

Abstract
Astrocytes are present in large numbers in the nervous system, are associated with synapses, and propagate ionic signals. Astrocytes influence neuronal physiology by responding to and releasing neurotransmitters, but the mechanisms that establish the close interaction between these cells are not defined. Here we use hippocampal neurons in culture to demonstrate that vasoactive intestinal polypeptide (VIP) promotes neuronal differentiation through activity-dependent neurotrophic factor (ADNF), a protein secreted by VIP-stimulated astroglia. ADNF is produced by glial cells and acts directly on neurons to promote glutamate responses and morphological development. ADNF causes secretion of neurotrophin 3 (NT-3), and both proteins regulate NMDA receptor subunit 2A (NR2A) and NR2B. These data suggest that the VIP–ADNF–NT-3 neuronal–glial pathway regulates glutamate responses from an early stage in the synaptic development of excitatory neurons and may also contribute to the known effects of VIP on learning and behavior in the adult nervous system.
Keywords: glia, synapse, hippocampus, ADNF, NT-3, VIP
An important role for glial cells in synapse function has long been suggested by the intimate relationship that exists between astrocytes and synaptic terminals in vivo (Peters et al., 1991; Ventura and Harris, 1999). Growing evidence indicates that astrocytes play active roles in the CNS (Dani et al., 1992; Porter and McCarthy, 1996). Astrocytes can signal to neurons by releasing soluble factors such as glutamate (Nedergaard, 1994; Parpura et al., 1994) or β-chemokines (Brenneman et al., 1999a,b) that regulate neuronal activity (Araque et al., 1998a,b;Meucci et al., 1998). In fact, astrocytes are now viewed as active partners of the presynaptic and postsynaptic terminals in the elaboration of tripartite synaptic structures (Araque et al., 1999). The temporal correlation between glial development and synaptogenesis also suggests an involvement of astrocytes in the formation of the first synapses. In the rat CNS, neurons form most of their synapses during the third postnatal week (Aghajanian and Bloom, 1967), after the differentiation of astrocytes has already been completed (Parnavelas et al., 1983). It has been proposed that glia-derived, soluble factors are necessary for the maturation of developing synapses in vitro(Pfrieger and Barres, 1997). However, the molecular mechanisms that regulate the interactions between neurons and glia are not well defined.
Secreted factors such as brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) have been shown to regulate synaptogenesis in the developing hippocampus (Vicario-Abejón et al., 1998), but these factors are neuron-derived proteins that do not require the presence of glia to act on neurons (Song et al., 1997). Among the known regulatory peptides found in the CNS, only vasoactive intestinal polypeptide (VIP) is thought to stimulate glia to generate neurotrophic factors (for review, see Brenneman et al., 1999b). The most potent of these glial signals is the activity-dependent neurotrophic factor (ADNF) (Brenneman and Gozes, 1996). ADNF is a secreted protein with a component that has an amino acid sequence similar to that of heat shock protein 60 (hsp60), with two serines in the active site of ADNF replacing two cysteines in the corresponding region of hsp60 (Cys-442 and Cys-447). In this study, we initially use a measure of synaptic connectivity to ask whether VIP and ADNF can act on the development of embryonic hippocampal neurons.
MATERIALS AND METHODS
Cultures. Monolayers of astrocytes were prepared from rat embryonic hippocampus and/or cortex at embryonic day 18 (E18; the day when a copulation plug was found was considered day E1). The tissue was dissected and treated with papain (Worthington Biochemicals, Lakewood, NJ). Cells suspended in a modified MEM (82-0234DJ; Life Technologies, Grand Island, NY) and 10% fetal bovine serum (FBS) were plated at low density (300 cells/mm2) on glass coverslips coated with Vitrogen (Collagen Biomaterials, Palo Alto, CA) and Poly-d-lysine (Sigma, St. Louis, MO) and incubated for 2 weeks at 37°C in 10% CO2.
Cultures of embryonic neurons were prepared from E16 or E18 rat hippocampus. The hippocampi were dissected and treated with papain as described above. For plating on a monolayer of astrocytes, cells were suspended in modified MEM as above, 5% horse serum (HS) and 1% FBS at 2000 cells/mm2. For glia-free neuronal cultures, cells suspended in Neurobasal + B27 (Life Technologies) were plated directly on glass coverslips coated as above at 1000 cells/mm2. The antimitotic agent floxuridine (Sigma) was added to all neuronal cultures from the day of plating to prevent astrocytic growth. The antiserum used to block VIP function was from Peninsula Laboratories (Belmont, CA) (1:500).
Electrophysiology. Patch-clamp recordings in the whole-cell configuration were used to record spontaneous and evoked synaptic activity as previously described (Vicario-Abejón et al., 1998) in E16 and E18 neurons cultured on a monolayer of astrocytes. The recorded cells were held at −60 mV, and afferent neurons were stimulated with a puff of 1 mm glutamate delivered by pressure through a glass micropipette. A measurement of synaptic connectivity was obtained by calculating the ratio of afferents that elicited an evoked response over the total number of stimulated cells in the field surrounding the recorded cell. The ratio was then expressed as the percentage of afferents connected to each cell from which we recorded.
In experiments in which E16 neurons in glia-free cultures were studied, field stimulation using hyperosmotic conditions were performed to assay the maturation of the presynaptic machinery. The presynaptic terminals were stimulated with 50 μl of a 300 mm sucrose solution added manually to the regular recording medium, whereas miniature postsynaptic currents were recorded during 10 sec intervals.
Immunostaining of cultured cells. Cells were fixed with 4% paraformaldehyde and 0.1 m PBS, pH 7.4, for 20 min. After treatment with 0.1% Triton X-100 and 10% normal goat serum, cells were incubated at 4°C with primary antibodies against VIP [1:300 (Peninsula Laboratories); Fig. 1C; similar results obtained with 1:200 (Chemicon, Temecula, CA) and 1:1000 (Sigma)], neuronal nuclei protein (NeuN, 1:300; Chemicon), NT-3 (1:600; Chemicon), glial fibrillary acidic protein (GFAP, 1:200; Dako, Carpinteria, CA), and synaptophysin (1:200, Sigma). Cells were then incubated with the corresponding Alexa Fluor 488-conjugated (Molecular Probes, Eugene, OR) or rhodamine-conjugated (1:100; Jackson ImmunoResearch, West Grove, PA) secondary antibodies.
Fig. 1.
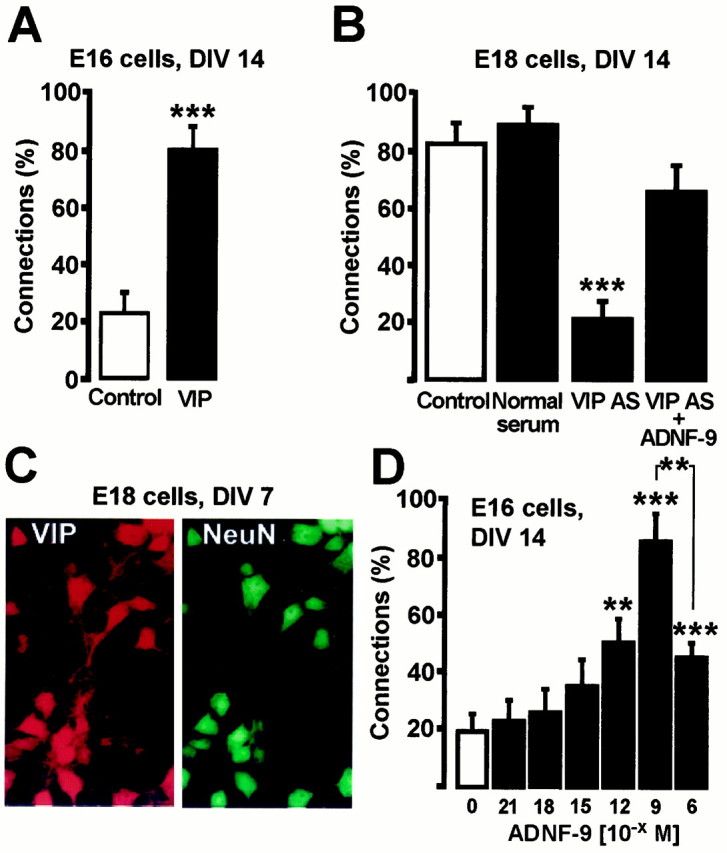
VIP and ADNF-9 regulate synaptogenesis in cultures of embryonic hippocampal neurons. A, Two-week treatment of E16 cultures with 10−10m VIP caused a marked increase in the level of synaptic connectivity. B, Two-week treatment with a VIP antiserum prevents the spontaneous formation of active synapses in E18 cultures, an effect that is reversed by co-treatment with 10−9m ADNF-9.C, Expression of VIP by E18 hippocampal neurons in culture. Cells were grown for 7 d, fixed, and immunostained for the expression of VIP and NeuN. Magnification, 63×. The same pattern was obtained after staining three independent cultures.D, Dose–response effect on connectivity in E16 cultures treated for 1 week with ADNF-9. **p < 0.01; ***p < 0.001, t test. Results are mean ± SEM of 10–23 cells from at least two independent cultures. DIV, Days in vitro.
Morphological analysis. To investigate the effects of the ADNF antiserum on neuronal morphology, E18 cells were fixed as above and immunostained for microtubule-associated protein 2 antibody (MAP2ab, 1:200; Sigma) as previously described (Vicario-Abejón et al., 1998). Cells were then incubated with a biotinylated secondary antibody (1:200) followed by avidin–biotin–horseradish peroxidase complex (Vectastain ABC kit; Vector Laboratories, Burlingame, CA) and developed using DAB. Optical sections were obtained with a Nikon (Melville, NY) Microphot-FXA microscope with a 20× objective and 4× supplementary magnification. The dendritic length and number of primary dendrites were measured using Neurolucida software (MicroBrightField, Cochester, VT). All measurements were performed blindly.
Quantification of synaptic puncta. The quantification of the number of synaptic puncta after synaptophysin immunostaining was done using an exhaustive photon reassignment algorithm, based on an experimentally determined point spread function (Carrington et al., 1990), to reassign mathematically out-of-focus fluorescence to the in-focus optical planes. Optical sections were obtained with a Nikon microscope with a 63×, 1.4 numerical aperture oil lens and 2.5× supplementary magnification. Images were recorded with a CELLscan fluorescence imaging system (Scanalytics, Fairfax, VA) equipped with a PXL-37 CCD camera (Photometrics, Tucson, AZ), a piezoelectricz-axis focus device, and a computer-controlled excitation light shutter. The number of puncta per micrometer was counted along the processes using the “deconvolved” images and the Neurolucida software. All measurements were performed blindly.
Reverse transcription-PCR. Total RNA from E16 and E18 cultures was isolated using the Trizol reagent (Life Technologies). Reverse transcription was performed using random primers and Superscript (Life Technologies). PCR were performed out by mixing PCR Supermix (Life Technologies) with 2 μl of cDNA of various dilutions with a 0.2 μm concentration of each gene-specific primer in a final volume of 30 μl. Samples were heated to 94°C and cycled 30 times through 94, 55, and 72°C for 30 sec each. To ensure that the amplification was in the linear range for each gene-specific primer pair, a set of titration reactions was done using serially diluted cDNA. The 18S rRNA (Ambion, Austin, TX) was used as an internal standard of amplification. PCR products were run on a 2% agarose gel and visualized using ethidium bromide staining. The primers used were 5′-GGTGAACAAGGTGATGTCCATC-3′ (sense) and 5′-GGCAGGGTGCTCTGGTAATTTTCCT-3′ (antisense) for NT-3, 5′-TTCGTGGAGACAGGAATCAGCGTC-3′ (sense) and 5′-GCCAGGAAGATGACAGCAAAGAAGG-3′ (antisense) for NMDA receptor subunit 2A (NR2A), 5′-GCGTCTGGTTTGAGATGATGCGAG-3′ (sense) and 5′-AGCCGTCACATTCTTGGTTCCTGG-3′ (antisense) for NR2B, and 5′-GCGTCTGGTTTGAGATGATGCGAG-3′ (sense) and 5′-AGCCGTCACATTCTTGGTTCCTGG-3′ (antisense) for NR1.
Western blotting. To assay for endogenous production of ADNF, conditioned medium from monolayers of astrocytes was prepared as previously described (Brenneman and Gozes, 1996). After dialysis and lyophilization, 5 mg of soluble proteins was separated by electrophoresis on a 10% polyacrylamide gel and electrotransfered to nitrocellulose filters. Membranes were treated with 10% milk and PBS-Tween 0.2% for 1 hr and incubated overnight at 4°C in 2% milk and PBS-Tween 0.2% and the ADNF-14 antiserum (AS; 1:1000). After incubation with peroxidase-conjugated secondary antibodies (Boehringer Mannheim, Indianapolis, IN) signals were revealed by chemiluminescence using an ECL kit (Amersham Pharmacia Biotech, Arlington Heights, IL).
To assay protein levels of NMDA receptors (NMDArs), cells from E16 or E18 hippocampus were plated on 6 cm dishes covered with a monolayer of astrocytes at 2000 cells/mm2. Cells were collected after 7 d in vitro, and total proteins were analyzed by SDS-PAGE. Ten microliters of crude cell homogenate were separated by electrophoresis on an 8% polyacrylamide gel and electrotransferred to 0.45 μm polyvinylidene fluoride membranes. Membranes were treated with 10% milk and PBS-Tween 0.2% for 1 hr and incubated overnight at 4°C in 2% milk and PBS-Tween 0.2% and primary antibodies against NR1, NR2A, and NR2B. All antibodies were from Upstate Biotechnology (Lake Placid, NY) and were used at 1:600. Secondary antibodies and signal detection were as above.
NT-3 release assay. Twenty-four hours before release experiments, hippocampal cultures were transferred from the serum-containing MEM to serum-free Neurobasal medium. Immediately before experimentation, cells were placed in 225 μl of fresh Neurobasal medium to maximize the concentration of released neurotrophins. After a 15 min exposure to various stimuli, supernatant was collected using a P-1000 micropipettor and assayed immediately. Release experiments were conducted in the presence of TTX (1 μm) to prevent activity-dependent release of NT-3. NT-3 levels were quantified using the E-max NT-3 immunoassay system (Promega, Madison, WI) according to the manufacturer's instructions. Raw data were analyzed using Microsoft Excel (Microsoft, Redmond, WA) to determine NT-3 concentrations, according to standard curves plotted from predetermined concentrations of NT-3. All data are averages of three separate measurements.
RESULTS
VIP regulates differentiation of hippocampal neurons
The formation of synapses in dissociated hippocampal neurons can be monitored as previously described (Vicario-Abejón et al., 1998, 2000). Briefly, the ability of neurons to respond to stimulation of afferent cells with postsynaptic currents (PSCs) is used to measure the density of connections in the culture. Postsynaptic cells are patch-clamped at resting potential (−60 mV), and 4–10 afferent cells in the field of view are stimulated with local application of brief pulses of glutamate. The proportion of occasions when stimulation of afferents results in the generation of PSCs within a 100 msec time window gives an estimate of synaptic connectivity. This test shows that neurons derived from the hippocampus of E16 rats require an exogenous supply of neurotrophins to develop synapses in vitro. In contrast, E18-derived neurons form functional synapses spontaneously.
To test the effect of VIP on neuronal differentiation, we first used cells prepared from day 16 hippocampus. These cells were cultured on a monolayer of astrocytes in the presence or absence of VIP. After 14 d in culture, only ∼20% of afferent cells evoked PSCs in untreated cultures (Fig.1A). In contrast, treatment with 10−10m VIP resulted in a threefold increase in the proportion of evoked PSCs (Fig. 1A), indicating that exogenous VIP promotes neuronal differentiation resulting in enhanced synaptic connectivity.
If endogenous VIP influences neuronal differentiation, then the spontaneous appearance of functional synapses in E18 neurons might be retarded by an anti-VIP antibody. E18 hippocampal neurons were cultured on a monolayer of astrocytes in the presence or absence of a polyclonal antibody against VIP and assayed for connectivity. After 2 weeks in culture, untreated E18 neurons and neurons treated with normal rabbit serum established extensive functional synaptic contacts. Connections were found on ∼80–90% of the occasions when afferent cells were stimulated. In contrast, cells treated with the VIP antibody displayed a much lower proportion of induced synaptic events (∼20%; Fig.1B), indicating that endogenous VIP is required to obtain maximal levels of connectivity. Immunostaining experiments using the same anti-VIP antibody used in blocking experiments showed a strong VIP expression in the vast majority of E18 hippocampal neurons between days 4 and 14 in culture. The image of a culture at day 7 in vitro is shown (Fig. 1C). Similar results were obtained using two other commercial anti-VIP antibodies (see Materials and Methods).
VIP is known to cause release of glia-derived growth factors. To test whether the effect of VIP on synapse formation involves the release of ADNF, 10−9m ADNF-9 was added for 2 weeks to E18 cultures in addition to the VIP antibody. ADNF-9 is a nine-amino acid peptide (SALLRSIPA) derived from ADNF that displays the full survival-promoting activity of the full-length ADNF protein (Brenneman et al., 1998). As shown in Figure 1B, the addition of ADNF-9 prevented the loss of connectivity observed in VIP antiserum-treated cultures. These results suggest that ADNF acts downstream of VIP in a signaling pathway that regulates differentiation in hippocampal neuron–glia co-cultures.
ADNF-9 regulates the functional and morphological maturation of developing hippocampal neurons
The connectivity assay was used to test whether ADNF-9 could directly stimulate neuronal differentiation. Addition of ADNF-9 (10−12–10−6m) to E16 cells cultured for 10 d significantly increased the number of connections in a dose-dependent manner (Fig.1D). The maximal inductive effect, a fourfold increase, was observed at 10−9m ADNF-9. This effect of ADNF-9 on connectivity was not accompanied by any changes in the survival or morphology of the cells, as assayed by staining of the neurons with an antibody against the microtubule-associated protein MAP2 (data not shown). The inductive effect of ADNF-9 was not mimicked by a synthetic peptide (CALLRCIPA) derived from the corresponding region of hsp60. Thus, the observed effect of ADNF on hippocampal synaptic transmission is a specific property of the SALLRSIPA motif at the concentration tested.
To test whether endogenous ADNF was required for the development of active synapses in E18 neurons, a polyclonal antibody was raised against a 14-amino acid ADNF-derived peptide (ADNF-14; VLGGGSALLRSIPA), that includes the ADNF-9 sequence. The resulting antiserum (ADNF-14 AS) diluted 1:500 did not show cross-reactivity with 5 μg of recombinant hsp60 as assayed by dot blot analysis and is therefore considered specific for ADNF. When total proteins were prepared from conditioned medium from VIP-treated cortical glial cultures and assayed by Western blotting, the ADNF-14 AS identified a major 60 kDa immunoreactive signal (Fig. 2A, lane 1). Thus, an ADNF-like protein, recognized by ADNF-14S, is secreted by the monolayers of astrocytes used as substrates for hippocampal neurons in these experiments. Using an affinity-purified antibody against ADNF-9, which specifically recognizes the active epitope of ADNF (Brenneman et al., 1998; Bassan et al., 1999), two additional results were obtained (Fig. 2A, lanes 2, 3): (1) an additional 14 kDa band was observed in conditioned medium from cortical glial cultures, as originally detected in purified ADNF preparations (Brenneman and Gozes, 1996); and (2) the 14 and 60 kDa immunoreactive protein bands were detected even in the absence of VIP stimulation, but the intensity of the signal was greatly enhanced after treatment of astroglial cells with 10−10m VIP. In summary, our Western blotting analyses indicate that detectable levels of the full-size and the truncated form of ADNF are present in the extracellular milieu of cortical glia and are increased dramatically after VIP stimulation.
Fig. 2.

Immunoneutralization of endogenous ADNF from neuron–glia co-cultures impairs the formation of synapses and the sensitivity to glutamate in E18 cultures. The antiserum ADNF-14 AS was raised against a 14-amino acid peptide corresponding to the active site of the ADNF protein. ADNF-14 AS was used to detect the presence of endogenous ADNF as well as to neutralize ADNF production in E18 neuron–glia co-cultures. A, ADNF-14 AS identifies a 60 kDa form of ADNF in conditioned medium derived from cultures of cortical astrocytes after incubation with 0.1 nm VIP for 3 hr at room temperature (lane 1). An affinity-purified antibody raised against ADNF-9 identifies a 60 kDa form and a 14 kDa form of ADNF in the extracellular milieu of both untreated (lane 2) and VIP-treated (lane 3) cortical astroglia. Note the increase of the ADNF signal after VIP stimulation.B, Two weeks of treatment with ADNF-14 AS prevented the formation of synapses in E18 cultures, an effect that can be reversed by co-treatment with 10−8m ADNF-9 or 20 ng/ml NT-3. C, Expression of NT-3 by E18 hippocampal neurons in culture. Cells were grown for 7 d, fixed, and immunostained for the expression of NT-3 and NeuN. Magnification, 63×. The same pattern was obtained after staining three independent cultures. D, Sample records of the response to topical applications of 1 mm glutamate in E18 neurons after 1 week in vitro. Direct glutamate stimulation induces a large inward current in control cells but not in cells treated for 1 week with ADNF-14 AS. Co-treatment of ADNF-14 AS-treated cells with 10−8mADNF-9 or 20 ng/ml NT-3 prevents the loss of sensitivity to glutamate.E, Summary of peak inward current densities generated by direct exposure to 1 mm glutamate in E18 cells. ***p < 0.001, t test. Results are mean ± SEM of 22–45 cells from at least two independent cultures.
ADNF-14 AS was used to neutralize endogenous ADNF in E18 neurons cultured for 14 d at a dilution of 1:1000. The ADNF-14 AS-treated cells failed to develop significant levels of connections (Fig.2B). However, after co-treatment with 10−8m ADNF-9, the proportion of connected cells had increased to 50% (Fig. 2B). These data indicate that endogenous ADNF can efficiently be neutralized by ADNF-14 AS, impairing the development of functional synapses in E18 cultures. We have previously shown that NT-3 and BDNF promote synapse formation in hippocampal neurons (Vicario-Abejón et al., 1998). Immunostaining experiments showed strong NT-3 expression in the vast majority of E18 hippocampal neurons between days 4 and 14 in culture. The image of a culture at day 7 in vitro is shown (Fig. 2C). Interestingly, co-treatment with 20 ng/ml NT-3 prevented the blocking of synaptogenesis by the ADNF-14 AS (Fig. 2B). In contrast, BDNF had no effect in this assay (data not shown). This last result suggests that ADNF and NT-3 share a common regulatory pathway involved in the electrophysiological differentiation of hippocampal neurons.
Not only did ADNF-14 AS-treated cells lose their ability to respond to stimulation of afferent cells, they also lost their ability to respond to direct applications of glutamate. After 1 week in culture, direct stimulation of an E18 neuron with 1 mm glutamate resulted in a large inward current, IGlu (Fig.2D). This response to glutamate was almost completely abolished in E18 cells that had been treated for 1 week with the ADNF-14 AS, unless the cells had been co-treated with 10−8m ADNF-9 or 20 ng/ml NT-3 (Fig.2D,E). This result suggests that the lack of synaptic connections may be secondary to the lack of functional postsynaptic receptors.
Treatment of E18 neurons with the ADNF-14 AS also resulted in dramatic morphological alterations. After 2 weeks in culture, ADNF-14 AS-treated neurons displayed approximately half the dendritic length of untreated neurons (Fig. 3). The ADNF-14 AS treatment did not have any significant effects on cell survival or on the number of dendrites (Fig. 3B). Interestingly, these morphological alterations were observed only when the ADNF-14 AS was added from the day of plating and were maintained only as long as the ADNF-14 AS was present in the culture medium. Addition of the ADNF-14 AS 3 d after plating did not alter the morphology of the cells significantly (Fig. 3B). When the ADNF-14 AS was added from days 1 to 7 and then withdrawn, E18 neurons at 14 d displayed the same morphological characteristics as the untreated control cells (Fig.3B), indicating the reversibility of the ADNF-14 AS block. These results suggest a narrow developmental time window when ADNF is needed for neuronal development.
Fig. 3.

Treatment of E18 cultures with anti-ADNF antiserum results in severe morphological alterations in developing neurons that can be prevented by co-treatment with ADNF-9 and NT-3.A, Samples of E18 hippocampal neuron morphologies. Cells were grown for 14 d in vitro(DIV) in the presence or absence of ADNF-14 AS, ADNF-9, or NT-3. Camera lucida drawings of dendrites and cell bodies were done on MAP2ab-positive neurons using a 63× objective. Drawings were digitally scanned and processed using Adobe Photoshop. ADNF-14 AS-treated neurons did not reach the size and morphological complexity of untreated neurons. ADNF-14 AS-treated cells co-treated with 10−8m ADNF-9 or 20 ng/ml NT-3 developed morphologies similar to the ones found in untreated cells. B, Effect of ADNF-14 AS treatment on numbers and morphology of neurons in E18 hippocampal cultures. ADNF-14 AS treatment during DIV 1–14 resulted in a marked reduction of the dendritic length but had no effect on the number of cells per field. ADNF-14 AS treatment started after DIV 3 (DIV 3–14) or restricted to the first week in culture (DIV 1–7) resulted in normal dendritic morphologies. Co-treatment with either ADNF-9 or NT-3 prevented the morphological changes observed in ADNF-14 AS-treated cells. Results are the mean ± SEM of data from 10 different fields in a single culture experiment and are representative of three such experiments. ***p < 0.001, t test.
Another consequence of the ADNF-14 AS treatment was a sharp decrease in the density of synaptic structures, as measured by immunostaining of the synaptic protein synaptophysin (Fig.4). Treatment of E18 cultures with ADNF-14 AS reduced the number of synaptophysin puncta per unit of dendritic length (Fig. 4E). Here again the effects of the ADNF-14 AS on the density of synaptic structures could be reversed by co-treatment with either 10−8m ADNF-9 or 20 ng/ml NT-3 (Fig. 4).
Fig. 4.

ADNF-14 AS treatment affects the density of synaptic structures in E18 neurons. E18 neurons were grown for 14 d in the presence or absence of ADNF-14 AS, ADNF-9, and NT-3. Cells were then fixed and immunostained for the expression of the synaptic vesicle protein synaptophysin (A–D). Scale bar, 10 μm. A, Untreated cells displayed a dense pattern of synaptophysin staining. B, In ADNF-14 AS-treated cells, only sparse synaptophysin puncta can be detected. Co-treatment of ADNF-14 AS-treated cultures with either ADNF-9 (C) or NT-3 (D) resulted in a normal synaptophysin expression pattern. The effect of ADNF-14 AS on the density of synaptic structures could not be attributed solely to a lower number of dendritic arborizations, because the number of puncta per micrometer was also reduced (E). Results are the mean ± SEM of data from four different fields in a single culture experiment and are representative of three such experiments. *p < 0.05; **p < 0.01; ***p < 0.001, t test.
These results indicate that the effect of the ADNF-14 AS treatment can result in severe alterations in the development of E18 neurons. The observed effects cannot be attributed to cell toxicity, because the ADNF-14 AS did not have any significant impact on cell numbers, and withdrawal of the antiserum after 1 week of treatment led to a complete morphological recovery. The morphological development of ADNF-14 AS-treated E18 neurons was arrested only when the antiserum was used immediately after plating, suggesting that endogenous ADNF induction of glutamate responses is essential for an early step in neuronal differentiation.
ADNF acts directly on neurons
“Glia-free” E16 and E18 neuronal cultures were used to test the hypothesis that ADNF acts directly on neurons. Instead of being grown on monolayers of astrocytes, neurons were plated directly on glass (Fig. 5A). Less than 2% of the cells in these cultures were GFAP-positive. E16 neurons were cultured for 2 weeks under these conditions and assayed using patch-clamp analysis. Synaptic contact was monitored in these cells by recording miniature postsynaptic currents (mPSCs) induced by high osmotic stimulation as previously described (Malgaroli and Tsien, 1992;Manabe et al., 1992; Araque et al., 1998b). In untreated E16 cultures, sucrose failed to induce significant miniature events (Fig.5B). In contrast, neurons treated for 2 weeks with 10−9m ADNF-9 responded to sucrose stimulation with large and frequent mPSCs (Fig. 5B). NT-3 at 20 ng/ml also increased the frequency of mPSCs, suggesting that both ADNF-9 and NT-3 can increase the probability of synaptic vesicle fusion by acting directly on E16 neurons. Direct stimulation of untreated E16 cultures with 1 mm glutamate resulted in large postsynaptic responses, but responses to direct application of 1 mm glutamate were significantly higher in cells treated with 10−9m ADNF or 20 ng/ml NT-3, indicating a clear increase in the sensitivity to glutamate in the treated neurons (data not shown).
Fig. 5.
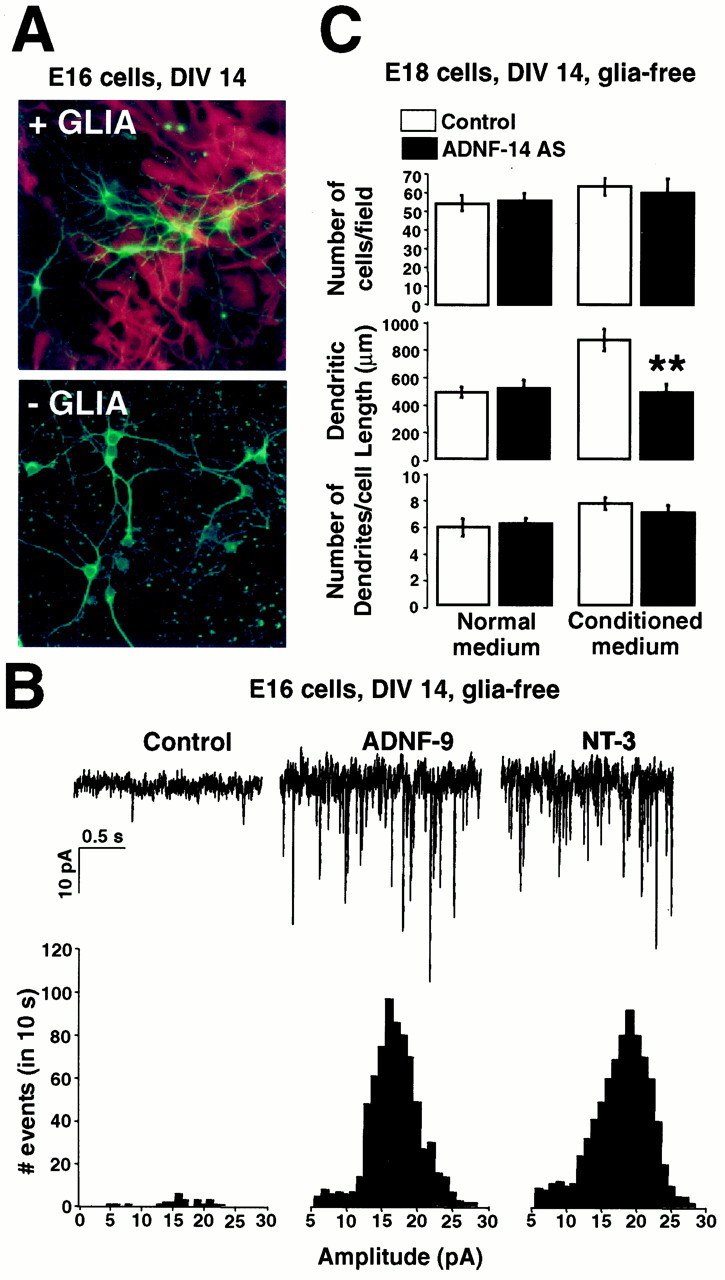
Physiological and morphological effects of exogenous and endogenous ADNF on glia-free neuronal cultures. To produce glia-free neuronal cultures, cells derived from hippocampus at E16 (A, B) and E18 (C) were plated on coated glass coverslips and grown for 2 weeks in the presence of the antimitotic agent floxuridine to prevent astrocytic growth.A, Neurons grown on glial monolayers or directly on glass were stained with the neuronal marker MAP2ab (green) and the glial marker GFAP (red). No GFAP staining was detected in glia-free cultures. B, Top trace, Spontaneous activity recorded from E16 glia-free cultures either untreated or treated for 2 weeks with 10−8m ADNF-9 or 20 ng/ml NT-3. Bottom histogram, Distribution of the frequency of mPSCs amplitudes. The frequency of the mPSCs was markedly enhanced in cultures treated with ADNF-9 or NT-3 compared with untreated cultures. C, Effect of ADNF-14 AS treatment on numbers and morphology of neurons in E18 glia-free cultures. Cultures were grown for 2 weeks in glia-derived conditioned medium (Conditioned medium) or fresh medium (Normal medium). CM markedly induced dendritic growth, an effect that was blocked by co-treatment with ADNF-14 AS. Treatment with ADNF-14 AS did not have any influence on the morphology of neurons grown in the absence of CM. The number of cells was comparable in all culture conditions. Results are the mean ± SEM of data from 10 different fields in a single culture experiment and are representative of three such experiments. **p < 0.01, t test. DIV, Days in vitro.
To test whether endogenous ADNF from astroglia promotes morphological differentiation, we exposed glia-deprived cultures of E18 neurons to conditioned medium (CM) harvested from monolayers of astrocytes, in the presence or absence of the ADNF-14 AS. E18 neurons cultured in glia-derived CM showed a 50% increase in total dendritic length when compared with cells that were not exposed to glia-derived signals (Fig. 5C). ADNF-14 AS added to the CM abolished this effect of CM (Fig. 5C). ADNF-14 AS treatment did not have any significant effect on the morphology of E18 cells cultured without CM. These results demonstrate that ADNF is present in the medium derived from glial but not neuronal cultures. The entire effect of CM on the morphological development of E18 neurons can be blocked by ADNF-14 AS. The dendritic length is similar in these cultures and in neurons that were grown in direct contact with astrocytes, suggesting that ADNF acts directly on neurons and is the major factor secreted by glia influencing neuronal morphology.
ADNF-9 regulates NT-3 secretion
Because a receptor for ADNF has yet to be characterized, the type of cytoplasmic signal that mediates its physiological effects is still unknown. The fact that exogenous applications of NT-3 allow developing neurons to overcome the effects of the ADNF-14 AS suggests the existence of a common regulatory pathway between ADNF and NT-3. The hypothesis that ADNF-9 influences the secretion of NT-3 was therefore tested. NT-3 release was measured in “immature” (E16, 5 din vitro) and “mature” (E18, 2 weeks in vitro) neuronal cultures using a colorimetric immunoassay system. Maximal release of NT-3 can be obtained in E16 (Fig.6A) and E18 (Fig.6B) cultures by exposing the cells to depolarizing levels (55 mm) of KCl. Short exposure (15 min) to ADNF-9 (10−13–10−5m) induced a significant release of NT-3 in both cultures. No release of NT-3 was observed when the cultures were exposed to a control peptide, characterized by an amino acid composition identical to ADNF-9 but a random sequence arrangement (LASLAPISR).
Fig. 6.

ADNF-9 regulates the release but not the expression of NT-3. A, B, Release of NT-3 in the extracellular milieu of neuron–glia co-cultures was assayed using a colorimetric immunoassay. E16 cells were assayed after 5 din vitro (DIV; A), and E18 cells were assayed after 2 weeks in vitro(B). High KCl concentrations (55 mm) were used to induce maximum NT-3 release. In both E16 and E18 cultures, ADNF-9 (15 min) induced release of NT-3 in a dose-dependent manner. The maximal releasing effect was obtained for an ADNF-9 concentration of 10−7m. A control peptide (SCRAMBLED) with a size and amino acid composition identical to ADNF-9 but a different amino acid sequence failed to induce any significant release of NT-3. C, Expression levels of NT-3 were assayed in E16 cultures after 3 and 7 d in vitro (DIV 3, DIV 7) using a semiquantitative reverse transcription-PCR method. The cells were exposed to 10−9m ADNF-9 for various times (12, 24, and 48 hr). Amplification of ribosomal RNA 18S was used as control. No significant effect of the ADNF-9 treatment on NT-3 expression was detected. Results are the mean ± SEM of data from three different measurements in a single culture experiment and are representative of three such experiments. ***p< 0.001, t test.
The ADNF-9 effect on NT-3 secretion is an immediate effect and may not require long-term changes in gene expression. Indeed, treatment of E16 neurons with 10−9m ADNF-9 for various periods (12–48 hr) and at different stages of maturation (3 and 7 d in vitro) did not influence NT-3 expression as assayed by a semiquantitative PCR analysis (Fig. 6C). These results demonstrate that ADNF-9 can efficiently trigger the release of NT-3 in cultures of neurons that have yet to develop functional synapses (early E16) as well as in spontaneously active and synaptically connected cells (late E18). NT-3 is therefore likely to mediate, at least in part, the observed effects of ADNF.
ADNF-9 and NT-3 regulate NR2A and NR2B levels in cultures of hippocampal neurons
The regulation of synaptogenesis by ADNF must involve either the expression or the function of glutamate receptors, because the ADNF-14 AS-treated cells are virtually insensitive to direct applications of glutamate. We therefore decided to study the expression patterns of the NMDAr subunits found in hippocampal neurons at the time of synapse formation (NR1, NR2A and NR2B; Okabe et al., 1998; Tovar and Westbrook, 1999). Treatment of E18 cultures for 1 week with ADNF-14 AS did not significantly change the protein levels of the NMDAr subunit NR1 as measured by Western blot (Fig.7A), but levels of the NR2A and NR2B subunits were sharply lower in the ADNF-14 AS-treated cells when compared with control cells. Co-treatment of E18 cultures with ADNF-14 AS and either 10−8m ADNF-9 or 20 ng/ml NT-3 prevented the drop in NR2A and NR2B protein expression.
Fig. 7.

Expression of NMDA receptor subtypes by hippocampal neurons in culture. A, B, Proteins from E18 (A) and E16 (B) hippocampal cultures were analyzed by immunoblotting. The amount of each receptor subunit was determined using antibodies specific to each subunit. In E18 neurons after 2 weeks in culture (A), ADNF-14 AS treatment resulted in a marked decrease in the expression of the NR2A and NR2B regulatory subunits when compared with control cells. This effect of ADNF-14 AS could be prevented by co-treatment with 10−8m ADNF-9 or 20 ng/ml NT-3. In E16 neurons after 1 week in culture (B), treatment with 10−9m ADNF-9 or 20 ng/ml NT-3 resulted in a marked increase in NR2A and NR2B expression.C, Expression levels of NT-3 were assayed in E16 cultures after 1 week in culture using a semiquantitative reverse transcription-PCR method. Treatments for 48 hr with 10−9m ADNF-9 or 20 ng/ml NT-3 failed to induce any significant changes in NR2A or NR2B mRNA levels. Amplification of ribosomal RNA 18S was used as control.
In E16 cells after 1 week in vitro, the expression of NR2A and NR2B subunits was barely detectable by immunoblotting (Fig.7B), but treatment with either 10−9m ADNF-9 or 20 ng/ml NT-3 resulted in NR2A and NR2B protein levels comparable with those found in E18 cells after 1 week in culture. This inductive effect of ADNF and NT-3 on NR2A and NR2B protein levels was not associated with any significant increase in NR2A and NR2B mRNA levels, as assayed by quantitative PCR (Fig.7C). Thus, ADNF and NT-3 exert a post-transcriptional control over the stability of the NR2A and NR2B subunits.
DISCUSSION
The initial goal of this study was to determine whether VIP-stimulated ADNF release regulates differentiation in hippocampal neurons. An assay for evoked synaptic responses was used to show that VIP treatment promotes and anti-VIP antiserum inhibits synaptic responses. Because VIP stimulates ADNF release, the effect of VIP may be mediated by ADNF. This possibility was confirmed by showing that an ADNF-derived peptide, ADNF-9, overcomes the effect of the anti-VIP antiserum.
Exposure of E16 hippocampal neurons to ADNF-9 (10−12–10−6m) increases the number of connections in a dose-dependent manner. The observed drop in biological response to ADNF-9 between 10−9 and 10−6m is attributed to a modulatory effect of non-neuronal cells. Indeed, ADNF-9 has been shown to exhibit attenuated pharmacological responses at high concentration in mixed neuronal–glial cultures but not in glia-depleted neuronal cultures (Brenneman et al., 1998). It is hypothesized that ADNF-9 interacts with non-neuronal cells to produce modulatory substance(s) that attenuate the biological response at high concentrations of the peptide.
Anti-ADNF antiserum ADNF-14 AS blocks synaptic responses and inhibits morphological development. These effects can be reversed by either ADNF-9 or NT-3. Morphological and electrophysiological measures suggest that ADNF and NT3 act directly on neurons. These results suggest that the release of VIP, ADNF, and NT-3 forms a signal pathway linking the differentiation of hippocampal neurons and glia.
We report three observations showing that response to and release of glutamate is the target of this pathway. First, cells grown on glia and treated with ADNF-14 AS show poor responses to glutamate that are restored by ADNF-9 and NT-3. Second, ADNF-9 and NT-3 control the sensitivity to direct glutamate stimulation, as well as the expression or stability of the NR2A and NR2B subunits of the NMDA receptor in neurons that have not yet established efficient synaptic connections. These findings suggest that there are postsynaptic consequences of ADNF-9 and NT-3 treatment. However, the third observation, that the frequency of mPSCs is increased in neurons treated with ADNF-9 or NT-3, suggests that the presynaptic site is also a target of this signaling system. Neurotrophins are known to increase the frequency of mEPSCs in dissociated hippocampal glutamatergic neurons (Vicario-Abejón et al., 1998; Schinder et al., 2000) and to promote excitatory synaptic transmission in hippocampal slices (Kang and Schuman, 1995; Patterson et al., 1996; Kafitz et al., 1999). Our results suggest that glial signals influence neurotrophin actions and result in both presynaptic and postsynaptic effects on glutamate responses. A simple model that accommodates these observations is proposed: VIP stimulates ADNF release by astrocytes. ADNF acts postsynaptically to cause the stabilization of the NR2A and NR2B subunits of the NMDA receptor as well as NT-3 release. The effects of ADNF on the frequency of vesicle fusion may be caused by the presynaptic action of NT-3. This model is still preliminary but draws attention to the central result of this manuscript that ADNF forms a link between VIP and NT-3 in a signal cascade that establishes the close interaction between neurons and astrocytes from the earliest stages of development.
The mechanisms induced by ADNF and NT-3 to control NMDA receptor stability are not currently explained by our experiments. The expression of NR2 proteins changes through hippocampal development, with the levels of NR2A and NR2B rising at the time when synapses are formed (Monyer et al., 1994; Okabe et al., 1998). The NR2 subunits carry long cytoplasmic domains that interact with proteins in the postsynaptic complex (Kennedy, 1997; Sheng and Pak, 1999). The NR2B subunit of the NMDA receptor predominates in the first fast synaptic transmission in dissociated hippocampal neurons (Tovar and Westbrook, 1999), and it seems likely that the stabilization of the NR2 subunits is an early step in the maturation of the postsynaptic site. Activation of glial cells has been shown to stimulate presynaptic responses in neurons (Araque et al., 1999). The model proposed here suggests that interactions between neurons and astrocytes mediated by VIP, ADNF, and NT-3 play an important role in the early stages of synaptic development.
The VIP–ADNF–NT-3 pathway has a striking effect on presynaptic (synapsin-positive puncta) and postsynaptic (dendritic length) morphology. In the absence of these signals the immature neurons develop the correct number of dendrites but then arrest, suggesting that the initial events in dendrite formation are controlled by a distinct mechanism. Hippocampal glutamatergic neurons can elaborate a complex dendritic morphology in the absence of fast synaptic transmission (Vicario-Abejón et al., 1998). These studies on dissociated hippocampal neurons are consistent with in vivoobservations showing that the basic organization of the neuropil does not require action potentials or evoked synaptic responses (Harris, 1984; Verhage et al., 2000). The interactions between neurons and glial cells reported here may play a role in establishing the initial “activity-independent” axonodendritic organization of the nervous system.
The stimulation of ADNF release by VIP has been described as a paracrine system protecting neurons against a wide range of toxic events (Gozes et al., 1997). Several properties of VIP have also suggested a control over neuronal development and synaptic plasticity in the adult. VIP-containing neurons are widely distributed in the cortex and the hippocampus (Magistretti and Morrison, 1988; Baldino et al., 1989). VIP synthesis is developmentally regulated, with most synthesis occurring postnatally and reaching a peak at the time of synapse formation (Gozes et al., 1987). In our embryonic hippocampal cultures, VIP is produced by virtually all the neurons during the first 2 weeks in vitro. In vivo, VIP production can be induced in most neurons of the adult rat hippocampus by running (Eilam et al., 1999), an activity that also results in increased firing patterns in this region of the brain (Czurkó et al., 1999) and improved learning performances (Fordyce and Wehner, 1993). Furthermore, physical activity (van Praag et al., 1999) as well as treatment with NT-3 (Kang and Schuman, 1995) or the VIP-related pituitary adenylate cyclase-activating polypeptide (Kondo et al., 1997) induce long-term potentiation in the dentate gyrus. Blockade of VIP function during early embryogenesis and the neonatal period causes CNS abnormalities ranging from microcephaly (Gressens et al., 1994) to impairment of neuronal morphology (Hill et al., 1994) and neurobehavioral development (Wu et al., 1997). VIP blockade in the adult rat brain (Glowa et al., 1992) or constitutive expression of a chimeric VIP gene in mice (Gozes et al., 1993) greatly impairs learning behaviors. Our data extend these results by suggesting that the action of VIP on brain development and function may be mediated in part by the actions of ADNF and NT-3 on presynaptic and postsynaptic parameters of glutamatergic neurons. Although this study is focused on the earliest stages of neuronal differentiation, the VIP–ADNF–NT-3 pathway linking neuronal activity and glial responses may also contribute to synaptic plasticity in the mature CNS.
Footnotes
This work was supported, in part by the US–Israel Binational Science Foundation. I.G. is the incumbent of the Avraham Gildor Chair for the Investigations of Growth Factors.
Correspondence should be addressed to Dr. Ronald D. G. McKay, National Institutes of Health, National Institute of Neurological Disorders and Stroke, Laboratory of Molecular Biology, Building 36, Room 3D02, Bethesda, MD 20892-4092. E-mail:mckay@codon.nih.gov.
REFERENCES
- 1.Aghajanian GK, Bloom FE. The formation of synaptic junctions in developing rat brain: a quantitative electron microscopic study. Brain Res. 1967;6:716–727. doi: 10.1016/0006-8993(67)90128-x. [DOI] [PubMed] [Google Scholar]
- 2.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998a;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- 3.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998b;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 5.Baldino F, Fitzpatrick-McElligott S, Gozes I, Card JP. Localization of VIP and PHI-27 messenger RNA in rat thalamic and cortical neurons. J Mol Neurosci. 1989;1:199–207. [PubMed] [Google Scholar]
- 6.Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blat C, Gibney G, Glazner G, Brenneman DE, Gozes I. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999;72:1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x. [DOI] [PubMed] [Google Scholar]
- 7.Brenneman DE, Gozes IJ. A femtomolar-acting neuroprotective peptide. J Clin Invest. 1996;97:2299–2307. doi: 10.1172/JCI118672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenneman DE, Hauser J, Neale E, Rubinraut S, Fridkin M, Davidson A, Gozes I. Activity-dependent neurotrophic factor: structure-activity relationships of femtomolar-acting peptides. J Pharmacol Exp Ther. 1998;285:619–627. [PubMed] [Google Scholar]
- 9.Brenneman DE, Hauser J, Spong CY, Phillips TM, Pert CB, Ruff M. VIP and d-ala-peptide T-amide release chemokines which prevent HIV-1 GP120-induced neuronal death. Brain Res. 1999a;838:27–36. doi: 10.1016/s0006-8993(99)01644-3. [DOI] [PubMed] [Google Scholar]
- 10.Brenneman DE, Hauser J, Philips TM, Davidson A, Bassan M, Gozes I. Vasoactive intestinal peptide. Link between electrical activity and glia-mediated neurotrophism. Ann NY Acad Sci. 1999b;897:17–26. doi: 10.1111/j.1749-6632.1999.tb07875.x. [DOI] [PubMed] [Google Scholar]
- 11.Carrington WA, Fogarty KE, Fay FS. 3D fluorescence imaging of single cells using image restoration. In: Foster K, Grinstein S, editors. Noninvasive techniques in cell biology. Wiley-Liss; New York: 1990. pp. 53–72. [Google Scholar]
- 12.Czurkó A, Hirase H, Csicsvari J, Buzsáki G. Sustained activation of hippocampal pyramidal cells by space clamping in a running wheel. Eur J Neurosci. 1999;11:344–352. doi: 10.1046/j.1460-9568.1999.00446.x. [DOI] [PubMed] [Google Scholar]
- 13.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 14.Eilam R, Davidson A, Gozes I, Segal M. Locomotor activity causes a rapid up-regulation of vasoactive intestinal peptide in rat hippocampus. Hippocampus. 1999;9:534–541. doi: 10.1002/(SICI)1098-1063(1999)9:5<534::AID-HIPO7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Fordyce DE, Wehner JM. Physical activity enhances spatial learning performance with an associated alteration in hippocampal protein kinase C activity in C57BL/6 and DBA/2 mice. Brain Res. 1993;619:111–119. doi: 10.1016/0006-8993(93)91602-o. [DOI] [PubMed] [Google Scholar]
- 16.Glowa JR, Panlilio LV, Brenneman DE, Gozes I, Fridkin M, Hill JM. Learning impairment following intracerebral administration of the HIV envelope protein gp120 or a VIP antagonist. Brain Res. 1992;570:49–53. doi: 10.1016/0006-8993(92)90562-n. [DOI] [PubMed] [Google Scholar]
- 17.Gozes I, Shani Y, Rostene WH. Developmental expression of the VIP-gene in brain and intestine. Mol Brain Res. 1987;2:137–148. doi: 10.1016/s0006-8993(87)80007-0. [DOI] [PubMed] [Google Scholar]
- 18.Gozes I, Glowa J, Brenneman DE, McCune SK, Lee E, Westphal H. Learning and sexual deficiencies in transgenic mice carrying a chimeric vasoactive intestinal peptide gene. J Mol Neurosci. 1993;4:185–193. doi: 10.1007/BF02782501. [DOI] [PubMed] [Google Scholar]
- 19.Gozes I, Davidson A, Gozes Y, Masculo R, Barth R, Warren D, Hauser J, Brenneman DE. Antiserum to activity-dependent neurotrophic factor produces neuronal cell death in CNS cultures: immunological and biological specificity. Dev Brain Res. 1997;99:167–175. doi: 10.1016/s0165-3806(96)00215-5. [DOI] [PubMed] [Google Scholar]
- 20.Gressens P, Hill JM, Paindaveine B, Gozes I, Fridkin M, Brenneman DE. Severe microcephaly induced by blockade of vasoactive intestinal peptide function in the primitive neuroepithelium of the mouse. J Clin Invest. 1994;94:2020–2027. doi: 10.1172/JCI117555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris WA. Axonal pathfinding in the absence of normal pathways and impulse activity. J Neurosci. 1984;4:1153–1162. doi: 10.1523/JNEUROSCI.04-04-01153.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill JM, Mervis RF, Politi J, McCune SK, Gozes I, Fridkin M, Brenneman DE. Blockade of VIP during neonatal development induces neuronal damage and increases VIP and VIP receptors in the brain. Ann NY Acad Sci. 1994;739:211–225. doi: 10.1111/j.1749-6632.1994.tb19823.x. [DOI] [PubMed] [Google Scholar]
- 23.Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- 24.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy MB. The postsynaptic density at glutamatergic synapses. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- 26.Kondo T, Tominaga T, Ichikawa M, Iijima T. Differential alteration of hippocampal synaptic strength induced by pituitary adenylate cyclase activating polypeptide-38 (PACAP-38). Neurosci Lett. 1997;221:189–192. doi: 10.1016/s0304-3940(96)13323-1. [DOI] [PubMed] [Google Scholar]
- 27.Magistretti PJ, Morrison JH. Noradrenaline- and vasoactive intestinal peptide-containing neuronal systems in neocortex: functional convergence with contrasting morphology. Neuroscience. 1988;24:367–378. doi: 10.1016/0306-4522(88)90338-7. [DOI] [PubMed] [Google Scholar]
- 28.Malgaroli A, Tsien RW. Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature. 1992;357:134–139. doi: 10.1038/357134a0. [DOI] [PubMed] [Google Scholar]
- 29.Manabe T, Renner P, Nicoll RA. Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature. 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- 30.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signalling and gp120 neurotoxicity. Proc Natl Acad Sci USA. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monyer H, Burnashev N, Laurie D, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 32.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 33.Okabe S, Collin C, Auerbach JM, Meiri N, Bengzon J, Kennedy MB, Segal M, McKay RDG. Hippocampal synaptic plasticity in mice overexpressing an embryonic subunit of the NMDA receptor. J Neurosci. 1998;18:4177–4188. doi: 10.1523/JNEUROSCI.18-11-04177.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parnavelas JG, Luder R, Pollard SG, Sullivan K, Libermam AR. A qualitative and quantitative ultrastructural study of glial cells in the developing visual cortex of the rat. Philos Trans R Soc Lond B Biol Sci. 1983;301:55–84. doi: 10.1098/rstb.1983.0022. [DOI] [PubMed] [Google Scholar]
- 35.Parpura V, Basarsky TA, Liu F, Jeftinja S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 36.Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 37.Peters A, Palay SL, Webster HF. The fine Structure of the nervous system: the neurons and supporting cells, Ed 3. Oxford UP; Oxford: 1991. [Google Scholar]
- 38.Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 39.Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schinder AF, Berninger B, Poo M-M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- 41.Sheng M, Pak DT. Glutamate receptor anchoring proteins and the molecular organization of excitatory synapses. Ann NY Acad Sci. 1999;868:483–493. doi: 10.1111/j.1749-6632.1999.tb11317.x. [DOI] [PubMed] [Google Scholar]
- 42.Song HJ, Ming JL, Poo M. M. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–279. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- 43.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA. 1999;96:13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 47.Vicario-Abejón C, Collin C, McKay RDG, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vicario-Abejón C, Collin C, Tsouflas P, McKay RDG. Hippocampal stem cells differentiate into excitatory and inhibitory neurons. Eur J Neurosci. 2000;12:677–688. doi: 10.1046/j.1460-9568.2000.00953.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu JY, Henins KA, Gressens P, Gozes I, Fridkin M, Brenneman DE, Hill JM. Neurobehavioral development of neonatal mice following blockade of VIP during the early embryonic period. Peptides. 1997;18:1131–1137. doi: 10.1016/s0196-9781(97)00146-0. [DOI] [PubMed] [Google Scholar]