNA from tissues was purified according toChomczynski and Sacchi (1987). For mRNA isolation, total RNA was further purified by the Dynabead method (Dynal, Hamburg, Germany). From NSC19 (neuroblastoma × spinal cord cells hybrid) cells, total RNA was isolated by the RNeasy preparation method (Qiagen, Hilden, Germany). For RT-PCR, either 1 μg of total RNA or 100 ng of mRNA in 20 μl total volume were subjected to RT using SuperScript reverse transcriptase (Life Technologies GmbH, Eggenstein, Germany). After RT, usually 1 μl of this mixture was used for PCR amplification in a total volume of 50 μl containing 10 pmol of each primer and 1 U of AmpliTaq (Perkin-Elmer GmbH, Überlingen, Germany). PCR conditions were as follows: 4 min at 94°C, 1 min annealing at 4°C below melting temperature, 1 min at 72°C, and 1 min at 92°C for 20–30 cycles. To verify specificity of RT-PCR, all amplificates were sequenced. The resulting amplification products were separated by electrophoresis on a 1% agarose gel and visualized with ethidium bromide. For quantitation, RT-PCR experiments were performed in 20–25 cycles. For quantitative evaluation of ADAM8 mRNA levels, the PCR products were blotted onto nitrocellulose, hybridized with an [α-32P]dATP-labeled ADAM8 probe and quantified using a Bio-Imager (Bio-Rad, Göttingen, Germany).
Bioassay for TNF-α. CNS tissue or cell pellets were homogenized in 10 vol of ice-cold PBS and clarified by centrifugation (10000 rpm, 10 min at 4°C), and the supernatants were used for the bioassay. TNF-α activity was determined in either supernatants from astrocyte cultures or in homogenates of tissues or cells measuring L929 cell cytotoxicity as described previously (Aggarwal et al., 1985). Recombinant mouse TNF-α was used as a standard.
Cell culture and transfection. Mouse cell lines L929 and NSC19 (Cashman et al., 1992) were maintained in DMEM supplemented with 10% fetal calf serum (FCS) in a humidified incubator with 5% CO2. For TNF-α treatment, 5 × 104 NSC19 cells were seeded onto six-well plates (30 mm in diameter each) and incubated for 48 hr with indicated concentrations. Transfection of IRF-1 oligonucleotides was done with FuGENE6 transfection reagent (Boehringer Mannheim) as described previously (Tao et al., 1999).
Preparation of granular cells from mouse cerebellum. Heads of 6-to 8-d-old mice were removed with scissors, and the cerebellums were prepared. Tissues were mechanically dissociated and treated with a 1% trypsin solution containing 0.05% DNase for 16 min at room temperature. After centrifugation at 100 × g for 5 min, tissue was dissociated further by using a Pasteur pipette. After an additional centrifugation step, the cell pellet was resuspended in basal medium (Eagle) medium containing insulin (12.5 μg/ml), thyroxin (4 nm), transferrin (100 μg/ml), and sodium selenite (30 nm). Cells were seeded onto culture dishes coated with poly-l-lysine and cultured for 3–5 d before experiments were performed.
Preparation of primary astrocyte cultures. Cerebellum, brainstem, and spinal cords were removed immediately from mice killed by decapitation. Tissues were kept in HEPES-buffered PBS and mechanically dissociated with scissors. The homogenate was further minced by triturating through an 18 ga needle several times and incubated with a 0.05% trypsin–EDTA solution for 20 min. After additional homogenization with an 18 ga needle, the cell pellet was resuspended in astrocyte basal medium (Clonetics, La Jolla, CA) containing 5% FCS, ferritin, insulin, transferrin, and steroid hormones.
Protein analysis. CNS and lung tissue was removed quickly and homogenized in buffer containing 100 mmTris-HCl, pH7.4, 1 mm EGTA, and Complete Inhibitor Mix (Boehringer Mannheim). After addition of Laemmli's buffer, the protein extracts were denatured at 100°C for 5 min and separated on a 10% SDS-polyacrylamide gel. Proteins were immobilized by capillary blot on a nitrocellulose membrane (Protran; Schleicher & Schüll, Dassel, Germany). The transfer was checked by reversible ponceau staining, and unspecific binding sites were blocked overnight with blocking buffer [5% nonfat milk powder in TTBS (20 mm Tris-HCl, pH 7.5, 500 mmNaCl, and 0.05% Tween 20]. Primary antibody solutions [pc-anti-ADAM8 at 1:2000 and mc-anti-β-Actin (Gimona et al., 1994) at 1:4000 diluted in blocking buffer] were incubated with the membranes for 4 hr at room temperature. After extensive washing (four times for 15 min each) with TTBS–0.5% nonfat milk powder, membranes were incubated with secondary antibodies, either goat anti-rabbit (1:4000; Sigma) or rat anti-mouse (1:10000; Jackson ImmunoResearch, West Grove, PA), both conjugated with horse radish peroxidase, for 45 min. After washing (three times for 10 min each) with TTBS, protein bands were detected with Lumi-LightPlus Western blotting substrate (Boehringer Mannheim) and Kodak X-OMAT film (Eastman Kodak, Rochester, NY).
RESULTS
Expression of ADAM8 in wild-type and WR CNS
We analyzed differential ADAM mRNA expression in various CNS regions of wild-type (WT) and WR mice by RT-PCR. With the exception of ADAM8, the mRNA levels of other ADAM proteinases tested were not significantly different between WT and WR mice (ADAMs 1, 4, 9, 10, 11, 12, 15, 17, 22, and 23). Interestingly, ADAM8 (mCD 156) (Yoshida et al., 1990) mRNA is expressed in the mouse CNS, hitherto described only for monocytic cells. For analysis of ADAM8 expression in the CNS,in situ hybridizations were performed using a mouse ADAM8-specific riboprobe (Fig. 1). In coronal sections of the mouse brain, ADAM8 mRNA expression was evenly distributed throughout neurons in the cortical layers (Fig.1C). Granular layer cells of the cerebellum (Fig.1B,D) and the dentate gyrus of the hippocampus (Fig. 1A,E) gave strong hybridization signals. We also found mRNA expression in motoneurons of the spinal cord (Fig. 1G).
Fig. 1.
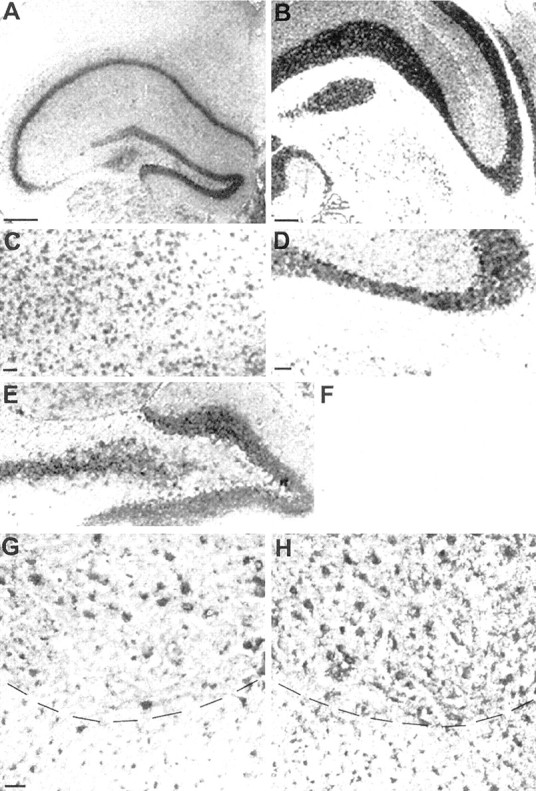
ADAM8 mRNA in the CNS of normal mice as shown byin situ hybridization (coronal and transversal sections; dorsal is up). A, Cerebral cortex, anterior part with hippocampus (lateral is left).B, Brainstem with part of cerebellum (lateral isright). C, Cortical layer (detail ofA). D, Granular layer of the cerebellum with Purkinje cells (detail of B). E, Gyrus dentatus (detail of A). F, Negative control (sense probe), detail of cerebellum, as in D.G, H, Cervical spinal cord, ventral part; comparison between wild type (G) and wobbler (H). Dashed lines indicate borders between gray (top) and white (bottom) substance. Scale bars: A,B, 500 μm; (in C) C,E, 100 μm; (in D) D,F, 100 μm; (in G) G,H, 50 μm.
Compared with WT controls, ADAM8 mRNA levels in WR mice were at least eight times higher in brainstem and 10 times higher in spinal cord, i.e., in the CNS regions that show neurodegeneration and reactive gliosis in the WR neuropathy (Fig.2A,B). In contrast, no significant change in mRNA expression was observed in cortical brain regions and in testis. We also performed in situ hybridizations on spinal cord sections from WT and WR mice. These data demonstrate that, compared with WT mice, ADAM8 expression in WR mice is stronger as a larger number of cells was stained (Fig.1G,H).
Fig. 2.
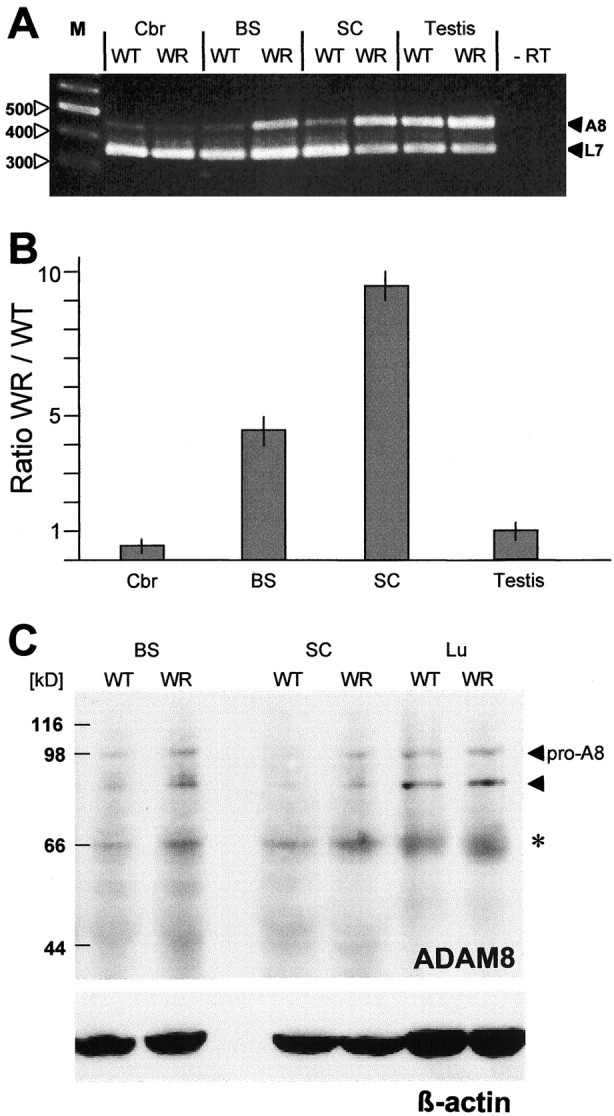
ADAM8 mRNA levels in the CNS and testis of WT and WR mice. A, RT-PCR analysis of ADAM8 mRNA in different brain regions of 40-d-old WR mice and their normal WT littermates. After reverse transcription, PCR was performed in 25 cycles as a duplex PCR with ADAM8 and L7 primers, the latter serving as an amplification standard. Chain lengths of marker bands are given on theleft. −RT, Negative control, no reverse transcriptase. B, Quantitation of ADAM8 mRNA in WT and WR mice, given as ratio WR/WT, derived by radioactive hybridization of amplificates normalized to the L7 reference signal. C, Immunodetection of ADAM8 in Western blots of WT and WR brainstem, spinal cord, and lung extracts. Membrane filters were incubated with polyclonal anti-ADAM8, and the bands were visualized by chemiluminescence. As loading control, a monoclonal β-actin-specific antibody was used to stain a parallel blot. The molecular mass marker positions are indicated on the left. Thepro-A8 on the right marks the position of the pro-ADAM8 at ∼100 kDa, and the filled triangle indicates the position of the processed form of ADAM8. The band indicated by an asteriskcorresponds to unspecific binding of the ADAM8 antibody.
Detection of ADAM8 protein in CNS extracts
We extracted total protein from different CNS regions and analyzed them by immunoblotting. As positive control for ADAM8 expression, we used lung extracts. Membranes were stained with two different antibodies, pc-anti-ADAM8 or mc-anti-ADAM8 (Fig. 2C) with identical results. In CNS and lung extracts, the putative proform of ADAM8 is detectable as a band with a molecular weight (MW) of ∼98 kDa, slightly larger than the calculated MW (∼90 kDa). This probably correlates with post-translational modification. Processing of ADAM8 by furin-like enzymes yields a molecular weight of ∼82 kDa (aa 71–74). This processed form of ADAM8 still contains the cysteine-switch region. No difference in ADAM8 levels was observed between lung extracts from WT and WR mice. In contrast, an increase in ADAM8 protein was observed in spinal cord and brainstem extracts of WR mice.
Tissue distribution of ADAM8 in WT and WR mice
To identify the cells expressing ADAM8, we stained spinal cord sections from WT and WR mice with ADAM8 antibodies and glial cell markers (Fig. 3). In normal mice, ADAM8 was detectable in neurons and in glia cells in the white matter of the spinal cord, which are oligodendrocytes, stained with CNPase (Fig.3B). In neurons, ADAM8 staining is seen in the cytoplasma and, in most cases, stronger toward the cell periphery (Fig.3A). In WR mice, neuronal staining for ADAM8 is enhanced and more diffuse, and more cells stained strongly for ADAM8, probably reactive glia cells (Fig.3D,J). To identify glial cell types expressing ADAM8, we used either anti-CD45 to detect activated microglia (Fig. 3B,E) or anti-GFAP (Fig. 3H,K) for astrocytes. A double exposure of glia-specific antibodies with anti-ADAM8 revealed that, in the WR spinal cord, both reactive glia cell types, astrocytes and microglia, express ADAM8 homogeneously (Fig.3F,L).
Fig. 3.

Localization of ADAM8 in glia cells in the spinal cord of 40-d-old WT (A–C, G–I) and WR (D–F, J–L) mice, as demonstrated by immunofluorescence. A–C, WT, anti-ADAM8 (green, A), anti-CD45 (red, B), or both (yellow, C); insetsin A–C, staining of cells in the white matter with anti-ADAM8 (in A), oligodendrocyte-specific antigen CNPase (in B) and merged (in C).D–F, WR, ADAM8 (green,D), CD45 (red, E), or both (yellow, F). Note that all microglia cells express ADAM8. G–I, Spinal cord of WT mouse, stained with anti-ADAM8 (G), GFAP (H), or with both (I). J–L, WR, stained for ADAM8 (J), GFAP (K), or both (L). Note that, in J–L, astrocytes appear more compacted than usual because of very short exposure times. GFAP staining in astrocytic processes is not visible. Scale bar (in A): A–L, 50 μm.
Cytokine expression in WR mice
To define mechanisms of glial cell activation in WR mice, we compared cytokine mRNA levels in mutant mice with their normal littermates (between 25 and 60 d old) by RT-PCR in several CNS regions. Typically, total RNA or mRNA from telencephalon, cerebellum, brainstem, and spinal cord was extracted, and RT-PCR experiments were performed in the linear amplification range (usually 20–25 cycles) with specific primers (Fig. 4). Significantly, mRNA levels of TNF-α and IL-1β were elevated in WR brainstem, spinal cord, and to a lesser extent, cerebellum. We have not seen significant differences in the telencephalon. In contrast, mRNA expression levels of IL-6, IL-18, and IFN-γ (Fig. 4) and of IL-2, IL-10, and IL-12 (data not shown) were not changed between WT and WR mice. To determine the concentrations of active TNF-α, we performed a cytotoxicity assay in L929 cells (Aggarwal et al., 1985). As shown in Table 1, concentrations of active TNF-α were significantly increased in WR spinal cord, whereas TNF-α concentrations in cerebrum tissues were not different. Furthermore, cell extracts and supernatants of primary astrocytes from WR mice compared with wild-type mice contained higher amounts of TNF-α. Using a neutralizing anti-TNF-α antibody, cytotoxicity was significantly reduced, indicating that the main proportion of cytotoxicity in WR mice is attributable to the presence of TNF-α.
Fig. 4.

Cytokine mRNA expression in 40-d-old wild-type and wobbler mice. The indicated cytokines were detected in an RT-PCR analysis with 25 cycles of PCR. Control, no reverse transcriptase (−RT), except for IL-1β and IFN-γ for which mRNA from mouse spleen was used as positive control to verify PCR conditions.
Table 1.
L929 cytotoxicity assay
| Age (d) | Brain | Spinal cord | Astrocytes | ||||
|---|---|---|---|---|---|---|---|
| WT | WR | WT | WR | WT | WR | ||
| 21 | <50 | <50 | <50 | 650 ± 48 | <50 (ce) | 250 ± 28 (ce) | |
| <50 (sn) | 150 ± 20 (sn) | ||||||
| 70 | <50 | 230 ± 30 | <50 | 1500 ± 60 | n.d. | n.d. | |
| +TNF-α antibody | |||||||
| 21 | <50 | <50 | <50 | 100 ± 25 | <50 | <50 | |
| 70 | <50 | <50 | <50 | 200 ± 30 | n.d. | n.d. | |
L929 cytotoxicity assay for determination of TNF-α levels (in picograms per milliliter) in extracts from brain and spinal cord and in cell extracts (ce) and supernatants (sn) from primary astrocytes, each from WT and WR mice, respectively. For neutralizing experiments, TNF-α antibody (1:50) was incubated with the extracts for 1 hr at 37°C before the assays. n.d., Not determined.
Cellular distribution of TNF-α in wild-type and WR mice
We determined the cellular distribution of TNF-α in the spinal cord of control and WR mice by double-immunolabeling experiments (Fig.5). TNF-α antibody was used in combination with cell type-specific markers CD45 (Fig.5A,B) and GFAP (Fig.5C,D). TNF-α expression is increased in degenerating neurons of the WR spinal cord (Fig.5B,D). A complete coincidence of TNF-α and the glial cell markers indicates that TNF-α like ADAM8 is strongly expressed in reactive glial cells of WR mice. Particularly around sites of degenerating neurons, a strong TNF-α staining was detected in astrocytes, which extended their processes toward blood vessels, consistent with the role of astrocytes in insulation of the blood–brain barrier.
Fig. 5.

Double immunolabeling of TNF-α and glia-specific markers CD45 (A, B) or GFAP (C, D) in the ventral spinal cord of 40-d-old WT (A, C) or WR (B, D) mice. Arrowheads inD indicate a degenerating motoneuron surrounded by astrocytes in the ventral horn of the spinal cord. Scale bar (inA): A–D, 20 μm.
ADAM8 mRNA can be induced by TNF-α
The striking similarity in protein expression patterns between TNF-α and ADAM8 in neurons and reactive glia cells suggested that ADAM8 expression may be modulated by TNF-α. To test this, we treated granular cells (primary neurons), the mouse motoneuron-like cell line NSC19 (Cashman et al., 1992), and cultured primary astrocytes from WT and WR mice with recombinant mouse TNF-α. Granular and NSC19 cells were treated in a concentration range of 1–3000 U/ml recombinant mouse TNF-α, harvested 24–48 hr later, measured for mRNA level of ADAM8 by RT-PCR, as visualized by ethidium bromide staining (Fig.6A) or quantified by slot blot hybridization (Fig. 7). Raising the TNF-α concentration up to 1500 U/ml resulted in a 12-fold increase in ADAM8 mRNA levels. With concentrations >1500 U/ml, ADAM8 mRNA levels in NSC19 cells declined. Whereas the survival of granular cells was reduced by TNF-α treatment, NSC19 cells were remarkably resistant to TNF-α. Even high doses of TNF-α (>3000 U/ml) did not interfere with the survival of NSC19 cells (Fig. 6B), monitored by proliferation assay based on MTT (data not shown). We have not observed an effect of IL-1β, lipopolysaccharide (LPS), and interferon-γ on ADAM8 expression in NSC19 cells and primary astrocytes.
Fig. 6.

ADAM8 transcriptional activation by mouse TNF-α in granular cells and NSC19 cells. A, Granular cells (GC) and NSC19 cells were incubated with indicated units of mouse TNF-α for 24 hr; the relative amounts of ADAM8, IRF-1, and L7 (reference) mRNA levels were determined by RT-PCR and visualized by ethidium bromide staining. B, ADAM8 immunofluorescence of NSC19 cells treated with indicated units of TNF-α. Note the staining toward the cell membrane. Even in the presence of 3000 U of TNF-α, NSC19 cells did not show signs of cell damage. Scale bar, 50 μm.
Fig. 7.

Upregulation of ADAM8 mRNA by exogenous TNF-α. Quantitation of ADAM8 mRNA levels in primary WT and WR astrocytes (A), granular cells (B), and NSC19 cells (C), as determined by RT-PCR.A, Induction of ADAM8 mRNA by TNF-α in primary astrocyte cultures from WT and WR mice. In all cases, values were normalized to those of untreated WT cells, 1.0 by definition.B, ADAM8 induction by TNF-α in granular cells.C, Dose response obtained with recombinant mouse (m) and human (h) TNF-α in NSC19 cells. D, ADAM8 mRNA induction by TNF-α in NSC19 cells after preincubation with cycloheximide (CHX); untreated NSC cells, 1.0 by definition. E, Induction of ADAM8 mRNA by TNF-α in the presence of either IRF-1 antisense (aIRF1) or sense (sIRF1) oligonucleotide. Values were normalized to untreated NSC19 cells with sIRF1. Data were given as mean values of at least three independent experiments. When the same experiment was performed on L929 cells, there was no stimulation of ADAM8 mRNA by TNF-α at the concentrations used above (data not shown).
Because astrocytes are the major source of TNF-α in WR mice, we investigated whether the upregulation of ADAM8 in astrocytes in situ can be mimicked in cell culture. Therefore, ADAM8 expression in WT and WR astrocytes was determined before and after TNF-α treatment. Without TNF-α treatment, WR astrocytes already showed an eightfold higher ADAM8 expression than WT astrocytes (Fig.7A). This is consistent with a constitutive TNF-α release by WR astrocytes, as demonstrated by the L929 bioassay (Table 1).
Upon treatment of WT and WR astrocytes with TNF-α, ADAM8 expression was enhanced in both WT and WR astrocytes, with maximal transcription levels of ∼1200 U/ml TNF-α. At TNF-α doses >1200 U/ml, transcription levels of ADAM8 decreased in WR astrocytes. Similar to NSC19 cells, astrocyte cultures did not show significant effects of various doses of TNF-α on cell survival.
Transcriptional activation of ADAM8 in neuronal cells
We performed experiments to unravel the possible mechanism of TNF-α-dependent ADAM8 activation in NSC19 cells and primary astrocytes. To address the question which TNF receptor is involved in ADAM8 activation, we treated NSC19 cells and astrocyte cultures with recombinant human TNF-α. It has been demonstrated with mouse receptors that human TNF-α binds only to TNF receptor type I (TNFRI) (p55) but not to TNFRII (p75) (Lewis et al., 1991). As shown in Figure7C, we obtained similar results with human and mouse TNF-α, suggesting a TNFRI-mediated ADAM8 activation.
TNF-α dependent ADAM8 induction in NSC19 cells was abolished by treating the cells with cycloheximide, a potent inhibitor of translation, indicating that additional de novo protein synthesis is necessary to activate ADAM8 transcription (Fig.7D). To identify downstream targets of TNFRI, we inspected the 5′ regulatory sequence of ADAM8 (Kataoka et al., 1997) for transcription factor binding sites that might be involved in TNF signaling. We found that IRF-1, a transcription factor involved in TNF-α response to interferon (Miyamoto et al., 1988), is upregulated upon treatment with TNF-α (Fig. 6A). These results argue for a role of IRF-1 in transcriptional activation of ADAM8. NSC19 cells were treated with IRF-1 oligonucleotides (sense or antisense) to inhibit translation of the IRF-1 protein (Horiuchi et al., 1997). With antisense IRF-1, we were able to block TNF-α induction of ADAM8 completely in the presence of 20 μmoligonucleotide, whereas the sense oligonucleotide had no effect on TNF-α mediated ADAM8 induction (Fig. 7E).
DISCUSSION
ADAM8 (CD 156) has originally been described as a transmembrane glycoprotein expressed in cells of the myelomonocytic lineage with the proposed function of leukocyte adhesion in the immune system (Yamamoto et al., 1999). Here we demonstrated that ADAM8 is expressed in the mammalian CNS, predominantly in neurons and oligodendrocytes. Compared with other ADAM proteases, such as ADAM9, expression levels in the CNS are low under nonpathological conditions. The dramatic upregulation of ADAM8 in reactive astrocytes and microglia suggests that ADAM8 has an essential function in neuron–glia interactions. Thus, in addition to ADAM12 and ADAM15, ADAM8 may play a role in somatic cell–cell interactions. In the integrin binding loop of ADAM8, the RGD sequence of ADAM15 is replaced by the motif KDM, which could act as an integrin receptor. A functional importance of the disintegrin domain of ADAM8 has been demonstrated in an antigen-induced autoimmune brain disease in which this domain as a recombinant protein was able to protect rats from autoimmune symptoms of experimental autoimmune encephalitis, neuritis, and uveitis (Schlüsener, 1998). This finding strongly argues for an essential role of ADAM8 in inflammatory responses within the organism.
In WR mice, the time course of neuropathological changes is in the temporal order of neurodegeneration, astrogliosis, and microglia activation (Rathke-Hartlieb et al., 1999). We provide evidence that, in the pathway of the events after neurodegeneration, the secretion of TNF-α causes the induction of ADAM8 in primary cells and cell lines of neuronal and glial origin.
ADAM8 induction in astrocytes is not an unspecific consequence of glial cell activation, e.g., by tissue dissociation and growth factors in the culture medium, because wild-type astrocytes in culture have a low intrinsic level of ADAM8 expression that is strongly elevated by exogenous TNF-α. On the other hand, induction of ADAM8 mRNA is nearly saturated in astrocytes from WR mice, and only a low additional elevation is possible in culture. Because these cells secrete TNF-α by themselves, this indicates an autocrine pathway.
The ability of human TNF-α to induce ADAM8 expression in mouse cells indicates that the TNF-α response in primary neurons, NSC cells, and in primary astrocytes is mediated by TNFRI (p55) (Lewis et al., 1991). The downstream target of TNFRI is IRF-1 (Miyamoto et al., 1988), a transcription factor for which a role in ischemic injury of the CNS has been described previously (Paschen et al., 1998). The ADAM8 promoter contains a binding site for IRF-1 in close proximity to a nuclear factor NF-I site and the TATA-box. This site has been identified as distinct from binding sites involved in LPS or IFN-γ-mediated stimulation of ADAM8 transcription (Kataoka et al., 1997). These data provide evidence that, within the CNS, induction of ADAM8 is linked to TNF-α and IRF-1 by a TNFRI-dependent signaling. A contribution of NF-κB in IRF-1 activation can be ruled out, because pyrrolidone-dithio-carbamate, a specific inhibitor of NF-κB (Kaltschmidt et al., 1999) had no effect on ADAM8 induction in NSC19 cells. These data argue for a pathway distinct from the NF-κB pathway in neuronal seizures involving TNF-α, TNFRI, and IRF-1.
Different cell types within the CNS have their own repertoire of target genes for TNF-α and may use different pathways to activate these genes. It is intriguing that activation of ADAM8 by TNF-α is observed in both neuronal and glial cells. In astrocytes, but not in neurons and microglia, another metalloprotease, MT1-MMP, is induced by TNF-α (Rathke-Hartlieb et al., 2000), supporting this notion.
TNF-α has been described as a signal triggering cell death, also in the CNS (Venters et al., 1999). As an important transcription factor in the CNS, NF-κB is involved in these processes (Nakai et al., 2000). Our data indicate that the observed TNF-α–IRF-1 pathway is independent of the activation of NF-κB. However, apoptosis is not observed in the CNS of WR mice (Popper et al., 1997) but is observed in another neurological mutant of the mouse, MND2. In this mutant, apoptosis of striatal neurons also leads to elevated expression of TNF-α and ADAM8, indicating that the mechanism of neuronal cell death, apoptotic or necrotic, is not critical for TNF-α and ADAM8 induction (S. Rathke-Hartlieb, U. Schlomann, M. Meisler, H. Jockusch, and J. W. Bartsch, unpublished results).
In summary, we have described a specific case of the “MMP–cytokine connection” as described in GFAP-TNF-α transgenic mice (Pagenstecher et al., 1998; Campbell and Pagenstecher, 1999), which seems to be of particular importance for pathological conditions within the CNS. Although the precise function of ADAM8 for neuron–glia interactions remains to be determined, ADAM8 expression could be a general feature of activated glia cells in CNS diseases. It will be of future interest to explore the function of ADAM8 in the formation of a glial scar (for review, see Stichel and Müller, 1998).
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 549/A4. We thank Drs. D. Männel (Regensburg, Germany) and H. Hauser (Braunschweig, Germany) for helpful comments, A. Becker and A. Perz for technical assistance, Dr. J. Frey (Bielefeld, Germany) for providing L929 cells and degenerated primers, Dr. S. Huettelmaier (Braunschweig, Germany) for help with antibody purification, and B. Schnegelsberg (Hamburg, Germany) for advice.
U.S. and S.R.-H. contributed equally to this work.
Correspondence should be addressed to Dr. J. W. Bartsch, Developmental Biology and Molecular Pathology, W7, University of Bielefeld, 33615 Bielefeld, Germany. E-mail:joerg.bartsch@biologie.uni-bielefeld.de.
REFERENCES
- 1.Aggarwal BB, Kohr WJ, Hass PE, Moffat B, Spencer SA, Henzel WJ, Bringman TS, Nedwin GE, Goeddel DV, Harkins RN. Human tumor necrosis factor. Production, purification, and characterization. J Biol Chem. 1985;260:2345–2354. [PubMed] [Google Scholar]
- 2.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 3.Blobel CP. Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and Notch. Cell. 1997;90:589–592. doi: 10.1016/s0092-8674(00)80519-x. [DOI] [PubMed] [Google Scholar]
- 4.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 5.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 6.Campbell IL, Pagenstecher A. Matrix metalloproteinases and their inhibitors in the nervous system: the good, the bad and the enigmatic. Trends Neurosci. 1999;22:285–287. doi: 10.1016/s0166-2236(99)01430-7. [DOI] [PubMed] [Google Scholar]
- 7.Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 8.Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 9.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 10.Duchen LW, Strich SJ. An hereditary motor neurone disease with progressive denervation of muscle in the mouse: the mutant “wobbler.”. J Neurol Neurosurg Psychiatry. 1968;31:535–542. doi: 10.1136/jnnp.31.6.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimona M, Vandekerckhove J, Goethals M, Herzog M, Lando Z, Small JV. Beta-actin specific monoclonal antibody. Cell Motil Cytoskeleton. 1994;27:108–116. doi: 10.1002/cm.970270203. [DOI] [PubMed] [Google Scholar]
- 12.Herren B, Raines EW, Ross R. Expression of a disintegrin-like protein in cultured human vascular cells and in vivo. FASEB J. 1997;11:173–180. doi: 10.1096/fasebj.11.2.9039960. [DOI] [PubMed] [Google Scholar]
- 13.Horiuchi M, Yamada T, Hayashida W, Dzau VJ. Interferon regulatory factor-1 up-regulates angiotensin II type 2 receptor and induces apoptosis. J Biol Chem. 1997;272:11952–11958. doi: 10.1074/jbc.272.18.11952. [DOI] [PubMed] [Google Scholar]
- 14.Izumi Y, Hirata M, Hasuwa H, Iwamoto R, Umata T, Miyado K, Tamai Y, Kurisaki T, Sehara-Fujisawa A, Ohno S, Mekada E. A metalloprotease-disintegrin, MDC9/meltrin-gamma/ADAM9 and PKCdelta are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J. 1998;17:7260–7272. doi: 10.1093/emboj/17.24.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:9409–9414. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kataoka M, Yoshiyama K, Matsuura K, Hijiya N, Higuchi Y, Yamamoto S. Structure of the murine CD156 gene, characterization of its promoter and chromosomal location. J Biol Chem. 1997;272:18209–18215. doi: 10.1074/jbc.272.29.18209. [DOI] [PubMed] [Google Scholar]
- 17.Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115–125. doi: 10.1016/s0165-5728(98)00273-2. [DOI] [PubMed] [Google Scholar]
- 18.Krätzschmar J, Lum L, Blobel CP. Metargidin, a membrane-anchored metalloprotease-disintegrin protein with an RGD integrin binding sequence. J Biol Chem. 1996;271:4593–4596. doi: 10.1074/jbc.271.9.4593. [DOI] [PubMed] [Google Scholar]
- 19.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 20.Laage S, Zobel G, Jockusch H. Astrocyte overgrowth in the brain stem and spinal cord of mice affected by spinal atrophy, wobbler. Dev Neurosci. 1988;10:190–198. doi: 10.1159/000111969. [DOI] [PubMed] [Google Scholar]
- 21.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis M, Tartaglia LA, Lee A, Bennett GL, Rice GC, Wong GH, Chen EY, Goeddel DV. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc Natl Acad Sci USA. 1991;88:2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mattson MP, Barger SW, Furukawa K, Bruce AJ, Wyss-Coray T, Mark RJ, Mucke L. Cellular signaling roles of TGF beta, TNF alpha and beta APP in brain injury responses and Alzheimer's disease. Brain Res Brain Res Rev. 1997;23:47–61. doi: 10.1016/s0165-0173(96)00014-8. [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell. 1988;54:903–913. doi: 10.1016/s0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- 25.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD. Cloning of a disintegrin metalloproteinase that processes precursor tumor necrosis factor-α. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 26.Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN. Kainic acid-induced apoptosis in rat striatum is associated with nuclear factor-kappaB activation. J Neurochem. 2000;74:647–658. doi: 10.1046/j.1471-4159.2000.740647.x. [DOI] [PubMed] [Google Scholar]
- 27.Pagenstecher A, Stalder AK, Kincaid CL, Shapiro SD, Campbell IL. Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am J Pathol. 1998;152:729–741. [PMC free article] [PubMed] [Google Scholar]
- 28.Paschen W, Gissel C, Althausen S, Doutheil J. Changes in interferon-regulatory factor-1 mRNA levels after transient ischemia in rat brain. NeuroReport. 1998;9:3147–3151. doi: 10.1097/00001756-199810050-00005. [DOI] [PubMed] [Google Scholar]
- 29.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1997;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 30.Popper P, Farber DB, Micevych PE, Minoofar K, Bronstein JM. TRPM-2 expression and TUNEL staining in neurodegenerative diseases: studies in wobbler and rd mice. Exp Neurol. 1997;143:246–254. doi: 10.1006/exnr.1996.6364. [DOI] [PubMed] [Google Scholar]
- 31.Rathke-Hartlieb S, Schmidt VC, Jockusch H, Schmitt-John T, Bartsch JW. Spatiotemporal progression of neurodegeneration and glia activation in the wobbler neuropathy of the mouse. NeuroReport. 1999;10:3411–3416. doi: 10.1097/00001756-199911080-00028. [DOI] [PubMed] [Google Scholar]
- 32.Rathke-Hartlieb S, Budde P, Ewert S, Schlomann U, Staege MS, Jockusch H, Bartsch JW, Frey J. Elevated expression of membrane type 1 metalloproteinase (MT1-MMP) in reactive astrocytes following neurodegeneration in mouse central nervous system. FEBS Lett. 2000;481:227–234. doi: 10.1016/s0014-5793(00)02011-1. [DOI] [PubMed] [Google Scholar]
- 33.Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 34.Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300:281–290. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlüsener HJ. The disintegrin domain of ADAM 8 enhances protection against rat experimental autoimmune encephalomyelitis, neuritis and uveitis by a polyvalent autoantigen vaccine. J Neuroimmunol. 1998;87:197–202. doi: 10.1016/s0165-5728(98)00080-0. [DOI] [PubMed] [Google Scholar]
- 36.Schlöndorff J, Blobel CP. Metallaprotease-disintegrins: modular proteins capable of promoting cell-cell interactions and triggering signals by protein-ectodomain shedding. J Cell Sci. 1999;112:3603–3617. doi: 10.1242/jcs.112.21.3603. [DOI] [PubMed] [Google Scholar]
- 37.Sedehizade F, Klocke R, Jockusch H. Expression of nerve-regulated genes in muscles of mouse mutants affected by spinal muscular atrophies and muscular dystrophies. Muscle Nerve. 1997;20:186–194. doi: 10.1002/(sici)1097-4598(199702)20:2<186::aid-mus8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Sharief MK. Cytokines in multiple sclerosis: pro-inflammation or pro-remyelination? Mult Scler. 1998;4:169–173. doi: 10.1177/135245859800400315. [DOI] [PubMed] [Google Scholar]
- 39.Stichel CC, Müller HW. The CNS lesion scar: new vistas on an old regeneration barrier. Cell Tissue Res. 1998;294:1–9. doi: 10.1007/s004410051151. [DOI] [PubMed] [Google Scholar]
- 40.Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- 41.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 42.Tao M, Miyano-Kurosaki N, Takai K, Takaku H. Specific inhibition of human telomerase activity by transfection reagent, FuGENE6-antisense phosphorothioate oligonucleotide complex in HeLa cells. FEBS Lett. 1999;454:312–316. doi: 10.1016/s0014-5793(99)00814-5. [DOI] [PubMed] [Google Scholar]
- 43.Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol. 1999;19:223–230. doi: 10.1023/a:1020568013953. [DOI] [PubMed] [Google Scholar]
- 44.Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNF-α is a silencer of survival signals. Trends Neurosci. 1999;23:175–178. doi: 10.1016/s0166-2236(99)01533-7. [DOI] [PubMed] [Google Scholar]
- 45.Wolfsberg TG, Straight PD, Gerena RL, Huovila AP, Primakoff P, Myles DG, White JM. ADAM, a widely distributed and developmentally regulated gene family encoding membrane proteins with a disintegrin and metalloprotease domain. Dev Biol. 1995;169:378–383. doi: 10.1006/dbio.1995.1152. [DOI] [PubMed] [Google Scholar]
- 46.Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, Fujisawa-Sehara A. A metalloprotease-disintegrin participating in myoblast fusion. Nature. 1995;377:652–656. doi: 10.1038/377652a0. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto S, Higuchi Y, Yoshiyama K, Shimizu E, Kataoka M, Hijiya N, Matsuura K. ADAM family proteins in the immune system. Immunol Today. 1999;20:278–284. doi: 10.1016/s0167-5699(99)01464-4. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida S, Setoguchi M, Higuchi Y, Akizuki S, Yamamoto S. Molecular cloning of cDNA encoding MS2 antigen, a novel cell surface antigen strongly expressed in murine monocytic lineage. Int Immunol. 1990;2:585–591. doi: 10.1093/intimm/2.6.585. [DOI] [PubMed] [Google Scholar]
- 49.Zhang XP, Kamata T, Yokoyama K, Puzon-McLaughlin W, Takada Y. Specific interaction of the recombinant disintegrin-like domain of MDC-15 (metargidin,ADAM-15) with integrin alphavbeta3. J Biol Chem. 1998;273:7345–7350. doi: 10.1074/jbc.273.13.7345. [DOI] [PubMed] [Google Scholar]
- 50.Zolkiewska A. Disintegrin-like/cysteine-rich region of ADAM12 is an active cell adhesion domain. Exp Cell Res. 1999;252:423–431. doi: 10.1006/excr.1999.4632. [DOI] [PubMed] [Google Scholar]