

Abstract
The onset and duration of sleep are thought to be primarily under the control of a homeostatic mechanism affected by previous periods of wake and sleep and a circadian timing mechanism that partitions wake and sleep into different portions of the day and night. The mouseClock mutation induces pronounced changes in overall circadian organization. We sought to determine whether this genetic disruption of circadian timing would affect sleep homeostasis. TheClock mutation affected a number of sleep parameters during entrainment to a 12 hr light/dark (LD 12:12) cycle, when animals were free-running in constant darkness (DD), and during recovery from 6 hr of sleep deprivation in LD 12:12. In particular, in LD 12:12, heterozygous and homozygous Clock mutants slept, respectively, ∼1 and ∼2 hr less than wild-type mice, and they had 25 and 51% smaller increases in rapid eye movement (REM) sleep during 24 hr recovery, respectively, than wild-type mice. The effects of the mutation on sleep are not readily attributable to differential entrainment to LD 12:12 because the baseline sleep differences between genotypes were also present when animals were free-running in DD. These results indicate that genetic alterations of the circadian clock system and/or its regulatory genes are likely to have widespread effects on a variety of sleep and wake parameters, including the homeostatic regulation of sleep.
Keywords: circadian, sleep, Clock mutation, gene, REM, NREM delta power, sleep homeostasis
Sleep regulation has been modeled as a two-process system consisting of a homeostatic process and a circadian timing process, which together determine the propensity, length, and incidence of episodes and intensity of sleep (Borbely, 1982). The homeostatic process is manifest in the dependence of sleep on wake, with a greater propensity for sleep after a longer wake time. The circadian control of sleep is manifest in the consolidation of periods of wake and sleep preferentially to specific phases of the light/dark (LD) cycle. More to the point, consolidation continues to occur on a near 24 hr basis when the environment is devoid of any time cues (Czeisler et al., 1980; Zulley et al., 1981). Furthermore, altering the circadian phase at which sleep occurs, as in temporal sleep displacement (Akerstedt and Gillberg, 1981) or a forced desynchrony protocol (Dijk and Czeisler, 1994), has also demonstrated clear circadian variation in sleep propensity and sleep architecture.
At the present time, it is not clear whether the circadian and the homeostatic processes are independent or whether they are interconnected with one another at the cellular and/or systems levels. Lack of knowledge about the relationship of these two processes to one another is in part attributable to the limited number of approaches that have been used to examine this relationship. The most widely used approach has been to lesion the master circadian pacemaker, the hypothalamic suprachiasmatic nuclei (SCN), and then to examine the effects of such lesions on the homeostatic process. Such an approach has yielded conflicting results in the two species that have been examined, the rat and the squirrel monkey (Mouret et al., 1978;Mistlberger et al., 1983, 1987; Tobler et al., 1983; Eastman et al., 1984; Borbely et al., 1989; Edgar et al., 1993; Klerman et al., 1999).
An alternative approach to lesion studies for determining the effects of circadian rhythmicity on the overall time spent in sleep and wake is to examine sleep characteristics in animals that have a genetically altered circadian clock system. Such an approach is now possible in mice. Clock, the first mammalian circadian gene identified, was found via a mutagenesis phenotypic screen (Vitaterna et al., 1994) and subsequently cloned (Antoch et al., 1997; King et al., 1997). TheClock mutation has effects on numerous aspects of circadian rhythmicity, including a lengthened and less stable circadian period in heterozygotes and homozygotes and loss of rhythmicity in constant conditions in homozygotes (Vitaterna et al., 1994). Although less stable, both heterozygous and homozygous Clock mutant animals remain entrained with normal phase to a 24 hr LD cycle (Vitaterna et al., 1994). To determine whether the Clockmutation affects the homeostatic regulation of sleep, we have compared sleep and electroencephalographic (EEG) activity in mice heterozygous (Clock/+) or homozygous (Clock/Clock) for theClock mutation with wild-type (+/+) animals under entrained baseline and recovery from a short period of sleep deprivation, as well as in free-running Clock/Clock and +/+ mice.
MATERIALS AND METHODS
All animals used in this experiment were coisogenic C57BL/6J male mice between 3 and 5 months of age born and maintained in the Association for Assessment and Accreditation of Laboratory Animal Care accredited Center for Experimental Animal Resources at Northwestern University. Different groups of animals were used for the experiments conducted under entrained and free-running conditions. For the entrained experiments, six homozygous Clock mutant mice, nine heterozygous Clock mice, and six wild-type mice were recorded. Mice were entrained to a 12 hr light/dark (LD 12:12) cycle with lights on at 5:00 A.M. and lights off at 5:00 P.M. For the free-running experiments, six Clock homozygotes and six wild types were maintained in constant darkness (DD) after implant surgery. All Clock genotypes were determined by PCR amplification of genomic DNA extracted from tail tip biopsies as described previously (Herzog et al., 1998). Food and water were available to all animals ad libitum. All procedures were approved in advance by the Animal Care and Use Committee of Northwestern University.
Activity recording. Activity was monitored using infrared (IR) motion sensors (A1 Securing and Electrical Ltd., Huyton Merseyside, UK) located directly above each cage. Activity patterns were recorded and analyzed with the Chronobiology Kit (Stanford Software Systems, Stanford, CA). IR activity monitoring was used to ensure entrainment to the light/dark cycle, monitor recovery from surgery, and provide a phenotypic measure of activity patterns for individual mice in DD.
Recording of sleep. Mice were anesthetized using methoxyflurane (Pittman-Moore Laboratories) inhalant anesthetic and implanted with EEG and electromyographic (EMG) electrodes for polysomnographic recording. For monitoring EEG signals, stainless steel recording screws (model 000–120; Small Parts Inc., Logansport, IN) were positioned 1 mm anterior to bregma, 0.5 mm right of the central suture, and contralaterally at 0.5 mm posterior to lambda and 1 mm left of the central suture. EMG activity was monitored using stainless steel, Teflon-coated wires bilaterally placed into both trapezius muscles. All electrodes were fastened to a 1 × 4 pin grid array, and the entire head implant was attached to the skull using cyanoacrylate. Recovery from surgery was considered complete when animals returned to presurgery activity levels, typically within 6 d.
In the LD experiment, mice were placed in a sleep-recording chamber and connected to a lightweight rotating tether that permitted free movement throughout the cage. Except for the recording tether, conditions in the recording chamber were identical to those in the home cage. Mice were allowed a 48 hr acclimation period to adjust to the tether. Baseline EEG and EMG waveforms were then collected continuously for 24 hr in each animal beginning at light onset. During the final 6 hr of the light period after the completion of baseline, mice were sleep deprived by placing them on 3.5 cm diameter platforms surrounded by room temperature water, 2 cm deep. EEG and EMG signals were observed during this period to try to detect and disrupt (by opening the recording chamber and gently touching the animal) any episodes of sleep. At the end of the sleep deprivation period (dark onset), mice were placed back in their home cages and allowed to sleep ad libitum. Polysomnographic data were then collected continuously for another 24 hr to monitor recovery sleep.
Procedures during the DD experiment were similar. All mice were placed in constant darkness immediately after surgery. This allowed 7–10 d of free-running activity (long enough to estimate free-running period) before sleep data were collected. EEG and EMG signals were recorded over one complete circadian cycle (activity onset–activity onset).
EEG signals were amplified ∼25,000×, with −6 dB/oct high-pass and low-pass filters set at 0.1 and 30 Hz (3 dB), respectively. EMG signals were amplified 50,000× and low-pass filtered at 100 Hz. Both signals were then digitized at 100 Hz/channel by an analog-to-digital converter (model DT-01EZ; Data Translation Inc., Marlboro, MA) and stored on an IBM AT-compatible computer. Waveforms were collected using ACQ, a software system designed specifically for gathering and analyzing rodent sleep data (Benington and Heller, 1994; Benington et al., 1994).
Scoring and analysis of sleep. After collection, all waveforms were classified independently by two sleep scorers (one blind to genotype, one not) into 10 sec epochs of either wake (low-voltage, high-frequency EEG; high-amplitude EMG), non-rapid eye movement (NREM) sleep (high-voltage, mixed-frequency EEG; low-amplitude EMG) or REM sleep (EEG with a predominance of theta activity; very low-amplitude EMG). Epochs that were unscorable because of electrical noise were considered artifact (<0.06% of epochs) and excluded from further analysis. Scorers agreed 95% on sleep versus wake epochs and 85% on NREM versus REM. Epochs with discrepant scores were reexamined by both scorers together to produce consensus scores. There were no significant differences between genotypes in a number of discrepant epochs (one-way ANOVA; F(2,18) = 0.53;p = 0.60).
Episodes of sleep or wake were defined as requiring six consecutive epochs for onset. Other aspects of sleep architecture, including number, duration, and continuity of wake, sleep, REM, and NREM episodes were also defined in terms of numbers of consecutive epochs in various stages as described previously (Naylor et al., 1998). Artifact episodes of <1 min were treated as a continuation of the previous wake–sleep episode. Artifact episodes 1 min or longer (five episodes among threeClock/Clock and six among three +/+ mice) caused any wake–sleep episodes begun before the artifact to be removed from analysis.
EEG power was calculated on EEG epochs classified as either NREM or REM sleep using a Fast Fourier Transform (FFT) from the MATLAB development platform. FFTs were calculated in 2 sec windows synchronized with the 10 sec scoring epochs. A 50 μV, 5 Hz calibration signal recorded for each mouse was used to calculate calibrated values for power density in the delta (0.5–5 Hz), theta (6–10 Hz), and sigma (11–15 Hz) bands during NREM and REM sleep.
Statistical analyses. As has been reported in other rodent studies (Trachsel et al., 1988; Naylor et al., 1998), the within-group variability for EEG delta power was high. This problem is particularly acute in mice, whose small skull size makes electrode placement difficult to replicate. To reduce within-group variability, the power measures for each mouse were normalized by dividing them by the mean delta power during REM for that mouse (Naylor et al., 1998). There were no significant differences across groups in calibrated REM delta power (F(2,18) = 1.85;p = 0.19); hence, this normalization would not differentially affect values for mice of different genotypes. Statistics reported below are for normalized values. Two types of measure are reported: mean power per NREM epoch, which might be considered a measure of sleep intensity, and total NREM energy (summed power over all NREM epochs), which can be considered a measure of sleep amount which takes both time and intensity into account.
All statistical comparisons were made using NCSS 97 software (NCSS, Kaysville, UT). Comparisons between genotypes were by one-way ANOVA followed, where appropriate, by Tukey–Kramer post hoctests. Significant differences were defined as p < 0.05. Group values were expressed as mean ± SEM.
RESULTS
Sleep–wake cycle during entrained conditions
As a first step in assessing the effects of the Clockmutation on sleep, vigilance states were scored from 24 hr of continuous recording in C57BL/6J coisogenic mice of all threeClock genotypes (+/+, Clock/+, andClock/Clock) in the same LD 12:12 cycle to which they had been exposed since birth. The three genotypes had indistinguishable EEG waveforms. As seen in Figure1A, the presence of the Clock mutation significantly decreased the total time spent asleep during the entire 24 hr LD cycle (F(2,18) = 13.73; p < 0.001). Whereas baseline sleep amounts seen in wild-type mice were similar to previously published results for this strain (Nagasaki et al., 1980; Richardson et al., 1985; Welsh et al., 1986),Clock heterozygotes slept 9% (or ∼1 hr) less per day andClock homozygotes slept 18% (or ∼2 hr) less per day than did wild-type mice.
Fig. 1.
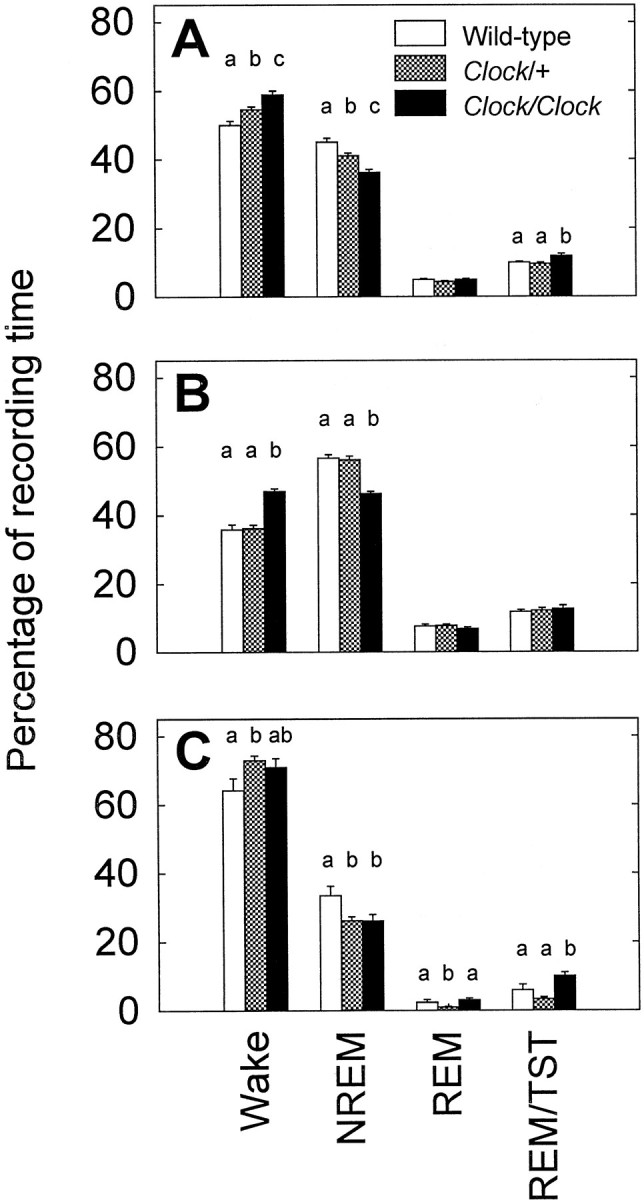
Percentage of recording time spent in the sleep states for all three genotypes. The four bar groups represent wake, NREM or REM sleep, and the percentage of sleep time spent in REM sleep: a measure of the REM/NREM ratio. A shows sleep during the entire 24 hr LD baseline period, whereas the other graphs further break this baseline period into the 12 hr light period (B) and the 12 hr dark period (C). a–c indicate significant pairwise differences between groups (p < 0.05, Tukey–Kramer post hoc tests).
The differences among Clock genotypes in total amounts of sleep were almost entirely attributable to differences in the amount of NREM sleep. Clock homozygotes had significantly lower NREM sleep times than wild-type mice during both the 12 hr light (Fig.1B) (F(2,18) = 30.31; p < 0.001) and 12 hr dark (Fig. 1C) (F(2,18) = 4.66; p < 0.05) periods, whereas Clock heterozygotes had significantly lower NREM sleep times than wild type only during the 12 hr dark period (F(2,18) = 4.66; p < 0.05). Also during the 12 hr dark period, REM sleep in Clockheterozygous mice was only 7 min compared with 17 min for wild-type mice and 22 min for Clock homozygous mice (F(2,18) = 5.31; p < 0.05).
To characterize further the lower sleep time observed in theClock mutants, sleep architecture was analyzed; in particular, we sought to determine whether differences in sleep episode length, number, or composition might underlie the sleep changes associated with the mutation. During the 12 hr light period, all genotypes had equal numbers of sleep episodes (wild type, 19.7 ± 1.7; Clock/+, 17.5 ± 1.0; Clock/Clock, 19.8 ± 1.6). The reduced sleep in Clock homozygotes during the light period corresponded with a significant reduction in the mean NREM episode length (wild type, 8.3 ± 0.4 min;Clock/+, 8.1 ± 0.4 min; Clock/Clock, 6.6 ± 0.2 min; F(2,18) = 5.76;p = 0.012), which, in turn, corresponded with marginally shorter sleep episodes (wild type, 26.3 ± 2.6 min;Clock/+, 29.0 ± 1.9 min; Clock/Clock, 21.6 ± 2.0 min; F(2,18) = 3.41;p = 0.055). The mean NREM episode length during the 12 hr dark period was also significantly lower in Clockhomozygotes (5.8 ± 0.2 min) compared with wild types (7.7 ± 0.6 min) or Clock heterozygotes (8.1 ± 0.4 min) (F(2,18) = 6.9; p < 0.01). However, the number of brief (<60 sec) arousals from sleep inClock homozygotes (200 ± 15) was nonsignificantly (F(2,18) = 1.3; p = 0.30) lower than in heterozygotes (211 ± 14) or wild-type mice (240 ± 23). Therefore, it is unlikely that the quality of the sleep in the Clock mutants was any worse than in wild types and, more importantly, unlikely that the mutation lowers the arousal threshold.
EEG spectral power in the delta frequency range is often used as a correlate of sleep drive or intensity. The total NREM delta energy (power summed over the entire 24 hr baseline recording period) was significantly lower in Clock homozygous compared with both wild-type and heterozygous mice (F(2,18) = 10.63; p < 0.001). However, there were no significant differences between genotypes in the theta and sigma bands (theta,F(2,18) = 2.15; p = 0.146; sigma, F(2,18) = 2.30;p = 0.128). NREM delta power also was not significantly decreased in Clock mutants (F(2,18) = 2.68; p = 0.096).
Sleep–wake cycle during free-running conditions
Clock homozygous mice exhibit a free-running period considerably longer than 24 hr (Vitaterna et al., 1994). To test the possibility that the reduced sleep of Clock mutants could be attributable to a difference in entrainment to a 24 hr LD cycle, we recorded sleep from free-running Clock/Clock and wild-type animals in DD. Clock homozygous mice demonstrated the expected increase in the free-running period of the activity rhythm (28.75 ± 0.09 hr) over wild types (23.68 ± 0.06 hr;p < 0.001). To be able to compare mice with different free-running periods, we normalized data that were in the form of sums over time by dividing by the individual period lengths to give proportional or rate measures. As was observed under entrained conditions, Clock homozygous mice spent a greater proportion of their circadian cycle awake than did wild-type mice (Fig.2). Likewise, the lower proportional sleep time under DD conditions was again associated with proportionally less NREM sleep.
Fig. 2.

Percentage of circadian period spent in various sleep states for wild-type and homozygous Clock mice housed in constant darkness. TST, Total sleep time.
When sleep architecture was analyzed, Clock homozygous mice again showed shorter NREM episode lengths (6.9 ± 0.4 min) than wild types (8.8 ± 0.5 min; p = 0.01). As in LD 12:12, there was no significant difference in the rate of brief arousals between genotypes. As in LD 12:12, there was somewhat lower total NREM delta energy production rate in the Clock mutants than in the wild-types mice when equated for period length, although the difference did not reach significance (p = 0.14). However, the total energy is calculated by summing the power over different total intervals for the different genotypes (determined by the individual's free-running period). In other words, an equivalent (actually 20% less) delta energy is accumulated over ∼28 hr in Clock/Clock mice as is accumulated over ∼24 hr in wild-type mice. As in LD 12:12, the mean NREM delta power was not different between the two DD groups.
Recovery sleep after sleep deprivation
To address the question of whether the reduced sleep observed inClock mutant mice might result from an inability to respond to sleep debt, we examined the response to a 6 hr period of sleep deprivation. Figure 3 shows the number of sleep epochs per hour for all three genotypes during both the baseline and recovery sleep periods. In all three genotypes, sleep time during the recovery 12 hr dark period was significantly greater than during the equivalent baseline period (p < 0.01). The total amount of sleep during the 12 hr dark period immediately after sleep deprivation was significantly lower in Clockhomozygous mice (39.1 ± 1.8%) compared with Clockheterozygotes (44.9 ± 1.6%) or wild-type mice (47.1 ± 1.6%) (F(2,18) = 5.41; p < 0.05). However, when the recovery sleep was treated as a percentage of each animal's individual baseline sleep amount, there were no significant differences across genotype in overall sleep or in NREM (Fig. 3). There were no significant differences among genotypes in the ratio of REM/NREM (+/+, 0.1615 ± 0.0047; Clock/+, 0.1451 ± 0.0089; Clock/Clock, 0.1664 ± 0.0134) in recovery sleep epochs (F(2,18) = 1.46;p = 0.26). Although there were no 24 hr baseline differences in REM, Clock homozygotes increased their time in REM sleep significantly less during the 24 hr recovery period (wild type, 33.5 ± 3.4 min over baseline; Clock/+, 25.1 ± 2.9; Clock/Clock, 16.4 ± 5.2%;F(2,18) = 4.28; p < 0.05). The smaller increase in Clock/Clock REM sleep was particularly evident during the 12 hr dark period immediately after sleep deprivation. During this time, Clock homozygotes had only 15.7 ± 3.4 min more REM sleep over their baseline REM amounts, whereas the other two genotypes showed nearly twice the increase (Clock heterozygotes, 31.0 ± 2.4 min; wild type, 28.9 ± 5.4 min; F(2,18) = 5.07; p < 0.05). As with the baseline period, differences in total NREM delta energy during recovery sleep were also noted. Clock homozygotes generated significantly less NREM delta energy than either wild-type or heterozygous mice (F(2,18) = 5.21; p < 0.05). Mean NREM delta power during recovery epochs, however, was not significantly different between any of the groups (F(2,18) = 0.94; p = 0.41; NS).
Fig. 3.
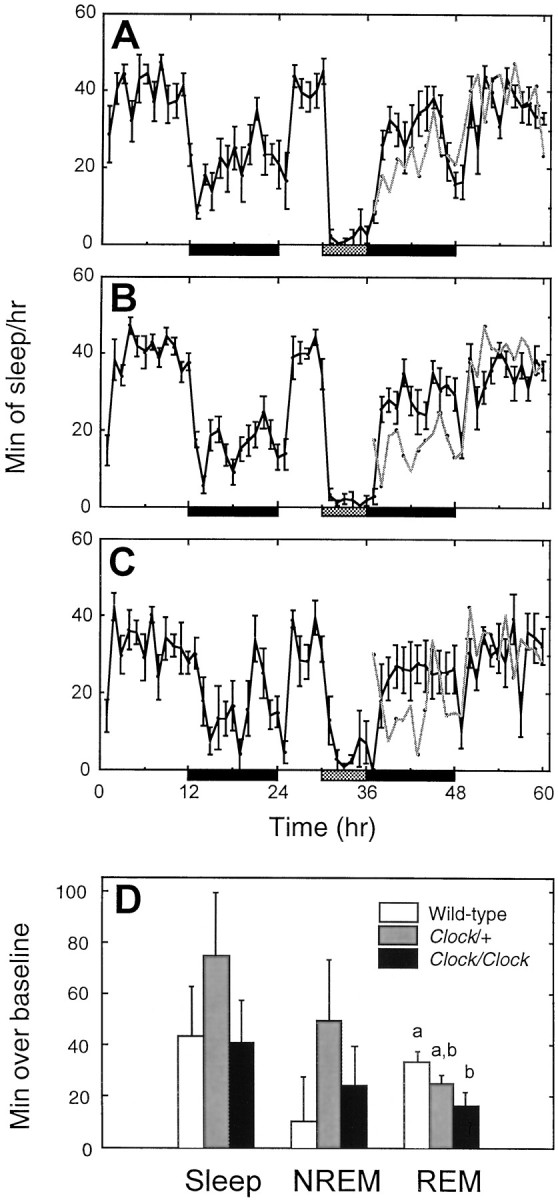
Response to sleep deprivation. Minutes of sleep per hour during baseline, sleep deprivation (gray bar), and recovery periods for wild-type (A), Clock heterozygous (B), and Clock homozygous (C) mice. Black bars represent times of lights off. For comparison, the baseline sleep amounts have been double-plotted in gray during the recovery period.D, The number of additional minutes of sleep over each animal's equivalent baseline for the entire 24 hr LD recovery period.a and b indicate significant pairwise differences between groups (p < 0.05, Tukey–Kramer post hoc tests).
DISCUSSION
The results demonstrate that the Clock mutation affects sleep–wake parameters in mice whether they are entrained to LD 12:12 or free-running in DD. These results indicate that mutation of a circadian clock gene influences not only sleep architecture but also the amount of sleep. The mutation does not, however, affect sleep intensity as measured by delta power.
The most striking effect of the Clock mutation on the sleep–wake cycle is that Clock heterozygous and homozygous mice slept 1 and 2 hr less, respectively, than wild-type mice during entrainment to LD 12:12. These differences in total sleep time were mainly in the form of reduced time in NREM sleep. The reduction in total NREM sleep in Clock mutant mice was associated with a reduction in the average NREM episode length and a decrease in the average sleep episode length. Of equal importance, these differences (as percentages of time spent awake and in NREM) were also present between free running wild types and Clock homozygotes. The finding that a mutation in the Clock gene changes the percentage of time spent asleep even when the animals are free-running indicates that the mutation is altering not only the timing of sleep (i.e., circadian sleep control) but also the amount of sleep (i.e., homeostatic sleep control).
If one accepts NREM delta power as indicative of sleep intensity (Borbely et al., 1989) (for review, see Rechtschaffen et al., 1999), then there is no indication that Clock mutants compensate for sleeping less by sleeping more intensely. Such compensation is seen in short- or long-sleeping humans, for example (Aeschbach et al., 1996). The Clock mutants could sleep less because they have some impairment preventing them from sleeping. Such an alteration should produce mice with a large cumulative sleep debt. However, there is no difference between genotypes in relative amount or intensity of recovery sleep after sleep deprivation, which indicates thatClock mutants are capable of increased sleep in response to increased sleep debt. Alternatively, the Clock mutants may sleep less because of altered homeostatic parameters, i.e., sleep “need” accumulates at a slower rate. Although our present results do not conclusively demonstrate such an alteration, they fail to support two other models (increased sleep intensity and an impaired homeostatic sleep response), which could account for the reduced sleep in Clock mutants.
Early studies in which the master circadian pacemaker, the SCN, was destroyed in the rat supported the hypothesis that the homeostatic process is independent of the circadian clock. Total destruction of the SCN abolished the normal circadian expression of the sleep–wake cycle but did not change the overall time spent asleep (Mouret et al., 1978;Eastman et al., 1984; Mistlberger et al., 1987). Moreover, recovery sleep after sleep deprivation, a measure of homeostatic response, was unchanged (Mistlberger et al., 1983; Tobler et al., 1983; Borbely et al., 1989). However, results of more recent SCN lesion studies in the squirrel monkey have not supported independent homeostatic and circadian processes. Complete lesions of the monkey SCN produced both a loss of circadian timing and a 4 hr increase in daily sleep time, suggesting that, at least in the monkey, the circadian and homeostatic processes do interact (Edgar et al., 1993). This finding led to Edgar's “opponent process” model, the hypothesis that the SCN clock produces an “alerting” signal that enhances wake and thereby actively opposes the homeostatic tendency for sleep (Edgar et al., 1993). Although they are in the opposite direction, the results of the present study are also consistent with the hypothesis that disruption of normal circadian clock function has major effects on the homeostatic control of sleep.
At least two general mechanisms can be proposed to explain how a mutation known to have a pronounced effect on the functioning of the circadian clock can lead to changes in the amount of NREM sleep. First, alteration in the circadian control either by lesioning the SCN or via the Clock mutation might expose a species-dependent bias of the circadian clock toward sleep or waking as in Edgar's opponent process model (Edgar et al., 1993). Alternatively, the effects of the Clock mutation on time spent asleep may be independent of SCN–circadian clock function. The finding thatClock mRNA is expressed throughout the brain, as well as in many other tissues (King et al., 1997; Steeves et al., 1999), raises the possibility that the effects of the Clock mutation on sleep may result from its action in other, non-SCN regions that influence the duration of sleep. The hypothesis that one or more of the genes controlling circadian timing has other functions has been proposed previously for the homolog of the mammalian Clockgene in Drosophila (Andretic et al., 1999). Whether or not the SCN mediates the effects of Clock on sleep, the finding that the Clock mutation does influence total sleep time indicates that the circadian and homeostatic processes underlying the regulation of sleep are linked together at the molecular level.
As with the anatomical issue, the Clock mutation may impact sleep homeostasis either directly or via any gene(s) whose transcription is directly or indirectly regulated by CLOCK.Clock is known to encode a member of the basic helix-loop-helix PAS (Per, ARNT, Ahr, Sim) family of transcription factors, with the site of the mutation in the putative transactivation domain (King et al., 1997). CLOCK and its known partner BMAL1 can induce transcription of mPer1 via binding to an E-box sequence (Gekakis et al., 1998). The Clock mutation results in reduced expression levels of mPer1, as well as other presumably nonclock genes, with regulatory sequences containing E-boxes, including vasopressin and the enzyme albumin D-element binding protein (DBP) (Shearman and Weaver, 1999; Ripperger et al., 2000). Although at this point the genes that constitute the sleep homeostat are unknown, analysis of which Clock-regulated genes ultimately impact sleep homeostasis may indicate a molecular pathway that would lead to the identification of such sleep genes.
Given that Clock regulates its expression level, it is then of interest that mice lacking the gene for the transcription factor DBP are reported to have altered NREM delta power (Franken et al., 2000). Gene-targeting studies have identified other genes that have effects on amounts, patterns, or proportions of time spent in different stages of sleep, including the prion protein (Tobler et al., 1996, 1997), the tumor necrosis factor 55 kDa receptor gene (Fang et al., 1997), the serotonin 1B receptor gene (Boutrel et al., 1999), the human insulin gene (Valatx et al., 1999), insulin-like growth factor 1, and growth hormone (Zhang et al., 1996). Mutations of the genes encoding the neuropeptide orexin (hypocretin) in the mouse (Chemelli et al., 1999) or its receptor in the dog (Lin et al., 1999) have been associated with sleep pathology similar to human narcolepsy. Genetic influences on sleep are also apparent from differences among inbred strains (Toth, 1996; Franken et al., 1998, 1999b), forming a basis for quantitative trait loci analysis (Tafti et al., 1997; Franken et al., 1999a) to ultimately identify loci influencing sleep. The effects of theClock mutation on sleep are of comparable impact with the effects of many of the targeted mutations or strain differences. Interestingly, some gene knock-outs with effects on sleep have shown modest effects on circadian period as well (Tobler et al., 1996;Lopez-Molina et al., 1997). Whether this reflects some fundamental genetic relationship between circadian periodicity and sleep homeostasis remains to be determined.
The present studies are the first to examine how genetic alteration of a known circadian clock component impacts the sleep–wake patterns in mice. It may well be that any pronounced genetic alteration of the circadian clock system will have effects on the amount and/or quality of the sleep–wake states, and it is now of obvious interest to determine to what extent manipulation of other proven and candidate mammalian circadian clock genes will affect sleep. Characterizing the sleep phenotypes in animals with genetically induced alterations in circadian clock function should prove to be informative for defining how the circadian clock and sleep–wake system regulate overall temporal organization in mammals.
Footnotes
This work was supported by National Institutes of Health Grants HL/MH-R01–59598, HL-T32–07909, and P01-AG-11412, and Army Research Office Grant DAAG55-98-1-0196. J.S.T. is an investigator in the Howard Hughes Medical Institute. We thank Dr. Christine Dugovic for her comments and advice in preparing this manuscript.
Correspondence should be addressed to Fred W. Turek, Department of Neurobiology and Physiology, Northwestern University, 2153 N. Campus Drive, Evanston, IL 60208. E-mail: fturek@nwu.edu.
REFERENCES
- 1.Aeschbach D, Cajochen C, Landolt H, Borbely AA. Homeostatic sleep regulation in habitual short sleepers and long sleepers. Am J Physiol. 1996;270:R41–R53. doi: 10.1152/ajpregu.1996.270.1.R41. [DOI] [PubMed] [Google Scholar]
- 2.Akerstedt T, Gillberg M. The circadian variation of experimentally displaced sleep. Sleep. 1981;4:159–169. doi: 10.1093/sleep/4.2.159. [DOI] [PubMed] [Google Scholar]
- 3.Andretic R, Chaney S, Hirsh J. Requirement of circadian genes for cocaine sensitization in Drosophila. Science. 1999;285:1066–1068. doi: 10.1126/science.285.5430.1066. [DOI] [PubMed] [Google Scholar]
- 4.Antoch MP, Song EJ, Chang AM, Vitaterna MH, Zhao Y, Wilsbacher LD, Sangoram AM, King DP, Pinto LH, Takahashi JS. Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell. 1997;89:655–667. doi: 10.1016/s0092-8674(00)80246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benington JH, Heller HC. REM-sleep timing is controlled homeostatically by accumulation of REM- sleep propensity in non-REM sleep. Am J Physiol. 1994;266:R1992–R2000. doi: 10.1152/ajpregu.1994.266.6.R1992. [DOI] [PubMed] [Google Scholar]
- 6.Benington JH, Kodali SK, Heller HC. Scoring transitions to REM sleep in rats based on the EEG phenomena of pre-REM sleep: an improved analysis of sleep structure. Sleep. 1994;17:28–36. doi: 10.1093/sleep/17.1.28. [DOI] [PubMed] [Google Scholar]
- 7.Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 8.Borbely AA, Achermann P, Trachsel L, Tobler I. Sleep initiation and initial sleep intensity: interactions of homeostatic and circadian mechanisms. J Biol Rhythms. 1989;4:149–160. [PubMed] [Google Scholar]
- 9.Boutrel B, Franc B, Hen R, Hamon M, Adrien J. Key role of 5-HT1B receptors in the regulation of paradoxical sleep as evidenced in 5-HT1B knock-out mice. J Neurosci. 1999;19:3204–3212. doi: 10.1523/JNEUROSCI.19-08-03204.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 11.Czeisler CA, Weitzman E, Moore-Ede MC, Zimmerman JC, Knauer RS. Human sleep: its duration and organization depend on its circadian phase. Science. 1980;210:1264–1267. doi: 10.1126/science.7434029. [DOI] [PubMed] [Google Scholar]
- 12.Dijk DJ, Czeisler CA. Paradoxical timing of the circadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans. Neurosci Lett. 1994;166:63–68. doi: 10.1016/0304-3940(94)90841-9. [DOI] [PubMed] [Google Scholar]
- 13.Eastman CI, Mistlberger RE, Rechtschaffen A. Suprachiasmatic nuclei lesions eliminate circadian temperature and sleep rhythms in the rat. Physiol Behav. 1984;32:357–368. doi: 10.1016/0031-9384(84)90248-8. [DOI] [PubMed] [Google Scholar]
- 14.Edgar DM, Dement WC, Fuller CA. Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep-wake regulation. J Neurosci. 1993;13:1065–1079. doi: 10.1523/JNEUROSCI.13-03-01065.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang J, Wang Y, Krueger JM. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFα treatment. J Neurosci. 1997;17:5949–5955. doi: 10.1523/JNEUROSCI.17-15-05949.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275:R1127–R1137. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- 17.Franken P, Chollet D, Malafosse A, Tafti M. QTL-Analysis of sleep and sleep EEG in BXD recombinant inbred mice. Sleep. 1999a;22 [Suppl]:112. [Google Scholar]
- 18.Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999b;22:155–169. [PubMed] [Google Scholar]
- 19.Franken P, Lopez-Molina L, Marcacci L, Schibler U, Tafti M. The transcription factor DBP affects circadian sleep consolidation and rhythmic EEG activity. J Neurosci. 2000;20:617–625. doi: 10.1523/JNEUROSCI.20-02-00617.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 21.Herzog ED, Takahashi JS, Block GD. Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat Neurosci. 1998;1:708–713. doi: 10.1038/3708. [DOI] [PubMed] [Google Scholar]
- 22.King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–653. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klerman EB, Boulos Z, Edgar DM, Mistlberger RE, Moore-Ede MC. Circadian and homeostatic influences on sleep in the squirrel monkey: sleep after sleep deprivation. Sleep. 1999;22:45–59. doi: 10.1093/sleep/22.1.45. [DOI] [PubMed] [Google Scholar]
- 24.Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Molina L, Conquet F, Dubois-Dauphin M, Schibler U. The DBP gene is expressed according to a circadian rhythm in the suprachiasmatic nucleus and influences circadian behavior. EMBO J. 1997;16:6762–6771. doi: 10.1093/emboj/16.22.6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mistlberger RE, Bergmann BM, Waldenar W, Rechtschaffen A. Recovery sleep following sleep deprivation in intact and suprachiasmatic nuclei-lesioned rats. Sleep. 1983;6:217–233. doi: 10.1093/sleep/6.3.217. [DOI] [PubMed] [Google Scholar]
- 27.Mistlberger RE, Bergmann BM, Rechtschaffen A. Relationships among wake episode lengths, contiguous sleep episode lengths, and electroencephalographic delta waves in rats with suprachiasmatic nuclei lesions. Sleep. 1987;10:12–24. [PubMed] [Google Scholar]
- 28.Mouret J, Coindet J, Debilly G, Chouvet G. Suprachiasmatic nuclei lesions in the rat: alterations in sleep circadian rhythms. Electroencephalogr Clin Neurophysiol. 1978;45:402–408. doi: 10.1016/0013-4694(78)90191-8. [DOI] [PubMed] [Google Scholar]
- 29.Nagasaki H, Kitahama K, Valatx JL, Jouvet M. Sleep-promoting effect of the sleep-promoting substance (SPS) and delta sleep-inducing peptide (DSIP) in the mouse. Brain Res. 1980;192:276–280. doi: 10.1016/0006-8993(80)91029-x. [DOI] [PubMed] [Google Scholar]
- 30.Naylor E, Buxton OM, Bergmann BM, Easton A, Zee PC, Turek FW. Effects of aging on sleep in the golden hamster. Sleep. 1998;21:687–693. doi: 10.1093/sleep/21.7.687. [DOI] [PubMed] [Google Scholar]
- 31.Rechtschaffen A, Bergmann BM, Gilliland MA, Bauer K. Effect of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep. 1999;22:11–31. doi: 10.1093/sleep/22.1.11. [DOI] [PubMed] [Google Scholar]
- 32.Richardson GS, Moore-Ede MC, Czeisler CA, Dement WC. Circadian rhythms of sleep and wakefulness in mice: analysis using long-term automated recording of sleep. Am J Physiol. 1985;248:R320–R330. doi: 10.1152/ajpregu.1985.248.3.R320. [DOI] [PubMed] [Google Scholar]
- 33.Ripperger JA, Shearman LP, Reppert SM, Schibler U. CLOCK, an essential pacemaker component, controls expression of the circadian transcription factor DBP. Genes Dev. 2000;14:679–689. [PMC free article] [PubMed] [Google Scholar]
- 34.Shearman LP, Weaver DR. Photic induction of Period gene expression is reduced in Clock mutant mice. NeuroReport. 1999;10:613–618. doi: 10.1097/00001756-199902250-00031. [DOI] [PubMed] [Google Scholar]
- 35.Steeves TD, King DP, Zhao Y, Sangoram AM, Du F, Bowcock AM, Moore RY, Takahashi JS. Molecular cloning and characterization of the human CLOCK gene: expression in the suprachiasmatic nuclei. Genomics. 1999;57:189–200. doi: 10.1006/geno.1998.5675. [DOI] [PubMed] [Google Scholar]
- 36.Tafti M, Franken P, Kitahama K, Malafosse A, Jouvet M, Valatx JL. Localization of candidate genomic regions influencing paradoxical sleep in mice. NeuroReport. 1997;8:3755–3758. doi: 10.1097/00001756-199712010-00019. [DOI] [PubMed] [Google Scholar]
- 37.Tobler I, Borbely AA, Groos G. The effect of sleep deprivation on sleep in rats with suprachiasmatic lesions. Neurosci Lett. 1983;42:49–54. doi: 10.1016/0304-3940(83)90420-2. [DOI] [PubMed] [Google Scholar]
- 38.Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–1879. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride PA, Manson JC. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 40.Toth LA. Strain differences in the somnogenic effects of interferon inducers in mice. J Interferon Cytokine Res. 1996;16:1065–1072. doi: 10.1089/jir.1996.16.1065. [DOI] [PubMed] [Google Scholar]
- 41.Trachsel L, Tobler I, Borbely AA. Electroencephalogram analysis of non-rapid eye movement sleep in rats. Am J Physiol. 1988;255:R27–R37. doi: 10.1152/ajpregu.1988.255.1.R27. [DOI] [PubMed] [Google Scholar]
- 42.Valatx JL, Douhet P, Bucchini D. Human insulin gene insertion in mice. Effects on the sleep-wake cycle? J Sleep Res. 1999;8 [Suppl 1]:65–68. doi: 10.1046/j.1365-2869.1999.00011.x. [DOI] [PubMed] [Google Scholar]
- 43.Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Welsh DK, Richardson GS, Dement WC. Effect of age on the circadian pattern of sleep and wakefulness in the mouse. J Gerontol. 1986;41:579–586. doi: 10.1093/geronj/41.5.579. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Obal F, Jr, Fang J, Collins BJ, Krueger JM. Non-rapid eye movement sleep is suppressed in transgenic mice with a deficiency in the somatotropic system. Neurosci Lett. 1996;220:97–100. doi: 10.1016/s0304-3940(96)13232-8. [DOI] [PubMed] [Google Scholar]
- 46.Zulley J, Wever R, Aschoff J. The dependence of onset and duration of sleep on the circadian rhythm of rectal temperature. Pflügers Arch. 1981;391:314–318. doi: 10.1007/BF00581514. [DOI] [PubMed] [Google Scholar]