

Abstract
Phorbol esters enhance release from a variety of cell types. The mechanism by which phorbol esters potentiate presynaptic release from central neurons is unclear, although effects of phorbol esters both on the readily releasable pool of vesicles and on presynaptic calcium channels have been shown. Using confocal microscopy and the fluorescent styryl dye FM 1-43, we have examined the effects of phorbol-12,13-dibutyrate (PDBu) on presynaptic vesicle turnover at individually identified synapses in dissociated cultures obtained from neonatal rat hippocampus. Using different dye staining and destaining protocols we were able to resolve two effects of PDBu. Potentiation of evoked release by PDBu was insensitive to calcium channel antagonists, suggesting that this effect results from an increased number of vesicles in the readily releasable pool. Since we observed no effect of PDBu on the size of the total recycling vesicle pool, we conclude that phorbol esters alter the equilibrium between reserve and readily releasable pools. An additional effect of PDBu on spontaneous release was observed. This effect was antagonized by nifedipine but not ω-conotoxin GVIA or ω-agatoxin IVA. We conclude that PDBu influences spontaneous and evoked release by two different mechanisms: through L-type calcium channels and through an increase in the proportion of recycling vesicles in the readily releasable pool. In addition to further clarifying the mechanism of action of phorbol esters, these results suggest that phorbol esters may be a useful tool with which to probe the function of the readily releasable pool of presynaptic vesicles at CNS synapses.
Keywords: hippocampus, neuron, L-type calcium channel, readily releasable pool, vesicle, FM 1-43, protein kinase, release probability
Phorbol ester treatment enhances exocytosis from many excitable and nonexcitable cell types. Several mechanisms have been suggested, including (1) an increase in the size of the readily releasable pool of vesicles (Gillis et al., 1996;Stevens and Sullivan, 1998), (2) a decreased rate of calcium clearance from the cytoplasm (Lin et al., 1994), (3) increased calcium influx through an effect on calcium channels (Stea et al., 1995), and (4) increased calcium influx by broadening the action potential through inhibition of potassium channels (Hoffman and Johnston, 1998).
Gillis et al. (1996) presented strong evidence that phorbols increase release from chromaffin cells by increasing the number of vesicles in the readily releasable pool. Altered calcium influx and clearance were not responsible, and phorbols did not affect the calcium sensitivity of release. These conclusions are consistent with those of Vitale et al. (1995), who observed disruption of the cortical actin ring and an increased number of docked vesicles after exposure of chromaffin cells to phorbol ester.
Release from neuroendocrine cells differs from synaptic transmission in several important respects, but studies of neuronal preparations have, likewise, implicated the readily releasable pool. Redman et al. (1997)reported phorbol-induced potentiation at the frog neuromuscular junction despite decreased presynaptic calcium influx. At hippocampal synapses, phorbol esters potentiate release in response to both electrical stimulation and application of hypertonic solutions (Malenka et al., 1996; Stevens and Sullivan, 1998). Because hypertonic solutions cause release in a calcium-independent manner, these data again suggest that phorbol esters potentiate release principally though an effect on the readily releasable pool.
In contrast, some authors have concluded that phorbols act via an increase in calcium channel activity. Parfitt and Madison (1993) found that phorbols increased the frequency of spontaneous miniature EPSPs through activation of L-type calcium channels at hippocampal terminals. Inhibition of N-type channels, which are partially responsible for evoked release, had no effect on the increase in spontaneous frequency induced by phorbols (Parfitt and Madison, 1993). However, L-type channels are not involved in evoked release from hippocampal synapses (Parfitt and Madison, 1993; Wheeler et al., 1994), and the effects of calcium channel antagonists on phorbol-induced potentiation of evoked release are unknown. Whether potentiation of evoked release by phorbol esters is partially attributable to effects on calcium channels is therefore unclear.
We have used established optical techniques to investigate the effects of phorbol esters on synaptic release from hippocampal terminals, distinguishing between effects on evoked and spontaneous release. LikeParfitt and Madison (1993), we find that phorbols increase spontaneous release after calcium influx through L-type calcium channels. However, we find that potentiation of action potential-evoked release is not sensitive to calcium channel antagonists and does not result from a change in the total number of recycling vesicles. We conclude that phorbol esters influence spontaneous and evoked release from hippocampal neurons by two different mechanisms: through a mechanism involving L-type calcium channels and through an increase in the proportion of presynaptic vesicles in the readily releasable pool.
MATERIALS AND METHODS
Preparation of dissociated cultures. Dissociated hippocampal cultures were prepared from postnatal day 2 Sprague Dawley rats. Hippocampi were dissected, and the dentate gyrus was removed. After treating for 15 min at room temperature in 10 mg/ml trypsin, the tissue was dissociated by trituration through the tip of a fire-polished, siliconized glass Pasteur pipette. Dissociated cells were collected by centrifugation at 800 × g at 4°C and plated onto Matrigel-coated coverslips in Neurobasal medium (Life Technologies, Gaithersburg, MD) supplemented with B-27 (Life Technologies), 28 mm glucose, 1.3 μm transferrin (Calbiochem, La Jolla, CA), 2 mm glutamine, 0.7 U/ml insulin (Sigma, St. Louis, MO), and 1% fetal calf serum (Hyclone, Logan, UT). Cells were maintained at 37°C in an atmosphere containing 5% CO2 until use after 10–16 d in vitro.
FM 1-43 staining and destaining. A coverslip was mounted in a custom-made, low-volume (60 μl) laminar perfusion chamber on the stage of an inverted microscope (IM 35; Zeiss, Thornwood, NY). This permitted continuous perfusion at ∼1 ml/min while imaging the cells using either transmitted light and Nomarski optics or in an epifluorescence configuration through the coverslip to which the cells adhered. Cells were perfused with a modified Tyrodes solution consisting of (in mm): 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, and 30 glucose, with (in μm) 10 CNQX, 50 APV, and 3 bicuculline added to reduce spontaneous and recurrent activity. All imaging experiments were performed at room temperature (22–23°C).
Stock solution of FM 1-43 at 3 mm in water was stored at 4°C. This was diluted into Tyrodes solution to a final concentration of 15 μm. Timing of the addition of FM 1-43 to the perfusing solution and the duration of stimulation depended on the experimental protocol used. Exocytosis was induced using trains of 1 msec field stimuli at 50 V/cm delivered through platinum electrodes positioned on opposite sides of the perfusion chamber. Control experiments using the intracellular calcium dye fluo-4 AM to detect action potential-induced calcium transients verified that stimulation invariably succeeded in firing neurons at the required frequency. After staining, the preparation was washed in dye-free medium for 10 min to reduce nonspecific staining before image acquisition. Subsequent stimulation resulted in destaining of the preparation.
Stock solutions of phorbol-12,13-dibutyrate (PDBu) were made at 1 mm in DMSO and stored at −20°C until use. PDBu was added to the perfusing Tyrodes solution to a final concentration of 1 μm. The resulting concentration of DMSO (0.1%, v/v) did not influence FM 1-43 staining or destaining in control experiments.
Imaging techniques. The sample was illuminated using the 488 nm line of an air-cooled argon ion laser at the minimal intensity commensurate with an acceptable signal-to-noise ratio of the fluorescent dye (60 μW at the back aperture of the objective). Laser light was focused onto the preparation using an oil immersion objective lens (40×, 1.3 numerical aperture; DApo UV; Olympus Optical, Tokyo, Japan), and emitted fluorescence was collected through an OG 520 nm long-pass filter. A Bio-Rad (Hercules, CA) MRC 500 confocal laser scanning microscope running dedicated software with custom modifications was used to acquire images. Images were stored digitally for off-line analysis using custom software (View; Dr. Noam Ziv, Rappaport Institute, Haifa, Israel) or a commercial equivalent (Metamorph; Universal Imaging, West Chester, PA). Five images were acquired after each staining or destaining step, and average fluorescence intensities were calculated offline. Fluorescence intensity measurements were taken from regions of interest of ∼1.5 × 1.5 μm, corresponding to individual puncta, each visibly separate from its nearest neighbors. Regions of staining >1.5 μm in diameter were excluded from analysis. All fluorescence intensities were corrected for nonspecific staining by subtracting the intensities measured after complete destaining of the preparation.
RESULTS
The fluorescent styryl dye FM 1-43 has proven a valuable tool with which one can monitor exocytosis and endocytosis of vesicles at presynaptic terminals (Murthy, 1999). When applied to neuronal preparations such as that shown in Figure1a, dye is trapped in endocytosed vesicles after synaptic activity (Betz and Bewick, 1992;Ryan et al., 1993; Betz et al., 1996). After removal of extracellular dye, a punctate staining pattern is observed (Fig. 1b;Henkel et al., 1996). Fluorescence intensity may be used as a quantitative measure of the number of vesicles endocytosed at each site of synaptic recycling (Ryan et al., 1997). FM 1-43 associates reversibly with the bilayer and therefore is released on subsequent exocytosis of labeled vesicles (Fig. 1c). Hence, after staining and subsequent removal of extracellular dye, the loss of fluorescence during a subsequent stimulus can be used to monitor vesicle release.
Fig. 1.

Example of FM 1-43 staining.a, Nomarski image of a pyramidal neuron in a mixed neuronal–glial dissociated culture. b, Fluorescence image of the same field acquired after FM 1-43 staining, using a 100-stimulus train at 10 Hz and a 40 sec exposure to FM 1-43. Discrete fluorescent puncta are visible, often at sites corresponding to dendritic interactions visible in the Nomarski image. c, Fluorescence image acquired after destaining using a 900-stimulus train at 10 Hz. Note that the punctate staining is primarily absent, leaving a dim background attributable to nonspecific membranous staining by FM 1-43. Scale bar, 10 μm.
PDBu potentiates FM 1-43 staining
We have examined the effect of 1 μm PDBu on FM 1-43 staining induced by brief stimulus trains consisting of 30 stimuli at 20 Hz. This stimulus train will turn over the readily releasable pool of synaptic vesicles (Murthy and Stevens, 1998). The protocol used is summarized in Figure 2a. After perfusion of FM 1-43 onto the preparation, a stimulus train consisting of 30 stimuli at 20 Hz was delivered to induce synaptic vesicle turnover. FM 1-43 was removed from the perfusion chamber 1 min later. After a 10 min wash period to reduce nonspecific staining, five images were acquired (Fig. 2a, images labeled a) and averaged offline. The fluorescence intensities of puncta in these images represent the number of vesicles endocytosed during the 1 min period in FM 1-43. Fluorescence was then released using a 900 stimulus train at 10 Hz (sufficient to turn over the entire vesicle pool; Ryan and Smith, 1995), and five images representing nonspecific staining were acquired (Fig. 2a, images labeled b). We will refer to each round of staining and subsequent destaining as one “trial.” After a 10 min rest period and an additional 5 min in 1 μm PDBu, a second, identical trial was conducted. Using this protocol, we compared vesicle turnover at identified synapses before and after exposure to PDBu. Control experiments were conducted in which both trials were performed without PDBu treatment.
Fig. 2.

Effect of PDBu on staining.a, Summary of the staining–destaining protocol used. b, Scatter plot showing fluorescence intensities for individual fluorescent puncta in two consecutive trials. PDBu resulted in increased FM 1-43 staining (filled symbols), whereas controls (open symbols) displayed no mean change in staining. c, Data represented as frequency histograms. Top panel, control data; bottom panel, effect of PDBu treatment.d, Frequency histogram showing the percentage increase in staining observed after PDBu exposure.
The fluorescence intensities of puncta during consecutive trials are plotted in Figure 2b. Each point represents the intensities of a single fluorescent punctum corrected for nonspecific staining, i.e., b − a (see Fig. 2a) for the first trial and c − d for the second. Control and PDBu treatment data sets were derived from sister cultures.
These data are presented as frequency histograms in Figure2c. Control synapses stained equally with FM 1-43 during the first and second trials (Fig. 2c, top panel). In contrast, exposure to PDBu between trials resulted in a pronounced increase in dye staining during the second trial (Fig. 2b, bottom panel; p < 0.0001, Wilcoxon signed rank test). The effect of PDBu, expressed as a percentage change in FM 1-43 loading, is plotted as a frequency histogram in Figure 2d. The data are positively skewed. The median effect was a 172% enhancement of dye staining (n = 1003 puncta; mean enhancement, 244%), compared with a 0.3% (median) loss of fluorescence staining in controls (n = 230 puncta). Potentiation by PDBu was observed throughout numerous culture preparations.
These data indicate that PDBu potentiated release. A brief train (30 stimuli at 20 Hz) was used to stain synaptic vesicles, because a train of this duration provides an estimate of the size of the readily releasable pool (Murthy and Stevens, 1998). Hence, the observed effect of PDBu probably resulted from an increase in the number of vesicles in the readily releasable pool, although other effects of PDBu cannot be excluded at this point. The possible mechanisms of action of PDBu include (1) mobilization of new vesicles to the readily releasable pool, (2) a redistribution of existing recycling vesicles into the readily releasable pool, (3) an increase in the rate of spontaneous release, and (4) an increased probability of release per vesicle, without an alteration in the number of readily releasable vesicles. These possibilities are considered in turn below.
PDBu does not alter total pool size
One possible mechanism of action of phorbol esters might be to increase the total number of vesicles available for release, termed the total recycling pool. The readily releasable and reserve pools of presynaptic vesicles (which together constitute the total recycling pool) are in dynamic equilibrium, with exchange occurring with a time constant of a few minutes (Ryan and Smith, 1995; Murthy and Stevens, 1999). The addition of vesicles to the reserve pool might therefore increase the size of the readily releasable pool, accounting for the effect of PDBu reported above.
The size of the total recycling pool is easily determined by labeling all the vesicles in a synapse with FM 1-43. To this end we repeated the above experiments using a different loading protocol. Preparations were exposed to 900 stimuli at 10 Hz during perfusion with FM 1-43. Dye was removed from the perfusion chamber 1 min after cessation of the stimulus train. This procedure ensures that all available synaptic vesicles are exocytosed and labeled with dye (Ryan and Smith, 1995;Ryan et al, 1996). Destaining was performed using another 900-stimulus train at 10 Hz. As before, the protocol was repeated after a 10 min wash period and a 5 min exposure to PDBu. This protocol is illustrated schematically in Figure 3a. The fluorescence intensities of each punctum during the two trials are plotted in Figure 3b, and the data are represented as frequency histograms in Figure 3c.
Fig. 3.
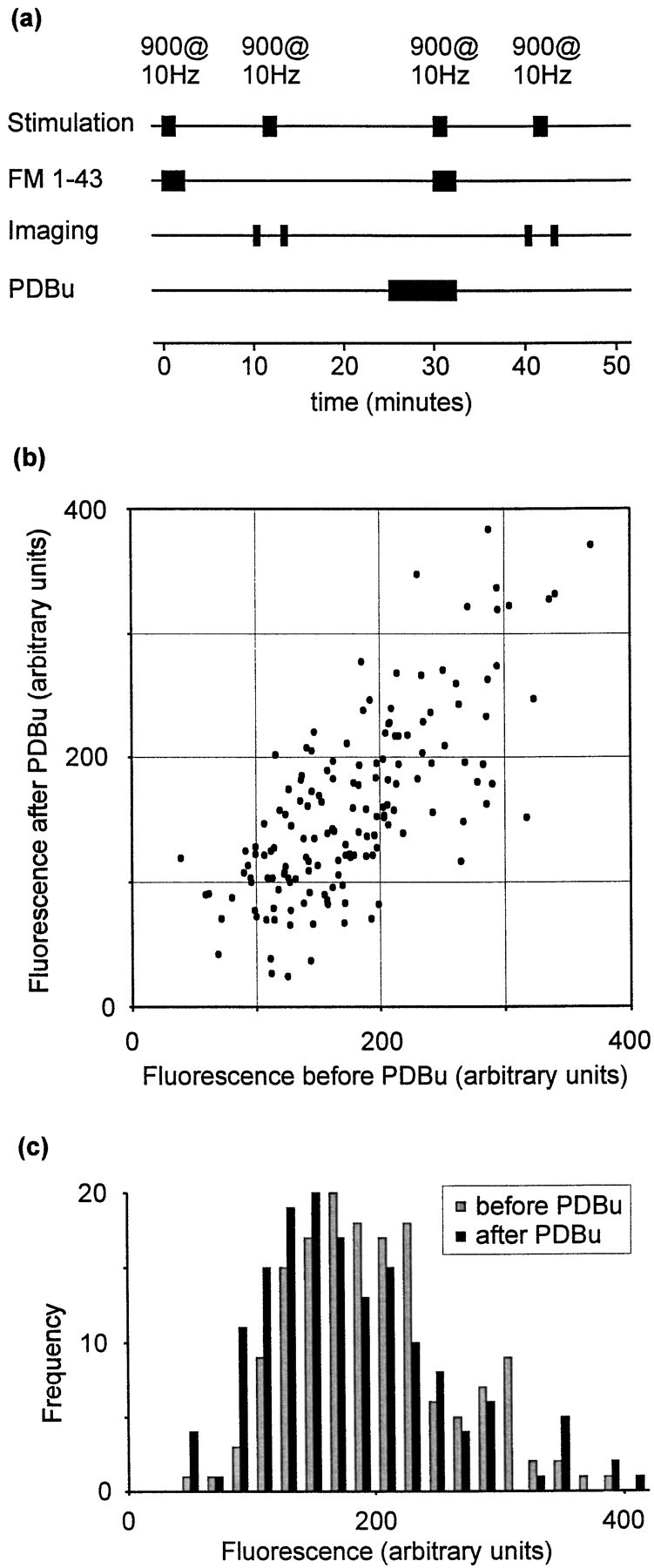
PDBu and total recycling pool size.a, Schematic illustration of the protocol used to examine the effect of phorbol treatment on total recycling pool size.b, Scatter plot comparing fluorescence staining before and after PDBu treatment. c, Data represented as a frequency histogram.
Using this protocol, no potentiation by phorbol ester was observed (n = 154 puncta), indicating that the effect of PDBu on staining in response to brief stimuli does not reflect an increase in the total number of vesicles available for release. [No change in dye staining was observed in control experiments without PDBu treatment (data not shown).] The above data exclude mobilization of new vesicles as a mechanism of action of PDBu.
Effect of PDBu on spontaneous release
The brief staining protocol described above (Fig. 2a) was designed to estimate the size of the readily releasable pool. However, in this protocol, synaptic terminals are exposed to FM 1-43 for 1 min. If spontaneous release occurs with sufficient frequency during this period, spontaneously released vesicles may contribute to the observed fluorescence staining. Since phorbol treatment increases spontaneous release from hippocampal terminals (Parfitt and Madison, 1993), this could potentially account for the observed effects of PDBu.
To determine whether spontaneous release is strongly affected by PDBu, we repeated the staining protocol (see Fig. 2a) with the exclusion of the 30-stimulus train (Fig.4a). This protocol measures the amount of spontaneous release occurring during the 1 min exposure to FM 1-43, in the absence of evoked release. (Note that a third round of staining and destaining was used to locate synapses regardless of their initial rates of spontaneous release; see legend to Fig.4a.) After phorbol treatment, spontaneous release was greatly increased. This is illustrated for individual fluorescent puncta in Figure 4b and as a frequency histogram in Figure4c. The median increase in fluorescence was 920% (n = 194; mean increase, 3230%).
Fig. 4.
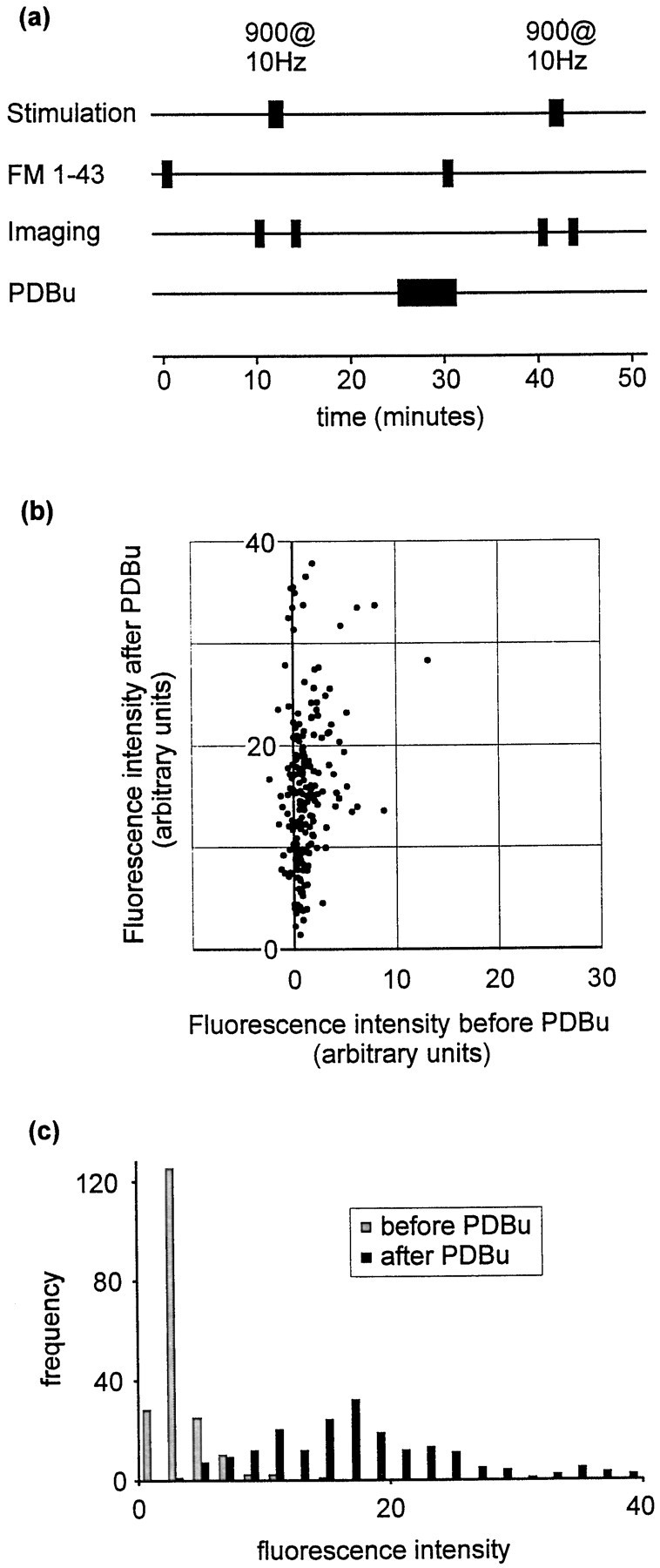
Strong effect of PDBu on spontaneous release.a, Protocol used to measure spontaneous release occurring during 1 min. FM 1-43 was applied for 1 min in the absence of stimulation. As in other protocols, destaining was performed using a 900-stimulus train at 10 Hz. For these experiments a third trial was also performed in which vesicles were stained using a 100-stimulus train at 10 Hz (trial not shown). The data from this third trial were used verify that the sites of fluorescence staining were positionally stable. This was necessary during these experiments, because many puncta exhibited very weak loading during the first trial, raising the possibility that a fluorescent punctum that was visible only during the second trial represented mobile fluorescence rather than a site at which recycling had been promoted by PDBu treatment. This precaution should therefore exclude the possibility that our selection procedure favored PDBu-sensitive synapses. b, Scatter plot showing fluorescence intensities before and after PDBu treatment.c, Data presented as a frequency histogram.
Comparison of spontaneous and evoked staining
Careful comparison of the above data derived from staining protocols with and without evoked release reveals that spontaneous release accounts for only part of the effect of PDBu observed using the staining protocol described in Figure 2a.
Median fluorescence intensities after spontaneous release-induced staining were 0.84 before and 15.1 after PDBu treatment (arbitrary fluorescence intensity units). Median intensities for the staining protocol (using 30 stimuli at 20 Hz and 1 min in FM 1-43) were 13.8 before and 34.7 after PDBu treatment. These effects of PDBu on spontaneous and evoked release are summarized in Figure5.
Fig. 5.

Summary of PDBu effect. Schematic showing the increase in fluorescence staining (30 stimuli at 20 Hz, 1 min in FM 1-43) before and after PDBu treatment. The total height of eachbar represents the observed fluorescence.Gray and black portions indicate the respective contributions of evoked and spontaneous release. Data are derived from Figures 2 and 5. PDBu potentiates spontaneous release by 920% and evoked release by 49%. The result is that the contribution of spontaneous release to total staining is much greater after phorbol treatment.
Since the fluorescence measured using the staining protocol is the sum of spontaneous and evoked release, one can calculate the amount of staining that is attributable to evoked release by subtracting the contribution of spontaneous release from the fluorescence observed using the staining protocol. Hence, evoked release accounts for 13.0 fluorescence units before and 19.3 units after PDBu treatment. PDBu therefore increased evoked staining from 13.0 to 19.3 arbitrary fluorescence units, a 49% increase.
These calculations indicate that an increased rate of spontaneous release accounts for part of the effect of PDBu. However, PDBu also influences evoked release (by altering the proportion of recycling vesicles in the readily releasable pool and/or the release probabilities of individual readily releasable vesicles). Were it possible to measure the effect of PDBu on evoked release alone, one would expect an increase in release of ∼50%.
Effect of PDBu on FM 1-43 destaining
Using a destaining protocol designed to minimize the influence of spontaneous release, we have measured the effect of PDBu on evoked release and compared the result with the 50% increase predicted from these calculations.
We began by staining vesicles with a more prolonged stimulus train consisting of 100 stimuli at 10 Hz. This protocol results in the release of ∼50% of the available vesicles at each synapse (Ryan and Smith, 1995). FM 1-43 was removed from the perfusing solution 30 sec after cessation of the stimulus train, by which time ∼60% of released vesicles are labeled (Ryan et al., 1995). The preparation was then washed in dye-free solution for 10 min, sufficient time for dye-labeled vesicles to partition equally between reserve and readily releasable pools (Ryan and Smith, 1995; Murthy and Stevens, 1999). Five fluorescence images were acquired (Fig.6a, images labeleda), and then the readily releasable pool was exocytosed using 30 stimuli at 20 Hz. After further image acquisition (Fig.6a, images labeled b), all remaining vesicles were released using a 900-stimulus train at 10 Hz, and then more images were acquired (Fig. 6a, images labeled c). To measure the percentage of vesicles released by 30 stimuli after PDBu treatment, another identical trial then was conducted. The preparation was treated with PDBu for 5 min, beginning 5 min after dye staining (i.e., after FM 1-43 was removed from the perfusion chamber). The 30-stimulus destaining train was delivered in the presence of PDBu during this second trial. This protocol is summarized in Figure6a. It is important to note that this protocol, in which PDBu is applied after the second round of staining, ensured that dye uptake was not influenced by PDBu treatment.
Fig. 6.

PDBu potentiates evoked release.a, Summary of the destaining protocol used to estimate the proportion of vesicles in the readily releasable pool. Note that PDBu was applied between staining and destaining steps during the second trial. b, Scatter plot comparing release before and after PDBu treatment at individual fluorescent puncta. Values represent the percentage of fluorescence staining released by 30 stimuli at 20 Hz during subsequent trials. c, Data represented as a frequency histogram. d, Frequency histogram showing the enhancement of release by phorbol ester.
The effect of PDBu is illustrated in Figure 6b. The data are presented as the percentage of vesicles released by 30 stimuli at 20 Hz during each trial. The percentage of vesicles released during each trial was calculated using mean fluorescence intensities manipulated in the following manner: first trial, percent released by 30 at 20 Hz = 100*((a − b)/(a −c)); second trial, percent released by 30 at 20 Hz = 100*((d − e)/(d −f)), where a–f represent mean fluorescence intensities of the five images acquired at time points indicated in Figure 6a.
These data are plotted as a frequency histogram in Figure6c. A significant effect of PDBu was observed (n = 772; p < 0.0001, Wilcoxon signed rank test). No effect was observed in control experiments in which PDBu treatment was excluded (data not shown). A frequency histogram showing the effect of phorbol ester, expressed as a percentage increase in the proportion of fluorescence released following PDBu treatment, is illustrated in Figure 6d. The data display a positive skew.
The median increase in evoked release by PDBu was 52% (mean increase, 140%). This figure is very similar to the 49% increase predicted above and strongly supports the assertion that the destaining protocol measures evoked release and the staining protocol measures a mixture of spontaneous and evoked release.
Effects of calcium channel antagonists
The above data suggest that phorbol esters enhance release by altering the proportion of recycling vesicles released by a brief stimulus train. Since phorbol esters increase release in response to hypertonic solutions (Stevens and Sullivan, 1998), this probably reflects a redistribution of vesicles from the reserve to the readily releasable pool. However, PDBu may act to increase release probability, perhaps by increasing calcium entry through voltage-gated calcium channels (Stea et al., 1995). In addition, the potentiating effect of phorbol esters on spontaneous release has previously been shown to require L-type calcium channel activity (Parfitt and Madison, 1993). To determine whether the effects reported above were the result of potentiation of presynaptic calcium currents by PDBu, we therefore examined the effects of PDBu on both evoked and spontaneous release in the presence of calcium channel antagonists.
We began by examining the roles of calcium channel subtypes in evoked release (without PDBu treatment). A total of 10 μmnifedipine had no significant effect on release (Fig.7a; p = 0.38, Wilcoxon signed rank test). In contrast, both 1 μm ω-conotoxin GVIA (ω-CTx-GVIA) and 500 nm ω-agatoxin IVA (ω-Aga-IVA) significantly inhibited evoked release (Fig. 7a; each p < 0.0001, Wilcoxon signed rank test). These concentrations of antagonists are sufficient to selectively eliminate L-, N-, and P/Q- type calcium channels, respectively, and the results are consistent with previous studies, which indicated that release from hippocampal terminals is mediated by N- and Q- but not L-type channels (Parfitt and Madison, 1993; Wheeler et al, 1994; Reuter, 1995).
Fig. 7.
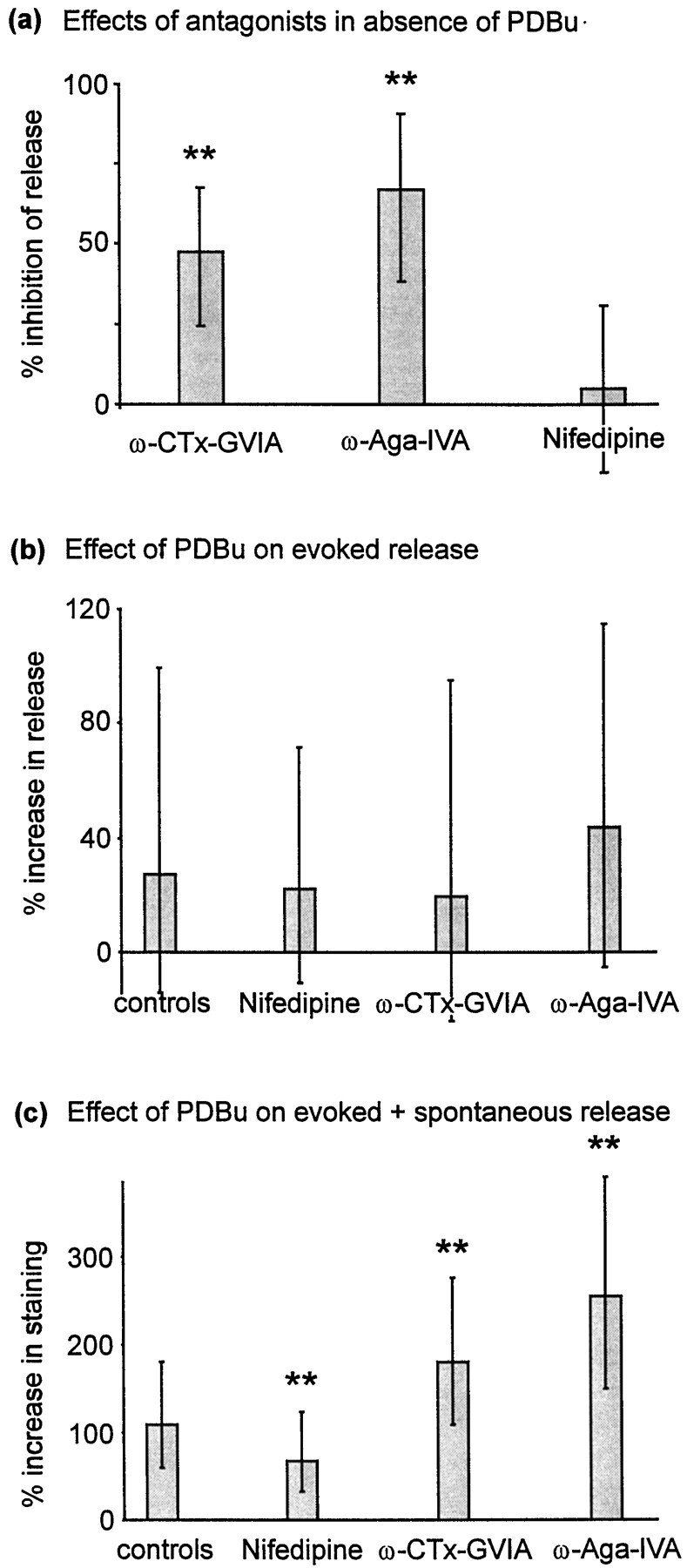
Effects of calcium channel antagonists.a, Effects of calcium channel antagonists on evoked release (no PDBu). Data were derived using a 60-stimulus (10 Hz) destaining protocol, after staining using a 100-stimulus train at 10 Hz. Preparations were subjected to two trials, the first a control and the second after or during application of antagonist. ω-CTx-GVIA (1 μm) was applied for 10 min after the second staining step. ω-Aga-IVA (500 nm) and nifedipine (10 μm) were each applied for 5 min before the second destaining step and remained in the perfusion chamber during destaining. n: nifedipine, 86; ω-CTx-GVIA, 344; ω-Aga-IVA, 100. b, Calcium channel antagonists failed to attenuate the effect of PDBu on evoked release measured using a 30-stimulus destaining assay (as in Fig. 6a). Data are derived from two consecutive trials, the first before and the second after PDBu exposure (as in Fig. 6). Controls were not treated with calcium channel antagonists (n = 391). Nifedipine-treated preparations (n = 279) were perfused throughout with 10 μm nifedipine, beginning 5 min before the start of the first trial. ω-CTx-GVIA effects were examined by pretreating the preparation with 1 μmω-CTx-GVIA for 10 min before the start of the first trial (n = 194). ω-Aga-IVA (500 nm) was applied for 5 min before each staining or destaining stimulus and was also present in the perfusion chamber throughout all destaining stimulus trains (n = 150). c, Influence of antagonists on PDBu-induced potentiation using a 30-stimulus staining protocol (as in Fig. 2a). Antagonists were applied as described above (controls,n = 531; 10 μm nifedipine perfused throughout, n = 326; 1 μmω-CTx-GVIA by pretreatment for 10 min, n = 105; 500 nm ω-Aga-IVA by pretreatment for 5 min and perfused throughout stimulation, n = 106). Throughout partsa–c, column heights represent medians, and error bars represent the quartile (25–75%) ranges of each distribution.
Using the 30-stimulus destaining protocol described above, we next examined whether PDBu was able to increase evoked release after calcium channel blockade. The resulting data are shown in Figure 7b. Although absolute staining was reduced after treatment with ω-CTx-GVIA and ω-Aga-IVA, potentiation by PDBu was not significantly different from controls after treatment with either of these toxins or with nifedipine (p > 0.05, Kruskal–Wallis test).
Since none of these antagonists either increased or decreased the effect of PDBu on evoked release, we conclude that PDBu does not potentiate evoked release by selectively increasing calcium entry through N-, P/Q-, or L- type voltage-gated calcium channels. In view of published literature showing differential effects of phorbol esters at these channel subtypes (Stea et al., 1995), it is also unlikely that PDBu acts to increase calcium influx equally at all channel subtypes. Our data therefore indicate that PDBu does not potentiate evoked release by influencing calcium channel activity. In addition, these data indirectly suggest that PDBu does not influence release probability by another mechanism, because the effect of PDBu on evoked release was not enhanced in the presence of any of these antagonists (see Discussion).
Data derived using the 30-stimulus staining protocol were more complex than those derived using the destaining protocol. Nifedipine reduced the median effect of PDBu from 109 to 68% (p < 0.0001, Mann–Whitney rank sum test), a 38% inhibition of the effect of PDBu. This is expected if nifedipine inhibits spontaneous but not evoked release and is therefore consistent with the data presented above (Figs. 5, 7b) and those of previous authors (Parfitt and Madison, 1993). In contrast, both ω-CTx-GVIA and ω-Aga-IVA significantly enhanced the effect of PDBu (each p < 0.0001, Mann–Whitney rank sum test). This probably reflects strong inhibition of evoked release by these toxins; after toxin treatment, a larger proportion of the remaining release (both before and after PDBu treatment) will result from spontaneous release. Since spontaneous release is much more strongly potentiated by PDBu than evoked release, the percentage increase in release after phorbol ester treatment should be greater after treatment with N- or Q-type channel antagonists.
From these data we conclude that phorbol esters influence evoked and spontaneous release by different mechanisms. Potentiation of evoked release probably results from a recruiting effect of phorbol esters, whereby vesicles from the reserve pool are transferred to the readily releasable pool. In contrast, the effect on spontaneous release reflects an increase in release probability involving L-type calcium channels.
DISCUSSION
Previous publications suggested that phorbols (1) increase the number of vesicles in the readily releasable pool in hippocampal neurons in dissociated culture (Stevens and Sullivan, 1998) and (2) increase L-type calcium channel activity in hippocampal slices (Parfitt and Madison, 1993). Our data indicate that both effects coexist in the same preparation, but that they produce two distinct effects: increases in evoked and spontaneous release respectively. In addition, we have demonstrated that phorbols do not supplement the recycling vesicle pool by mobilizing new vesicles, because the total number of recycling vesicles is not altered by PDBu. We conclude that phorbols potentiate evoked release by altering the distribution of vesicles between readily releasable and reserve pools.
We observed a mean increase in evoked release of 140% after phorbol ester treatment, comparable with that reported by Stevens and Sullivan (1998), who used hypertonic solutions to induce release. This similarity is important, because we were unable to definitively discount the possibility that the observed potentiation resulted from increased release probability per vesicle. In contrast to action potential-induced release, hypertonic solutions release vesicles in a calcium-independent manner, so alterations in release probability per vesicle will have little effect on this measure of readily releasable pool size (Rosenmund and Stevens, 1996). The fact that our data show potentiation comparable with that measured using hypertonic solutions therefore suggests that the potentiation of evoked release that we observed reflects a change in the number of vesicles in the readily releasable pool rather than an increased release probability per vesicle.
Using electrophysiological techniques, Murthy and Stevens (1999)reported that ∼30% of recycling vesicles were within the readily releasable pool. Unfortunately, the temporal resolution afforded by FM dye imaging techniques prevents estimation of the readily releasable pool size in the manner of electrophysiological experiments, in which one can calculate the number of vesicles released before depletion. From electrophysiological data, one would expect 30 stimuli delivered at 20 Hz to be sufficient to exocytose the entire readily releasable pool (Murthy and Stevens, 1998). However, in our experiments the average proportion of vesicles released by this stimulus was slightly less than 30% (see Fig. 6c). Our protocol may therefore slightly underestimate the proportion of recycling vesicles in the readily releasable pool.
The fact that 30 stimuli at 20 Hz release almost the entire readily releasable pool before phorbol exposure further suggests that the effects of PDBu were the result of an increase in the number of vesicles in the readily releasable pool. Since most of the readily releasable pool was released by this stimulus under control conditions, it is unlikely that a large potentiation by PDBu could be observed without mobilization of extra vesicles to the readily releasable pool. In view of this “ceiling” on the effect of PDBu that could result without the mobilization of additional vesicles, it is unlikely that an increase in release probability per vesicle could account for the large potentiation observed.
Furthermore, if potentiation by PDBu was limited by this ceiling effect, one might expect to see increased potentiation of evoked release after reducing release probability with calcium channel antagonists. No such effect was observed (Fig. 7b). Together these data suggest that phorbols enhance evoked release by altering the proportion of recycling vesicles in the readily releasable pool rather than increasing release probability per vesicle.
Destaining protocol as a measure of evoked release
Using both direct and indirect protocols, we concluded that PDBu treatment enhances evoked release by ∼50%. Changes in spontaneous release rates may have influenced our measurements of evoked release given the pronounced effect of PDBu on spontaneous release. However, spontaneous loss of fluorescence was extremely slow, even after PDBu treatment (data not shown). Consequently, intensities of puncta in the stained condition were reduced only ∼9% (median) by a 5 min treatment with PDBu (data not shown). This probably reflects a very low rate of spontaneous release before PDBu application, such that even pronounced potentiation of spontaneous release by PDBu produced only modest rates of spontaneous destaining. Although the destaining protocol is therefore not entirely unaffected by spontaneous release, even large changes in spontaneous release rates do not influence the data on evoked release derived in this manner. Furthermore, the effects of spontaneous release on data derived with the evoked release protocol would have been mostly eliminated by the analysis method used, in which the effects of the destaining stimulus were expressed as a proportion of the fluorescence intensity in the stained condition.
Although an experimental approach that definitively separated evoked and spontaneous release was not available, it is therefore likely that the destaining protocol used (Fig. 6) separated evoked and spontaneous release sufficiently to yield accurate data describing the effect of PDBu on evoked release. Consequently, destaining measurements (Fig. 6) and calculations based on staining protocols (Figs. 2, 4, 5) yielded similar estimates for the effect of PDBu on evoked release (median potentiations of 49 and 52%, respectively).
Molecular mechanism of action of phorbol esters
The molecular targets of PDBu are unknown, but it has been suggested that phorbols increase the size of the readily releasable pool through actions at two steps in the secretion pathway (Bittner and Holz, 1993). This may reflect interactions with both protein kinase C and another presynaptic phorbol ester-activated protein such as Munc 13-1 (Goda et al., 1996; Betz et al., 1998). In murine hippocampus, Munc 13-1 is thought to be involved in a calcium-independent priming step that gates entry into the readily releasable pool (Augustin et al., 1999). At the Calyx of Held, PDBu potentiates release through actions on both protein kinase C and Munc 13-1 (Hori et al., 1999). In light of these reports, our data suggest that PDBu might act through three mechanisms; a protein kinase C-mediated increase in vesicle docking, a Munc 13-1-mediated increase in vesicle priming, and an action on L-type calcium channels (probably through protein kinase C).
Serial reconstruction of hippocampal synapses from electron micrographs has revealed that on average no more than half the docking sites at an active zone are occupied (Harris and Sultan, 1995; Schikorski and Stevens, 1997). It is possible, therefore, that active zones could accommodate an increased number of docked vesicles. Increased docking (presumably via protein kinase C activation) is therefore one potential mechanism by which phorbol esters might increase the size of the readily releasable pool. The molecular targets by which protein kinase C may induce vesicle docking are unknown, but there are numerous presynaptic proteins that are phosphorylated after activation of protein kinase C. These include cytoskeletal proteins that could influence vesicle trafficking (Vitale et al., 1995; Ryan, 1999) as well as docking/release proteins such as VAMP, SNAP25, Munc 18, and the synapsins (Browning and Dudek, 1992; Turner et al, 1999).
Other potential synaptic effects of phorbol esters have been identified, including effects on potassium channels, on calcium clearance, and on the release machinery itself. Although we have not specifically addressed these effects, it seems unlikely that these mechanisms are responsible for our observations, because all three relate to calcium metabolism. Were alterations in calcium metabolism a factor underlying the effects of PDBu, one would expect inhibition of calcium influx to influence the effects of PDBu. Since calcium channel antagonists did not alter the potentiating effect of PDBu on evoked release, it is unlikely that PDBu acts through these alternative routes at hippocampal synapses.
Model to account for the effects of phorbol esters
The fact that phorbol esters influence spontaneous release through L-type calcium channel activation is surprising in view of the fact that L-type calcium channels do not mediate presynaptic release from hippocampal terminals (Parfitt and Madison, 1993; Wheeler et al., 1994). Such channels are, however, distributed throughout hippocampal neurons and influence the resting intracellular calcium concentration through tonic activity (Magee et al., 1996). The effect of phorbol esters on spontaneous release may therefore reflect a modest rise in intracellular calcium concentration throughout the neuron rather than a localized effect within the presynaptic terminal.
We conclude that phorbol esters influence release from hippocampal terminals by several mechanisms. Spontaneous release is increased, probably through an extrasynaptic effect of phorbol esters on L-type calcium channels. This does not influence evoked release. In addition, phorbol esters probably activate both protein kinase C and Munc 13-1, causing increased vesicle docking and priming, respectively. Together these effects on docking and priming are observed functionally as an increased number of vesicles in the readily releasable pool. We have also shown that these effects do not involve mobilization of new vesicles into the recycling vesicle pool. Hence phorbol esters must induce redistribution of vesicles from the reserve to the readily releasable pool. Our data therefore identify an increase in the proportion of recycling vesicles in the readily releasable pool as the functional mechanism by which phorbol esters potentiate release at hippocampal synapses. In addition to clarifying the mechanism of action of phorbol esters, our data indicate that phorbol ester treatment may be used to selectively probe the functions of different presynaptic vesicle pools.
Footnotes
This work was supported by National Institute of Mental Health Silvio Conte Centre for Neuroscience Research Grant MH48108 to S.J.S. We thank Ron Holz, Murali Prakriya, and Stephen M Smith for critical reading of this manuscript.
Correspondence should be addressed to Jack Waters at the above address. E-mail: jwaters@leland.stanford.edu.
REFERENCES
- 1.Augustin I, Rosenmund C, Südhof TC, Brose N. Munc 13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- 2.Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- 3.Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Curr Opin Neurobiol. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- 4.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc 13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 5.Bittner MA, Holz RW. Protein kinase C and clostridial neurotoxins affect discrete and related steps in the secretory pathway. Cell Mol Neurobiol. 1993;13:649–664. doi: 10.1007/BF00711564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browning MD, Dudek EM. Activators of protein kinase C increase the phosphorylation of the synapsins at sites phosphorylated by cAMP-dependent and Ca2+/calmodulin-dependent protein kinases in the rat hippocampal slice. Synapse. 1992;10:62–70. doi: 10.1002/syn.890100109. [DOI] [PubMed] [Google Scholar]
- 7.Gillis KD, Möβner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 8.Goda Y, Stevens CF, Tonegawa S. Phorbol ester effects at hippocampal synapses act independently of the gamma isoform of PKC. Learn Mem. 1996;3:182–187. doi: 10.1101/lm.3.2-3.182. [DOI] [PubMed] [Google Scholar]
- 9.Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacology. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-s. [DOI] [PubMed] [Google Scholar]
- 10.Henkel AW, Lübke J, Betz WJ. FM1-43 dye ultrastructural localization in and release from frog motor nerve terminals. Proc Natl Acad Sci USA. 1996;93:1918–1923. doi: 10.1073/pnas.93.5.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hori T, Takai Y, Takahashi T. Presynaptic mechanism for phorbol ester-induced synaptic potentiation. J Neurosci. 1999;19:7262–7267. doi: 10.1523/JNEUROSCI.19-17-07262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin LF, Kao LS, Westhead EW. Agents that promote protein phosphorylation inhibit the activity of the Na+/Ca2+ exchanger and prolong Ca2+ transients in bovine chromaffin cells. J Neurochem. 1994;63:1941–1947. doi: 10.1046/j.1471-4159.1994.63051941.x. [DOI] [PubMed] [Google Scholar]
- 14.Magee JC, Avery RB, Christie BR, Johnston D. Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:3460–3470. doi: 10.1152/jn.1996.76.5.3460. [DOI] [PubMed] [Google Scholar]
- 15.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1996;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 16.Murthy VN. Optical detection of synaptic vesicle exocytosis and endocytosis. Curr Opin Neurobiol. 1999;9:314–320. doi: 10.1016/s0959-4388(99)80046-4. [DOI] [PubMed] [Google Scholar]
- 17.Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–500. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- 18.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 19.Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol (Lond) 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redman RS, Searl TJ, Hirsh JK, Silinski EM. Opposing effects of phorbol esters on transmitter release and calcium currents at frog motor nerve endings. J Physiol (Lond) 1997;501.1:41–48. doi: 10.1111/j.1469-7793.1997.041bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reuter H. Measurements of exocytosis from single presynaptic nerve terminals reveal heterogeneous inhibition by Ca2+ channel blockers. Neuron. 1995;14:773–779. doi: 10.1016/0896-6273(95)90221-x. [DOI] [PubMed] [Google Scholar]
- 22.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 23.Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–1323. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryan TA, Smith SJ. Vesicle pool mobilization during action potential firing at hippocampal synapses. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 25.Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 26.Ryan TA, Smith SJ, Reuter H. The timing of synaptic vesicle endocytosis. Proc Natl Acad Sci USA. 1996;93:5567–5571. doi: 10.1073/pnas.93.11.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryan TA, Reuter H, Smith SJ. Optical detection of a quantal presynaptic membrane turnover. Nature. 1997;388:478–482. doi: 10.1038/41335. [DOI] [PubMed] [Google Scholar]
- 28.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 30.Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- 31.Turner KM, Burgogne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- 32.Vitale ML, Seward EP, Trifaró J-M. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- 33.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]