

Abstract
The subthalamic nucleus (STN) is a key nucleus in the basal ganglia motor circuit that provides the major glutamatergic excitatory input to the basal ganglia output nuclei. The STN plays an important role in normal motor function, as well as in pathological conditions such as Parkinson's disease (PD) and related disorders. Development of a complete understanding of the roles of the STN in motor control and the pathophysiological changes in STN that underlie PD will require a detailed understanding of the mechanisms involved in regulation of excitability of STN neurons. Here, we report that activation of group I metabotropic glutamate receptors (mGluRs) induces a direct excitation of STN neurons that is characterized by depolarization, increased firing frequency, and increased burst-firing activity. In addition, activation of group I mGluRs induces a selective potentiation of NMDA-evoked currents. Immunohistochemical studies at the light and electron microscopic levels indicate that both subtypes of group I mGluRs (mGluR1a and mGluR5) are localized postsynaptically in the dendrites of STN neurons. Interestingly, pharmacological studies suggest that each of the mGluR-mediated effects is attributable to activation of mGluR5, not mGluR1, despite the presence of both subtypes in STN neurons. These results suggest that mGluR5 may play an important role in the net excitatory drive to the STN from glutamatergic afferents. Furthermore, these studies raise the exciting possibility that selective ligands for mGluR5 may provide a novel approach for the treatment of a variety of movement disorders that involve changes in STN activity.
Keywords: metabotropic glutamate receptor, subthalamic nucleus, basal ganglia, Parkinson's disease, burst firing, NMDA receptor, mGluR1, mGluR5
The basal ganglia (BG) are a set of subcortical nuclei that play a critical role in motor control and are a primary site of pathology in a number of movement disorders, including Parkinson's disease (PD), Tourette's syndrome, and Huntington's disease. Recent studies reveal that a key nucleus in the BG motor circuit, the subthalamic nucleus (STN), plays an especially important role in BG function. The STN is an excitatory glutamatergic nucleus in the BG and provides the major excitatory input to the BG output nuclei, the substantia nigra pars reticulata (SNr) and the internal globus pallidus. Normal motor function requires an intricate balance between excitation of the output nuclei by glutamatergic neurons from the STN and inhibition of the output nuclei by GABAergic projections from the striatum (for review, see Wichmann and DeLong, 1997).
Interestingly, recent studies suggest that the major pathophysiological change that occurs in response to loss of nigrostriatal dopamine neurons in PD patients is an increase in activity of STN neurons. The resultant increase in synaptic excitation of GABAergic projection neurons in the output nuclei leads to a “shutdown” of thalamocortical projections and produces the motor impairment characteristic of PD (DeLong, 1990). Conversely, hyperkinetic disorders such as Huntington's disease (Reiner et al., 1988; Albin et al., 1990) and Tourette's syndrome (Albin et al., 1989; Leckman et al., 1997) are associated with decreases in STN activity. These discoveries have led to a major interest in development of novel strategies to treat these disorders by altering neuronal STN activity or STN-induced excitation of BG output nuclei. Interestingly, surgical lesions (Bergman et al., 1990; Aziz et al., 1991; Guirdi et al., 1996) and high-frequency stimulation of the STN (Benazzouz et al., 1993; Limousin et al., 1995a,b) are highly effective in treatment of PD. Development of a detailed understanding of the mechanisms involved in regulation of STN activity could lead to development of novel therapeutic agents that alter STN activity without surgical intervention.
Recent studies suggest that metabotropic glutamate receptors (mGluRs) play an important role in regulating excitability of neurons in a wide variety of brain regions, including BG structures (Conn and Pin, 1997). If mGluRs are involved in regulating excitation of STN neurons, this could provide a critical component of regulation of STN activity by glutamatergic afferents. Thus, it will be important to determine whether mGluRs are postsynaptically localized in these neurons and whether activation of mGluRs alters STN activity. To date, eight mGluR subtypes have been cloned from mammalian brain and are classified into three major groups based on sequence homologies, second messenger coupling, and pharmacological profiles (for review, see Conn and Pin, 1997). Group I mGluRs (mGluR1 and mGluR5) couple primarily to Gq, whereas group II (mGluR2 and mGluR3) and group III mGluRs (mGluRs 4, 6, 7, and 8) couple to Gi/Go. We now report that activation of the group I mGluR mGluR5 has a dramatic excitatory effect and selectively increases NMDA receptor currents in STN neurons.
MATERIALS AND METHODS
Slice preparation for electrophysiology. Experiments were performed in STN neurons from 10- to 14-d-old Sprague Dawley rats. Rats were decapitated, the brains were removed, and a block of tissue containing the STN was isolated on ice. The tissue was mounted and immersed in an oxygenated sucrose-artificial CSF (ACSF) solution containing (in mm): 3 KCl, 1.9 MgSO4, 1.2 KH2PO4, 2 CaCl2, 187 sucrose, 20 glucose, 26 NaHCO3, 0.5 pyruvate, and 0.005 glutathione, equilibrated with 95% O2 and 5% CO2, pH 7.4. Saggital slices (300 μm) were prepared using a manual VibroSlice (Stoelting, Chicago, IL) and then incubated at room temperature in ACSF containing (in mm): 124 NaCl, 2.5 KCl, 1.3 MgSO4, 1 NaH2PO4, 2 CaCl2, 20 glucose, 26 NaHCO3, 0.5 pyruvate, and 0.005 glutathione, equilibrated with 95% O2 and 5% CO2, pH 7.4. In experiments requiring potassium channel blockade the ACSF had the following composition (in mm): 105.4 NaCl, 19.6 NaOAc, 2.5 KCl, 1.3 MgCl2, 0.1 CaCl2, 0.1 CdCl2, 2 BaCl2, 6 CsCl, 20 glucose, 26 NaHCO3, 3 4-aminopyridine (4-AP), and 25 tetraethylammonium-Cl.
Electrophysiological recordings. After a 2 hr incubation, the slices were transferred to a recording chamber mounted on the stage of an Olympus Optical (Tokyo, Japan) microscope and continuously perfused at 1–2 ml/min with oxygenated ACSF containing 50 μm picrotoxin and 0.5 μm tetrodotoxin (except when firing was studied). Recordings were made with visualized patch-clamp techniques using Nomarski optics with a water immersion 40× objective. Whole-cell patch-clamp recordings were made using patch electrodes pulled from borosilicate glass on a Narishige (Tokyo, Japan) vertical puller. Electrodes were filled with (in mm): 140 potassium gluconate, 10 HEPES, 10 NaCl, 0.2 EGTA, 2 MgATP, and 0.2 NaGTP. Internal solutions used in experiments requiring potassium channel block contained 140 mm cesium methylsulfonate in place of potassium gluconate. Signals were recorded using a Warner PC-501A patch-clamp amplifier (Warner Instrument Corp., Hamden, CT) and a pClamp data acquisition and analysis system (Axon Instruments, Foster City, CA).
For measurement of NMDA and kainate-evoked currents, NMDA (100 μm–1 mm) with glycine (100 μm) or kainate (100 μm) was pressure-ejected into the slice from a low-resistance pipette using a Picospritzer (General Valve, Fairfield, NJ) at pressures ranging from 5 to 20 psi and for durations of 50–200 msec. Currents were recorded from a holding potential of −60 mV. Slices were bathed in ACSF containing 0.5 μmtetrodotoxin to block synaptic transmission. Agonists and antagonists of mGluRs were then applied by bath infusion for 5 min. NMDA receptor (NMDAR) and kainate receptor current amplitude was measured from baseline to peak of the current.
Animal perfusion and preparation of tissue for immunohistochemistry. Five male Sprague Dawley rats were deeply anesthetized with ketamine (20 mg/kg) and transcardially perfused with cold, oxygenated Ringer's solution followed by 500 ml of 4% paraformaldehyde and 0.1% glutaraldehyde in phosphate buffer (PB, 0.1m), pH 7.4, followed by 300 ml of cold PB. The brain was removed from the skull and stored in PBS (0.01 m), pH 7.4, before being sliced on a vibrating microtome into 60 μm transverse sections. These sections were then treated with 1.0% sodium borohydride for 20 min and rinsed in PBS.
Immunohistochemistry. The sections were preincubated at room temperature in a solution containing 10% normal goat serum (NGS), 1.0% bovine serum albumin (BSA), and 0.3% Triton X-100 in PBS for 1 hr. They were then transferred to solutions containing each of four primary antibodies raised against synthetic peptides corresponding to either the C terminus of mGluR1a (Chemicon, Temecula, CA) or to residues 1116–1130 of mGluR1a (Dr. Carmelo Romano, Washington University School of Medicine, St. Louis, MO) or to the C terminus of mGluR5 (Upstate Biotechnology, Lake Placid, NY; Dr. Carmelo Romano). Antibodies were diluted at 0.5–1.0 mg/ml in a solution containing 1.0% NGS, 1.0% BSA, and 0.3% Triton X-100 in PBS. The tissue was incubated in this solution overnight at room temperature. The sections were rinsed in PBS and incubated for 1 hr at room temperature in a secondary antibody solution containing biotinylated goat-anti-rabbit IgGs (Vector Laboratories, Burlingame, CA) diluted 1:200 in the primary antibody diluent solution. After rinsing, sections were put in a solution containing 1:100 avidin–biotin–peroxidase complex (Vector). The tissue was then washed in PBS and 0.05 m Tris buffer before being transferred to a solution containing 0.01 mimidazole, 0.0005% hydrogen peroxide, and 0.025% 3,3-diaminobenzidine tetrahydrochloride (DAB; Sigma, St. Louis, MO) in Tris for 7–10 min. Sections were then mounted on gelatin-coated slides, dried, and coverslipped with Permount.
For immunohistochemical analysis at the electron microscopic level, the sections were treated with cryoprotectant for 20 min and transferred to a −80°C freezer for an additional 20 min. The sections were then thawed and treated with successively decreasing concentrations of cryoprotectant and finally PBS. The immunocytochemical procedure was the same as that used for studies at the light level, except that Triton X-100 was not used, and the incubation in the primary antibody was performed at 4°C for 48 hr. After DAB revelation, the sections were processed for the electron microscope. They were first washed in 0.1 m PB for 30 min and then post-fixed in 1.0% osmium tetroxide for 20 min. Afterward, they were washed in PB and dehydrated by a series of increasing concentrations of ethanol (50, 70, 90, and 100%). Uranyl acetate (1.0%) was added to the 70% ethanol to enhance contrast in the tissue. Next, the sections were exposed to propylene oxide and embedded in epoxy resin (Durcupan; Fluka, Buchs, Switzerland) for 12 hr. They were then mounted on slides, coverslipped, and heated at 60°C for 48 hr.
Five blocks (three for mGluR1a and two for mGluR5) were cut from the STN and mounted on resin carriers to allow for the collection of ultrathin sections using an ultramicrotome (Ultracut 2; Leica, Nussloch, Germany). The ultrathin sections were collected on single-slot copper grids, stained with lead citrate for 5 min to enhance contrast, and examined on a Zeiss (Thornwood, NY) EM-10C electron microscope. Electron micrographs were taken at 10,000 and 31,500× magnifications to characterize the nature of immunoreactive elements in the STN.
Drugs. All drugs were obtained from Tocris Cookson (Ballwin, MO), except that (+)-2-aminobicyclo[3.1.0]-hexane-2,6-dicarboxylate monohydrate (LY354740) was a gift from D. Schoepp and J. Monn (Eli Lilly, Indianapolis, IN), methylphenylethynylpyridine (MPEP) and 7-hydroxyiminocyclopropan-[b]chromen-1a-carboxylic acid ethyl ester (CPCCOEt) were gifts from R. Kuhn (Novartis, Basel, Switzerland), and (S)-(+)-2-(3′-carboxy-bicyclo[1.1.1]pentyl)-glycine (CBPG) was purchased from Alexis (San Diego, CA). All other materials were obtained from Sigma.
Data analysis. Values are expressed as mean ± SEM. Statistical significance was assessed using Student's ttest.
RESULTS
Group I mGluRs mediate depolarization of STN neurons
Previous studies suggest that STN neurons express multiple mGluR subtypes, including receptors belonging to each of the major groups of mGluRs (groups I–III) (Testa et al., 1994, 1998; Bradley et al., 1998,1999). We took advantage of highly selective agonists of each of the mGluR groups to determine whether activation of these receptors has effects on membrane properties of STN neurons. Unless otherwise stated, all studies were performed in the presence of tetrodotoxin (TTX; 0.5 μm) to block action potential firing. The group I mGluR-selective agonist 3,5-dihydroxyphenylglycine (DHPG) (Schoepp et al., 1994) (100 μm) induced a robust depolarization of STN neurons (17.1 ± 1.1 mV; p< 0.001; Fig. 1A,C) that was accompanied by an increase in membrane input resistance (Fig.1B). In voltage-clamp mode, this could be seen as an inward current with an accompanying decrease in membrane conductance (data not shown). DHPG-induced depolarization was seen in 23 of 24 cells, indicating a relatively homogeneous population of neurons. The dose–response relationship for DHPG-induced depolarization of STN neurons revealed an EC50 of ∼30 μm (Fig. 1D), which is consistent with the EC50 value of DHPG at activation of group I mGluRs (Schoepp et al., 1994).
Fig. 1.

Group I mGluR-mediated depolarization of STN neurons. A, Representative current-clamp traces of membrane potential changes in response to DHPG (100 μm), LY354740 (100 nm), and l-AP-4 (1 mm) from a holding potential of −60 mV. B, Corresponding change in membrane input resistance accompanying the change in membrane potential. C, Mean data ± SEM of membrane potential changes, showing a significant depolarization by the group I-selective agonist DHPG (**p < 0.001).D, Dose–response curve of DHPG-mediated changes in membrane potential.
In contrast with DHPG, the group II selective mGluR agonist LY354740 (100 nm) (Monn et al., 1997) had no effect on membrane potential (Fig. 1A,C) or input resistance (Fig.1B). Likewise, the group III-selective agonistl(+)-2-amino-4-phosphonobutyric acid (l-AP-4) (1 mm) (Conn and Pin, 1997) had no effect on the membrane potential or input resistance of STN neurons when examined in the presence of the NMDA receptor antagonistsd-AP-5 (20 μm) and MK801 (10 μm) (Fig.1A–C). l-AP-4 (1 mm) did induce a slight depolarization of STN neurons when applied in the absence of NMDA receptor antagonists (data not shown). This is consistent with previous reports thatl-AP-4 is a weak NMDA receptor agonist (Davies and Watkins, 1982).
In other neuronal populations, activation of group I mGluRs can induce cell depolarization by inhibiting a leak potassium current (Guérineau et al., 1994) or by increasing an inward cation current (Crépel et al., 1994; Guérineau et al., 1995; Pozzo Miller et al., 1995). The finding that the DHPG-induced depolarization or inward current in STN neurons is accompanied by a decrease in membrane conductance suggests that this effect is more likely mediated by inhibition of a leak potassium current. To determine whether the DHPG-induced current has a reversal potential consistent with mediation by inhibition of a potassium current, we performed an analysis of theI–V relationship of the DHPG-induced current in voltage-clamp mode. I–V relationships were determined in the presence and absence of 100 μm DHPG by a series of voltage steps ranging from −120 to −30 mV in increments of 10 mV. I–V plots were determined in a total of five cells. A representative leak-subtracted I–V plot in the presence and absence of DHPG (100 μm) is shown in Figure2A. A subtraction of the predrug I–V plot from that in the presence of DHPG yielded an I–V plot of the DHPG-induced current alone (Fig.2B). DHPG induced a net outward current at hyperpolarized potentials (greater than −80 mV) and an inward current at potentials in the range of the resting potential (Fig.2B). The reversal potential of the DHPG-induced current is approximately −80 mV.
Fig. 2.

Ionic basis of DHPG-induced current.A, Leak-subtracted I–V plot in normal K+ conditions in the presence and absence of DHPG (100 μm). B, SubtractedI–V plot representing DHPG-induced current alone, showing reversal potential of approximately −80 mV. C, Representative leak-subtracted I–V plot in the presence of potassium channel block and cesium. D, SubtractedI–V plot representing DHPG-induced current alone, showing reversal potential of −30 mV. E, Mean data ± SEM of DHPG-induced inward current amplitude (picoamperes) in voltage-clamp mode in normal potassium conditions, potassium block and cesium, and in the presence of cadmium (100 μm) (**p < 0.01).
A reversal potential in a hyperpolarized range coupled with the reduction in membrane conductance is consistent with the hypothesis that DHPG-induced membrane depolarization is partially attributable to inhibition of a leak potassium current. However, the reversal potential is somewhat more depolarized than the predicted equilibrium potential for potassium (−105 mV). This, coupled with the nonlinearI–V curve of the DHPG-induced current, suggests that other factors may also play a role in this membrane depolarization. In some other systems, group I mGluR activation can also activate inward cation currents (Crépel et al., 1994; Guérineau et al., 1995;Pozzo Miller et al., 1995). It is possible that a similar effect occurs in STN neurons. To determine whether the DHPG-induced current is solely a potassium current, we performed ion substitution experiments. Voltage–clamp experiments were performed under conditions of potassium channel block by replacement of a K+ ion with a Cs+ ion and inclusion of 4-AP and tetraethylammonium in the external bathing solution. DHPG-induced inward current amplitude at −60 mV was significantly reduced (10.8 ± 2.3 pA; n = 6) compared with that in control conditions (36.2 ± 8.1 pA; n = 4;p < 0.01; Fig. 2E). However, the current was not completely blocked, and the residual current is probably mediated by ions other than potassium. Consistent with this, the leak-subtracted I–V plot in the presence of potassium channel block has a reversal potential of −30 mV and shows that DHPG causes an inward current at potentials more negative than −30 mV and outward current at potentials more positive than −30 mV (n = 3; Fig. 2C,D).
The simplest interpretation of the data presented above is that DHPG induces depolarization of STN neurons by actions on postsynaptically localized group I mGluRs. However, it is possible that DHPG acts by inducing release of another neurotransmitter that then depolarizes STN neurons. Because all of the studies presented above were performed in the presence of TTX, it is unlikely that DHPG acts by increasing cell firing and thereby increasing neurotransmitter release. However, this does not rule out the possibility that DHPG directly depolarizes presynaptic terminals, which could lead to calcium influx and neurotransmitter release. To rule out this possibility, we performed experiments in the presence of cadmium (100 μm) in the bathing solution to block Ca+ channel activity. Consistent with an effective block of neurotransmitter release, this concentration of cadmium completely eliminated EPSCs elicited in the STN by stimulation of the internal capsule (data not shown). However, the amplitude of the DHPG-induced current was not significantly different than that seen in control (31.1 ± 6.9 pA; n = 4; Fig. 2E).
When measured in the absence of TTX, the DHPG-induced depolarization was accompanied by a dramatic increase in action potential firing (Fig.3A). This is consistent with a recent report that group I mGluR agonists increase extracellular single unit firing of STN neurons (Abbott et al., 1997). This increase in cell firing was completely eliminated by hyperpolarizing current injection to hold the membrane potential at the predrug level (Fig.3A). This suggests that the increase in firing frequency was strictly attributable to the DHPG-induced depolarization rather than being partially mediated by other changes in membrane properties that allow the cells to fire at a higher frequency. There was no effect of DHPG on other membrane properties of the cell, including spike width, spike amplitude, and the shape or amplitude of afterhyperpolarizations (data not shown). However, DHPG did induce an increase in the incidence of burst firing, a property of STN neurons previously described by Beurrier et al. (1999) (Fig. 3B). Oscillatory activity underlying burst firing was not seen in any of 15 cells examined at a resting potential of −60 mV before DHPG treatment but was seen in 7 of 26 cells (∼27%) during the DHPG-induced depolarization. In the absence of TTX, oscillatory activity was accompanied by burst firing (Fig. 3B). When studied in the presence of TTX, the DHPG-induced oscillatory activity underlying burst firing was seen, indicating that such activity may not be dependent on synaptic transmission and may be an intrinsic membrane property of STN neurons (Fig. 3C). Furthermore, burst firing was not seen in any cells treated with the group II agonist LY354740 (n= 6) or the group III mGluR agonist l-AP-4 (n = 9) when the cell membrane was held at −60 mV before drug application.
Fig. 3.

Postsynaptic effects of group I mGluR activation in STN neurons. A, Representative current-clamp traces of firing rate before drug application (at −50 mV) and dramatic increase in the presence of DHPG (100 μm) that is countered by current injection to return membrane potential to the predrug level. B, DHPG-mediated switch to burst-firing mode (from a holding potential of −60 mV), which is countered by hyperpolarizing current injection to maintain membrane potential at the predrug level. C, DHPG-mediated membrane oscillations in the presence of TTX are also countered by hyperpolarizing current injection. Action potentials are truncated in A andB. Scale bars in C also apply toB.
Group I mGluRs potentiate NMDA-evoked currents
The data presented above suggest that activation of group I mGluRs can exert a direct excitatory effect on STN neurons that could contribute to the overall excitatory drive to this important nucleus in the BG motor circuit. In some other brain regions, activation of mGluRs can also potentiate excitatory synaptic responses by potentiating currents through glutamate-gated cation channels. To determine the effects of mGluR agonists on the responses of STN neurons to activation of ionotropic glutamate receptors, we used pressure-evoked application of constant amounts of NMDA or kainate onto the cell. Stable baseline NMDA- or kainate-evoked currents were obtained before perfusion of the slice with mGluR agonists. Representative NMDA-evoked current traces are shown before, during, and after mGluR agonist application (Fig.4A). DHPG (100 μm) caused a reversible potentiation of NMDA-evoked current amplitude (39.1 ± 9.2%; n = 10; p < 0.05; Fig. 4B). In contrast, the group II- and group III-selective mGluR agonists LY354740 andl-AP-4 had no effect on NMDA-evoked currents (Fig. 4A,B). None of the group-selective mGluR agonists had any effects on kainate-evoked currents in STN neurons (Fig. 4C).
Fig. 4.
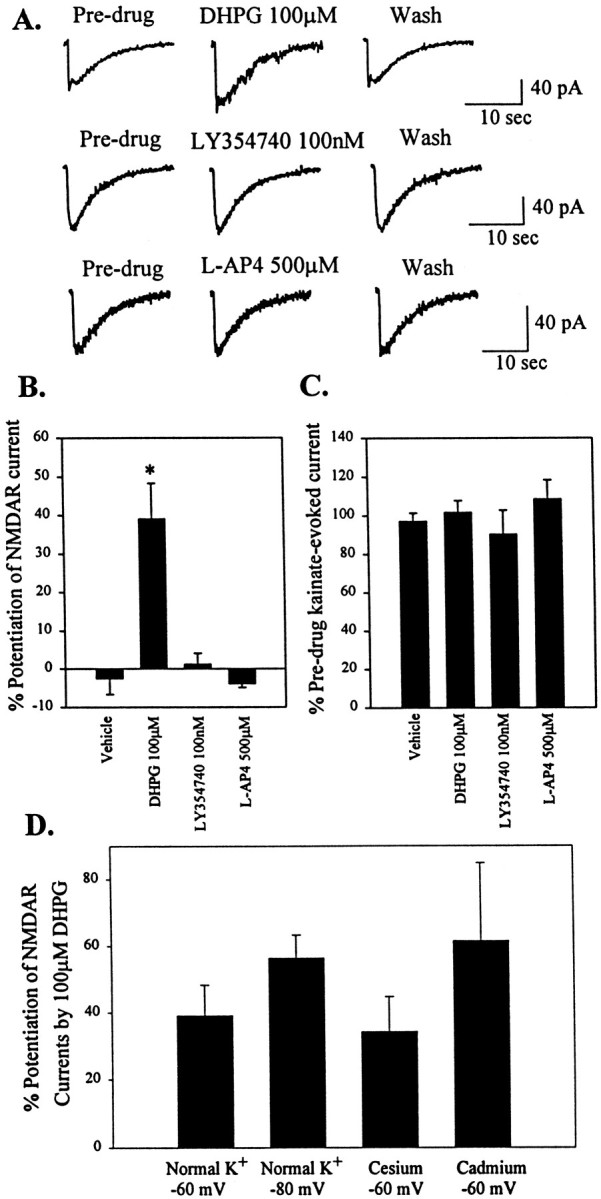
Activation of group I mGluRs potentiates NMDA-evoked currents in STN neurons but has no effect on kainate-evoked currents. A, Representative voltage-clamp traces of NMDA-evoked currents in predrug, agonist, and wash conditions. Only the group I-selective agonist caused a reversible potentiation of NMDA-evoked currents. The group II and III agonists had no effect on NMDA-evoked currents. B, Mean data ± SEM of percent potentiation of NMDA-evoked currents by DHPG over predrug current amplitude. DHPG caused a significant potentiation compared with vehicle (*p < 0.05). C, Mean data ± SEM of percent predrug kainate-evoked current amplitude showing no difference compared with vehicle. D, Mean data ± SEM of percent potentiation of NMDAR currents by DHPG in normal K+ at −60 and −80 mV, cesium and potassium channel block at −60 mV, and in the presence of Cd2+ (100 μm).
One concern that should be considered with studies of modulation of NMDA-evoked currents in brain slices is that it is impossible to obtain complete voltage control of the entire dendritic region of the STN neurons. Thus, it is possible that DHPG-induced potentiation of the NMDA-evoked current is attributable to depolarization of dendritic regions in which we have not achieved adequate voltage control. If so, the DHPG-induced depolarization may relieve the voltage-dependent magnesium block of the NMDA receptor and thereby cause NMDA-evoked currents to be potentiated. To test for this possibility, we performed a series of experiments to determine the effect of DHPG on NMDA-evoked currents under conditions in which the DHPG-induced depolarization is blocked. First, experiments were performed in normal K+ concentrations when holding at the reversal potential of the DHPG-induced current (−80 mV). Also, we determined the effect of DHPG in the presence of conditions that block voltage-dependent potassium channels and thereby reduce the DHPG-induced depolarization. Under both conditions, DHPG-induced inward current was either reduced or absent. In contrast, neither manipulation significantly altered DHPG-induced potentiation of NMDA-evoked currents (Fig. 4D). In addition, experiments were performed in the presence of cadmium (100 μm) to ensure that the response was not caused by calcium-dependent release of another neurotransmitter from presynaptic terminals. As with the studies of DHPG-induced inward currents, cadmium had no significant effect on DHPG-induced potentiation of NMDA-evoked currents (Fig. 4D).
mGluR1 and mGluR5 are postsynaptically localized in STN neurons
DHPG is an agonist at both mGluR1 and mGluR5, suggesting that either of these mGluR subtypes could mediate the responses described above. We performed immunocytochemical studies with mGluR1a and mGluR5 antibodies at the electron microscopic level to determine whether both of these receptors are localized at postsynaptic sites in STN neurons.
At the light microscopic level, the STN showed strong neuropil labeling for mGluR1a and mGluR5 (Figs.5A, 6A). In general, the immunoreactivity was found predominantly in dendritic processes, whereas the level of labeling in cell bodies was very low. In sections immunostained for mGluR1a, labeled dendrites were found in the cerebral peduncle (Fig. 5A), which is consistent with previous Golgi studies showing that the dendrites of neurons located along the ventral border of the STN extend ventrally into the cerebral peduncle (Iwahori, 1978; Afsharpour, 1985).
Fig. 5.
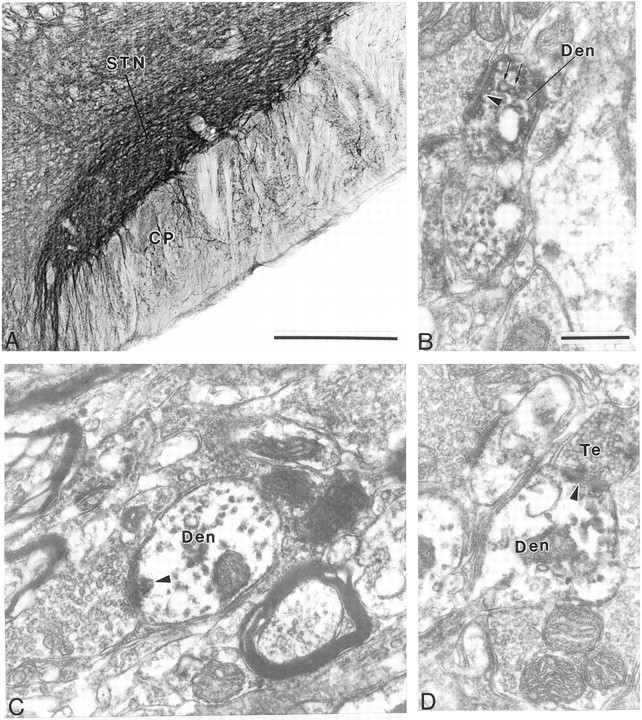
Immunostaining for mGluR1a in the STN. A, Low-power light micrograph of mGluR1a in the STN. B, C, High-power electron micrographs of mGluR1a-immunoreactive dendrites (Den) that form asymmetric synapses (arrowheads) with unlabeled terminals. Note that the dendrite in B contains vesicles (arrows). D, High-power electron micrograph of mGluR1a-immunoreactive terminal that forms an asymmetric synapse (arrowhead) with an immunoreactive dendrite.CP, Cerebral peduncle. Scale bars: A, 500 μm; B–D, 0.5 μm.
To determine the exact nature of the immunoreactive neuronal elements, we performed further analysis at the electron microscopic level. Both antibodies primarily labeled dendritic processes, which formed symmetric and asymmetric synapses with unlabeled axon terminals (Figs.5B–D, 6B–D). In general, the labeling was seen throughout the dendrites, instead of being associated selectively with the plasma membrane (Figs. 5B–D,6B–D). It is worth noting that such a pattern of labeling was also detected for mGluR5 in the SNr using both immunogold and immunoperoxidase techniques (Hubert and Smith, 1999). In addition to dendrites, mGluR1a immunoreactivity was occasionally seen in a few myelinated and unmyelinated axonal segments as well as a few axon terminals (Fig. 5D). Cell bodies displayed light intracytoplasmic labeling with either antibody.
Fig. 6.
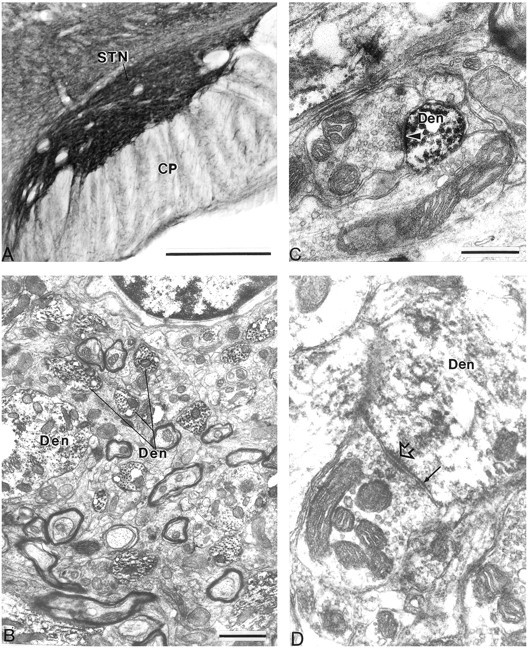
Immunostaining for mGluR5 in the STN. A, Low-power light micrograph of mGluR5 immunostaining in the STN. B, Low-power electron micrograph of mGluR5-immunoreactive dendrites (Den).C, High-power electron micrograph of a small mGluR5-immunoreactive dendrite that forms an asymmetric synapse (arrowhead) with the unlabeled terminal.D, High-power electron micrograph of a large mGluR5-immunoreactive dendrite that forms a symmetric synapse (arrow) with an unlabeled terminal. The open arrowhead points to a puncta adherentia. CP, Cerebral peduncle. Scale bars: A, 500 μm;B, 1 μm; C, D, 0.5 μm.
Immunohistochemical studies were repeated with additional anti-mGluR1a (Romano et al., 1996) and anti-mGluR5 (Reid et al., 1995; Romano et al., 1995) antibodies and showed similar results confirming our findings of the postsynaptic localization of both of these receptor subtypes in the STN (data not shown). This is consistent with previousin situ hybridization studies (Testa et al., 1994) as well as studies of mGluR localization at the light level (Testa et al., 1998).
Effects of DHPG in STN neurons are mediated by mGluR5
The postsynaptic localization of both mGluR1a and mGluR5 in STN neurons suggests that either or both of these receptors could be involved in mediating the effects of DHPG. We used newly available pharmacological tools that distinguish between these two group I mGluR subtypes to further characterize the group I-mediated effects in STN. Interestingly MPEP (10 μm), a highly selective noncompetitive antagonist at mGluR5 (Bowes et al., 1999; Gasparini et al., 1999), blocked DHPG-induced membrane depolarization (4.2 ± 0.27 mV; p < 0.001; n = 3; Fig. 7A,B). In contrast, the mGluR1-selective noncompetitive antagonist CPCCOEt (100 μm) (Annoura et al., 1996; Casabona et al., 1997; Litschig et al., 1999), had no effect on DHPG-mediated depolarization of STN neurons (18.8 ± 3.0 mV; n = 3; Fig. 7A,B) at concentrations that have been shown to be effective at blocking mGluR1a in recombinant (Litschig et al., 1999) and native (Casabona et al., 1997) systems. These data suggest that DHPG-induced depolarization of STN neurons is mediated by mGluR5 rather than mGluR1. Consistent with this, CBPG (100 μm), a partial agonist of mGluR5 with mGluR1 antagonist activity (Mannaioni et al., 1999), mimics DHPG-induced depolarization of STN neurons. As with DHPG, the response to CBPG is blocked by MPEP but not by CPCCOEt (Fig. 7B).
Fig. 7.

mGluR5 mediates group I mGluR-evoked depolarization of STN neurons. A, Membrane potential traces showing depolarization with DHPG (100 μm), which is blocked by the mGluR5-selective antagonist MPEP (10 μm). Membrane depolarization is not blocked by the mGluR1-selective antagonist CPCCOEt (100 μm).B, Mean data ± SEM of change in membrane potential showing a significant inhibition of DHPG-mediated depolarization of STN neurons by MPEP (10 μm) compared with DHPG alone (**p < 0.001). MPEP also significantly blocks depolarization mediated by the mGluR5-selective agonist CBPG (100 μm) (*p < 0.05).
Pharmacological analysis of mGluR-mediated potentiation of NMDA receptor currents suggests that this response is also mediated by mGluR5. As with the depolarization, MPEP blocks DHPG-induced potentiation of NMDA-evoked currents (2.3 ± 3.2%;p < 0.05; n = 5), whereas CPCCOEt is without effect on this potentiation (44.2 ± 18.9%;n = 6; Fig. 8A,B).
Fig. 8.

mGluR5 mediates group I mGluR-induced potentiation of NMDA-evoked currents. A, Current traces of NMDA-evoked currents before, during, and after application of DHPG (100 μm). The potentiation is blocked by MPEP (10 μm) but not CPCCOEt (100 μm).B, Mean data ± SEM of percent potentiation of NMDA-evoked currents by DHPG over predrug conditions. MPEP (10 μm) significantly blocks potentiation of NMDA-evoked current compared with DHPG alone (*p < 0.05).
DISCUSSION
The data presented reveal that group I mGluRs are postsynaptically localized on neurons in the STN and that activation of these receptors leads to a direct depolarization of STN neurons. In most cells, the DHPG-induced depolarization was accompanied by an increase in firing frequency with no obvious effects on the recently characterized stable oscillations of STN neuronal firing (Bevan and Wilson, 1999). However, in approximately one-third of the cells examined, DHPG induced a switch in the firing pattern from the characteristic single-spike firing mode to a burst-firing mode that was recently characterized in detail byBeurrier et al. (1999). In addition, DHPG induced a selective increase in NMDA receptor currents in STN neurons. These combined effects of group I mGluR activation could provide an important component of the net excitatory drive elicited by activity of the major excitatory afferents to the STN from the cortex or thalamus. However, it is important to note that activation of group I mGluRs has also been shown to induce presynaptic effects on glutamate release in some brain regions (Gereau and Conn, 1995; Manzoni and Bockaert, 1995;Rodriguez-Moreno et al., 1998). If group I mGluRs have similar effects in the STN, the net effect of group I mGluR activation in this region will ultimately depend on a combination of presynaptic and postsynaptic effects.
Immunohistochemistry studies revealed that both mGluR1a and mGluR5 are postsynaptically localized in STN neurons. Interestingly, pharmacological analysis suggested that each of the responses studied was mediated by mGluR5, with little or no contribution of mGluR1. This finding suggests that although STN neurons contain both group I mGluR subtypes, there is a segregation of function of these two receptors. It is possible that mGluR1 also plays important roles in regulating STN functions that were not measured in the present studies. Future studies of the roles of mGluR1 in these cells may shed important light on the functions of expression of multiple subtypes of closely related receptors by a single neuronal population.
Implications of mGluR5 actions for treatment of PD
One of the most interesting implications of the finding that mGluR5 is involved in regulating activity and NMDA receptor currents in STN neurons is the possibility that antagonists of this receptor could provide novel therapeutic agents that could be useful for treatment of PD. Traditional dopamine replacement strategies for PD treatment tend to lose efficacy over time, and patients begin to experience serious adverse effects, including motor fluctuations (Poewe and Granata, 1997). Because of this, a great deal of effort has been focused on developing a detailed understanding of the circuitry and function of the BG in the hopes of developing novel therapeutic approaches for the treatment of PD. Interestingly, a large number of animal and clinical studies reveal that loss of nigrostriatal dopamine neurons results in an increase in activity of the STN and that an increase in STN-induced excitation of the output nuclei is ultimately responsible for the motor symptoms of PD (for review, see DeLong, 1990; Wichmann and DeLong, 1997). These findings suggest that pharmacological agents that reduce the excitatory drive to the STN or otherwise reduce STN activity could provide a therapeutic effect in PD patients.
The data reported here suggest that mGluR5 may be a particularly interesting candidate as a receptor that could regulate STN output. Of particular interest is the finding that mGluR5 activation increases burst firing of STN neurons. For instance, previous studies suggest that a transition of STN neurons from single-spike activity to a burst-firing mode is one of the characteristics of parkinsonian states in rats and nonhuman primates (Hollerman and Grace, 1992; Bergman et al., 1994; Hassani et al., 1996) as well as parkinsonian patients (Benazzouz et al., 1996; Rodriguez et al., 1997). The finding that membrane oscillations underlying burst firing occur in the presence of TTX is consistent with the findings of Beurrier et al. (1999) and suggests that burst firing is, in part, an intrinsic property of STN neurons. However, these data do not rule out the possibility that synaptic mechanisms also participate in induction of burst firing (Plenz and Kitai, 1999). Consistent with the hypothesis that group I mGluRs can increase the output of STN in vivo, Kaatz and Albin (1995) recently reported that injection of group I mGluR agonists into the STN induces rotational behavior. Furthermore, Kronthaler and Schmidt reported that (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (1996) and (25,35,45)-α-carboxycyclopropyl glycine (1998), agonists of both group I and group II mGluRs, induce catalepsy in rats. These effects are not mimicked by a highly selective agonist of group II mGluRs (Marino et al., 1999b; Bradley et al., 2000), suggesting that it is likely mediated by mGluRs belonging to group I. Taken together, these findings raise the exciting possibility that mGluR5 antagonists could reduce STN activity and thereby provide a therapeutic benefit to PD patients.
For antagonists of group I mGluRs to be effective in the treatment of PD, mGluR5 must be physiologically activated by endogenous glutamate release onto STN neurons from various glutamatergic afferents. We made several attempts to elicit slow mGluR-mediated synaptic responses using single-pulse stimuli as well as stimulus trains of varying frequencies and durations. Unfortunately, we were unable to reliably elicit mGluR-mediated EPSPs or EPSCs in our slices (our unpublished findings). Although disappointing, this is not surprising, because mGluR-mediated slow EPSPs have also been difficult to measure in other brain regions, except those in which there is a laminar or other organization in which glutamatergic afferent pathways are not severed by slice preparation. In addition to severing afferent projections during slice preparation, mGluR-mediated slow EPSPs are often difficult to measure because of the small size of the events as well as the distance between the recording site in the soma and the distal dendrites where the slow EPSP is likely generated. Because of this, failure to measure a slow EPSP does not imply that mGluRs are not synaptically activated in vivo. Also, we often elicited slow EPSPs that were not blocked by antagonists of known receptors and could have occluded an mGluR-mediated slow EPSP (our unpublished findings). Ultimately, the question of whether mGluR5 in the STN is activated by endogenous glutamate may requirein vivo electrophysiological and/or behavioral studies in which mGluR5 antagonists are injected into this structure.
Potential therapeutic effects of mGluR5 agonist actions in the STN
In addition to the potential utility of mGluR5 antagonists in treatment of PD, it is important to point out that the actions of mGluR5 agonists could provide a therapeutic benefit in some other motor disorders, such as Tourette's syndrome and Huntington's disease. Tourette's syndrome is a relatively common neuropsychiatric disorder that is characterized by motor and phonic tics that can include sudden repetitive movements, gestures, or utterances. According to current models, Tourette's syndrome is associated with an increase in striatal dopamine or in the dopamine sensitivity of striatal neurons that has effects that are opposite of those seen in PD patients (Albin et al., 1989; Leckman et al., 1997). Huntington's disease is another hyperkinetic disorder that is thought to be caused by a selective loss of striatal spiny neurons that gives rise to the indirect pathway and, consequently, a decrease in STN activity (Reiner et al., 1988; Albin et al., 1990). On the basis of this, it is possible that selective mGluR5 agonists could provide a therapeutic benefit to patients suffering from these hyperkinetic disorders by increasing activity of STN neurons.
Roles of group I mGluRs in other basal ganglia nuclei
Interestingly, agonists of group I mGluRs have actions in other areas of the basal ganglia motor circuit that could complement their actions in the STN. For instance, group I mGluRs, and especially mGluR5, are heavily localized in the striatum (Shigemoto et al., 1993;Tallaksen-Greene et al., 1998), where agonists of these receptors induce excitatory effects similar to those described here in the STN (Calabresi et al., 1993; Colwell and Levine, 1994; Pisani et al., 1997). Furthermore, recent behavioral studies reveal that injection of group I mGluR agonists into the striatum induces turning behavior that is accompanied by an increase in activity of neurons in the STN and BG output nuclei (Sacaan et al., 1991, 1992; Kaatz and Albin, 1995; Kearney et al., 1997). Group I mGluRs are also present in the SNr (Hubert and Smith, 1999). Recent physiological studies from our laboratory suggest that activation of these receptors has direct excitatory effects and decreases evoked IPSCs (Marino et al., 1999a) in SNr neurons. Taken together, these data suggest that group I mGluRs function at multiple levels of the BG circuit to lead to a net increase in activity of neurons in the output nuclei. Thus, in addition to the STN, both antagonists and agonists of group I mGluRs could act at the levels of the striatum and SNr to provide a therapeutic benefit in the treatment of PD or hyperkinetic disorders, respectively.
Footnotes
This work was supported by grants from the National Institutes of Health National Institute of Neurological Disorders and Stroke, the Tourette's Syndrome Foundation, and the US Army Medical Research and Material Command. We thank Dr. Carmelo Romano (Washington University) for supplying anti-mGluR1a and anti-mGluR5 antibodies, Dr. Darryle Schoepp and Dr. James Monn (Eli Lilly) for supplying LY354740, Dr. Rainer Kuhn (Novartis) for supplying MPEP and CPCCOEt, and Stephanie Carter for valuable technical assistance.
Correspondence should be addressed to Dr. P. Jeffrey Conn, Emory University, Department of Pharmacology, Rollins Research Center, 1510 Clifton Road, Atlanta, GA 30322. E-mail: Pconn@emory.edu.
REFERENCES
- 1.Abbott A, Wigmore MA, Lacey MG. Excitation of rat subthalamic nucleus neurones in vitro by activation of a group I metabotropic glutamate receptor. Brain Res. 1997;766:162–167. doi: 10.1016/s0006-8993(97)00550-7. [DOI] [PubMed] [Google Scholar]
- 2.Afsharpour S. Light microscopic analysis of Golgi-impregnated rat subthalamic nucleus neurons. J Comp Neurol. 1985;236:1–13. doi: 10.1002/cne.902360102. [DOI] [PubMed] [Google Scholar]
- 3.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 4.Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington's disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol. 1990;27:357–365. doi: 10.1002/ana.410270403. [DOI] [PubMed] [Google Scholar]
- 5.Annoura H, Fukunaga A, Uesugi M, Tatsouka T, Horikawa Y. A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropchromen-1a-carboxylates. Bioorg Med Chem Lett. 1996;6:763–766. [Google Scholar]
- 6.Aziz TZ, Peggs D, Sambrook MA, Crossman AR. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6:288–292. doi: 10.1002/mds.870060404. [DOI] [PubMed] [Google Scholar]
- 7.Benazzouz A, Gross C, Féger J, Boraud T, Bioulac B. Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur J Neurosci. 1993;5:382–389. doi: 10.1111/j.1460-9568.1993.tb00505.x. [DOI] [PubMed] [Google Scholar]
- 8.Benazzouz A, Piallat B, Pollak P, Limousin P, Gao DM, Krack P, Benabid AL (1996) Single unit recordings of subthalamic nucleus and pars reticulata of substantia nigra in akineto-rigid parkinsonian patients. Soc Neurosci Abstr 225.
- 9.Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1346–1348. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- 10.Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- 11.Beurrier C, Congar P, Bioulac B, Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci. 1999;19:599–609. doi: 10.1523/JNEUROSCI.19-02-00599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bevan MD, Wilson CJ. Mechanisms underlying spontaneous oscillation and rythmic firing in rat subthalamic neurons. J Neurosci. 1999;19:7617–7628. doi: 10.1523/JNEUROSCI.19-17-07617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowes M, Panesar M, Gentry C, Urban L, Gasparini F, Kuhn R, Walker K. Anti-hyperalgesic effects of the novel metabotropic glutamate receptor 5 antagonist, 2-methyl-6-(phenylethynyl)-pyridine, in rat models of inflammatory pain. Br J Pharmacol. 1999;125:250.P. [Google Scholar]
- 14.Bradley SR, Rees HD, Yi H, Levey AI, Conn PJ. Distribution and developmental regulation of metabotropic glutamate receptor 7a in rat brain. J Neurochem. 1998;71:636–645. doi: 10.1046/j.1471-4159.1998.71020636.x. [DOI] [PubMed] [Google Scholar]
- 15.Bradley SR, Standaert DG, Rhodes KJ, Rees HD, Testa CM, Levey AI, Conn PJ. Immunohistochemical localization of subtype 4a metabotropic glutamate receptor in the rat and mouse basal ganglia. J Comp Neurol. 1999;407:33–46. [PubMed] [Google Scholar]
- 16.Bradley SR, Marino MJ, Wittmann M, Rouse ST, Awad H, Levey AI, Conn PJ. Activation of group II metabotropic glutamate receptors inhibits synaptic excitation of the substantia nigra pars reticulata. J Neurosci. 2000;20:3085–3094. doi: 10.1523/JNEUROSCI.20-09-03085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Heterogeneity of metabotropic glutamate receptors in the striatum: electrophysiological evidence. Eur J Neurosci. 1993;5:1370–1377. doi: 10.1111/j.1460-9568.1993.tb00923.x. [DOI] [PubMed] [Google Scholar]
- 18.Casabona G, Knoepfel T, Kuhn R, Gasparini F, Baumann P, Sortino MA, Copani A, Nicoletti F. Expression and coupling to polyphosphoinositide hydrolysis of group I metabotropic glutamate receptors in early postnatal and adult rat brain. Eur J Neurosci. 1997;9:12–17. doi: 10.1111/j.1460-9568.1997.tb01348.x. [DOI] [PubMed] [Google Scholar]
- 19.Colwell CS, Levine MS. Metabotropic glutamate receptors modulate N-methyl-d-aspartate receptor function in neostriatal neurons. Neuroscience. 1994;61:497–507. doi: 10.1016/0306-4522(94)90429-4. [DOI] [PubMed] [Google Scholar]
- 20.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 21.Crépel V, Aniksztein L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated non-specific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 22.Davies J, Watkins JC. Actions of D and L forms of 2-amino-5-phosphonovalerate and 2-amino-4-phosphonobutyrate in the cat spinal cord. Brain Res. 1982;235:378–386. doi: 10.1016/0006-8993(82)91017-4. [DOI] [PubMed] [Google Scholar]
- 23.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 24.Gasparini F, Lingenhol K, Stoehr N, Flor P, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 25.Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guérineau NC, Gähwiler BH, Gerber U. Reduction of resting K+ current by metabotropic glutamate and muscarinic receptors in rat CA3 cells: mediation by G-proteins. J Physiol (Lond) 1994;474:27–33. doi: 10.1113/jphysiol.1994.sp019999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guérineau NC, Bossu J-L, Gähwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guirdi J, Herrero MT, Luquin MR, Guillén J, Ruberg M, Laguna J, Vila M, Javoy-Agid Y, Hirsch E, Obeso A. Subthalamotomy in parkinsonian monkeys. Behavioural and biochemical analysis. Brain. 1996;119:1717–1727. doi: 10.1093/brain/119.5.1717. [DOI] [PubMed] [Google Scholar]
- 29.Hassani OK, Mouroux M, Féger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- 30.Hollerman JR, Grace AA. Subthalamic nucleus cell firing in the 6-OHDA-treated rat: basal activity and response to haloperidol. Brain Res. 1992;590:291–299. doi: 10.1016/0006-8993(92)91108-q. [DOI] [PubMed] [Google Scholar]
- 31.Hubert GW, Smith Y. Subsynaptic localization of group I metabotropic glutamate receptors (mGluRs) in the ventral midbrain of rats. Soc Neurosci Abstr. 1999;25:1156. [Google Scholar]
- 32.Iwahori N. A Golgi study on the subthalamic nucleus of the cat. J Comp Neurol. 1978;182:383–397. doi: 10.1002/cne.901820303. [DOI] [PubMed] [Google Scholar]
- 33.Kaatz KW, Albin RL. Intrastriatal and intrasubthalamic stimulation of metabotropic glutamate receptors: a behavioral and Fos immunohistochemical study. Neuroscience. 1995;66:55–65. doi: 10.1016/0306-4522(94)00568-p. [DOI] [PubMed] [Google Scholar]
- 34.Kearney JAF, Frey KA, Albin RL. Metabotropic glutamate agonist-induced rotation: a pharmacological, FOS immunohistochemical, and [14C]-2-deoxyglucose autoradiographic study. J Neurosci. 1997;17:4415–4425. doi: 10.1523/JNEUROSCI.17-11-04415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kronthaler UO, Schmidt WJ. 1S,3R-ACPD has cataleptogenic effects and reverses MK-801- and less pronounced, d,l-amphetamine-induced locomotion. Eur J Pharmacol. 1996;316:129–136. doi: 10.1016/s0014-2999(96)00673-5. [DOI] [PubMed] [Google Scholar]
- 36.Kronthaler UO, Schmidt WJ. The mGluR group II agonist (2S,3S,4S)-alpha-carboxycyclopropyl-glycine induces catalepsy in the rat, which is pronouncedly antagonised by dizocilpine and d,l-amphetamine. Neurosci Lett. 1998;253:25–28. doi: 10.1016/s0304-3940(98)00593-x. [DOI] [PubMed] [Google Scholar]
- 37.Leckman JF, Peterson BS, Anderson GM, Arnsten AF, Pauls DL, Cohen DJ. Pathogenesis of Tourette's syndrome. J Child Psychol Psychiatry. 1997;38:119–142. doi: 10.1111/j.1469-7610.1997.tb01508.x. [DOI] [PubMed] [Google Scholar]
- 38.Limousin P, Pollak P, Benazzouz A, Hoffman D, Broussolle E, Perret JE, Benabid AL. Bilateral subthalamic nucleus stimulation for severe parkinson's disease. Mov Disord. 1995a;10:672–674. doi: 10.1002/mds.870100523. [DOI] [PubMed] [Google Scholar]
- 39.Limousin P, Pollak P, Benazzouz A, Hoffman D, Le Bas JF, Broussolle E, Perret JE, Benabid AL. Effect on parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet. 1995b;345:91–95. doi: 10.1016/s0140-6736(95)90062-4. [DOI] [PubMed] [Google Scholar]
- 40.Litschig S, Gasparini F, Ruegg D, Stoehr N, Flor PJ, Vranesic I, Prezeau L, Pin JP, Thomsen C, Kuhn R. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- 41.Mannaioni G, Attucci S, Missanelli A, Pellicciari R, Corradetti R, Moroni F. Biochemical and electrophysiological studies on (S)-(+)-2-(3′-carboxybicyclo[1.1.1]pentyl)-glycine (CBPG), a novel mGlu5 receptor agonist endowed with mGlu1 receptor antagonist activity. Neurpharmacology. 1999;38:917–926. doi: 10.1016/s0028-3908(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 42.Manzoni O, Bockaert J. Metabotropic glutamate receptors inhibiting excitatory synapses in the CA1 area of rat hippocampus. Eur J Neurosci. 1995;7:2518–2523. doi: 10.1111/j.1460-9568.1995.tb01051.x. [DOI] [PubMed] [Google Scholar]
- 43.Marino MJ, Bradley SR, Wittmann M, Conn PJ. Direct excitation of GABAergic projection neurons of the rat substantia nigra pars reticulata by activation of the mGluR1 metabotropic glutamate receptor. Soc Neurosci Abstr. 1999a;25:446. doi: 10.1523/JNEUROSCI.21-18-07001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marino MJ, Bradley SR, Wittmann M, Rouse ST, Levey AI, Conn PJ. Potential antiparkinsonian actions of metabotropic glutamate receptors in the substantia nigra pars reticulata. Proceedings of the Third International Meeting on Metabotropic Glutamate Receptors, 93. Neuropharmacology. 1999b;38:A12. [Google Scholar]
- 45.Monn JA, Valli MJ, Massey SM, Wright RA, Salhoff CR, Johnson BG, Howe T, Alt CA, Rhodes GA, Robey RL, Griffey KR, Tizzano JP, Kallman MJ, Helton DR, Schoepp DD. Design, synthesis, and pharmacological characterization of (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740): a potent, selective, and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic properties. J Med Chem. 1997;40:528–537. doi: 10.1021/jm9606756. [DOI] [PubMed] [Google Scholar]
- 46.Pisani A, Calabresi P, Centonze D, Bernardi G. Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol. 1997;120:1007–1014. doi: 10.1038/sj.bjp.0700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plenz D, Kitai ST. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400:677–682. doi: 10.1038/23281. [DOI] [PubMed] [Google Scholar]
- 48.Poewe W, Granata R. Pharmacological treatment of Parkinson's disease. In: Watss RL, editor. Movement disorders: neurological principles and practice. McGraw-Hill; New York: 1997. pp. 201–219. [Google Scholar]
- 49.Pozzo Miller LD, Petrozzino JJ, Connor JA. G protein-coupled receptors mediate a fast excitatory postsynaptic current in CA3 pyramidal neurons in hippocampal slices. J Neurosci. 1995;15:8320–8330. doi: 10.1523/JNEUROSCI.15-12-08320.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reid SNM, Romano C, Hughes T, Daw NW. Immunohistochemical study of two phosphoinositide-linked metabotropic glutamate receptors (mGluR1a and mGluR5) in the cat visual cortex before, during, and after the peak of the critical period for eye-specific conditions. J Comp Neurol. 1995;355:470–477. doi: 10.1002/cne.903550311. [DOI] [PubMed] [Google Scholar]
- 51.Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington's disease. Proc Natl Acad Sci USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez MC, Gorospe A, Mozo A, Guirdi J, Ramos E, Linazasoro G, Obeso JA. Characteristics of neuronal activity in the subthalamic nucleus (STN) and substantia nigra pars reticulata (SNr) in Parkinson's disease (PD). Soc Neurosci Abstr. 1997;23:470. [Google Scholar]
- 53.Rodriguez-Moreno A, Sistiaga A, Lerma J, Sanchez-Prieto J. Switch from facilitation to inhibition of excitatory synaptic transmission by group I mGluR desensitization. Neuron. 1998;21:1477–1486. doi: 10.1016/s0896-6273(00)80665-0. [DOI] [PubMed] [Google Scholar]
- 54.Romano C, Sesma MA, McDonald CT, O'Malley K, Van Den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 55.Romano C, Yang WL, O'Malley KL. Metabotropic glutamate receptor 5 is a disulfide-linked dimer. J Biol Chem. 1996;271:28612–28616. doi: 10.1074/jbc.271.45.28612. [DOI] [PubMed] [Google Scholar]
- 56.Sacaan AI, Monn JA, Scheopp DD. Intrastriatal injection of a selective metabotropic excitatory amino acid receptor agonist induces contralateral turning in the rat. J Pharmacol Exp Ther. 1991;259:1366–1370. [PubMed] [Google Scholar]
- 57.Sacaan AI, Bymaster FP, Schoepp DD. Metabotropic glutamate receptor activation produces extrapyramidal motor system activation that is mediated by striatal dopamine. J Neurochem. 1992;59:245–251. doi: 10.1111/j.1471-4159.1992.tb08897.x. [DOI] [PubMed] [Google Scholar]
- 58.Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR. 3,3-Dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J Neurochem. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- 59.Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- 60.Tallaksen-Greene SJ, Kaatz KW, Romano C, Albin RL. Localization of mGluR1a-like immunoreactivity and mGluR5-like immunoreactivity in identified populations of striatal neurons. Brain Res. 1998;780:210–217. doi: 10.1016/s0006-8993(97)01141-4. [DOI] [PubMed] [Google Scholar]
- 61.Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Testa CM, Friberg IK, Weiss SW, Standaert DG. Immunohistochemical localization of metabotropic glutamate receptors mGluR1a and mGluR2/3 in the rat basal ganglia. J Comp Neurol. 1998;390:5–19. [PubMed] [Google Scholar]
- 63.Wichmann T, DeLong MR. Physiology of the basal ganglia and pathophysiology of movement disorders of basal ganglia origin. In: Watts RL, editor. Movement disorders: neurological principles and practice. McGraw-Hill; New York: 1997. pp. 87–97. [Google Scholar]