

Abstract
Birth asphyxia can cause moderate to severe brain injury. It is unclear to what degree apoptotic or necrotic mechanisms of cell death account for damage after neonatal hypoxia–ischemia (HI). In a 7-d-old rat HI model, we determined the contributions of apoptosis and necrosis to neuronal injury in adjacent Nissl-stained, hematoxylin and eosin-stained, and terminal deoxynucleotidyl transferase-mediated UTP nick end-labeled sections. We found an apoptotic–necrotic continuum in the morphology of injured neurons in all regions examined. Eosinophilic necrotic neurons, typical in adult models, were rarely observed in neonatal HI. Electron microscopic analysis showed “classic” apoptotic and necrotic neurons and “hybrid” cells with intermediate characteristics. The time course of apoptotic injury varied regionally. In CA3, dentate gyrus, medial habenula, and laterodorsal thalamus, the density of apoptotic cells was highest at 24–72 hr after HI and then declined. In contrast, densities remained elevated from 12 hr to 7 d after HI in most cortical areas and in the basal ganglia. Temporal and regional patterns of neuronal death were compared with expression of caspase-3, a cysteine protease involved in the execution phase of apoptosis. Immunocytochemical and Western blot analyses showed increased caspase-3 expression in damaged hemispheres 24 hr to 7 d after HI. A p17 peptide fragment, which results from the proteolytic activation of the caspase-3 precursor, was detected in hippocampus, thalamus, and striatum but not in cerebral cortex. The continued expression of activated caspase-3 and the persistence of cells with an apoptotic morphology for days after HI suggests a prolonged role for apoptosis in neonatal hypoxic ischemic brain injury.
Keywords: apoptosis, necrosis, hypoxia-ischemia, cysteine proteases, caspase-3 cleavage, cell death continuum, cerebral palsy, newborn brain injury, developmental brain
Birth asphyxia can cause cerebral hypoxic ischemic injury, resulting in severe neurological sequelae and death. Survivors of perinatal asphyxia frequently have moderate to severe brain injury for which there currently is no promising therapy (Johnston, 1997). The results of morphological, histochemical, and molecular studies indicate that apoptotic and necrotic mechanisms account for neuronal death after cerebral hypoxia–ischemia (HI) in different neonatal animal models (Mehmet et al., 1994;Charriaut-Marlangue et al., 1996a,b; Chopp and Li, 1996; Macaya, 1996;Yue et al., 1997; Banasiak and Haddad, 1998; Pulera et al., 1998;Renolleau et al., 1998). For example, a neonatal ischemia-reperfusion model showed terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL)-positive nuclei from 4 hr to 30 d after reperfusion (Renolleau et al., 1998). A human study demonstrated apoptotic and necrotic forms of cell death after hypoxic injury, whereas in some brains from stillbirths, only apoptotic figures were observed (Scott and Hegyi, 1997). The form of cell death also may depend on the severity of ischemic injury (Kerr et al., 1972; Bonfoco et al., 1995). Necrosis predominates in more severe cases, whereas apoptosis occurs in areas with milder ischemic injury, often days after the initial insult (Stroemer and Rothwell, 1998).
In general, hypoxic ischemic damage in the immature brain evolves more rapidly than its adult counterpart (Rice et al., 1981; Towfighi et al., 1995). In vitro evidence indicates an increased susceptibility to apoptosis in immature cortical neurons (McDonald et al., 1997). However, the relative contribution of apoptotic and necrotic mechanisms to cell death in neonatal ischemia is unknown. In the present study, we characterized the time course of hypoxic ischemic brain injury in neonates in selected regions. We used a 7-d-old rat HI model in which the pattern of brain injury resembles that of hypoxic ischemic injury in term human infants (Johnston, 1983; Vannucci, 1990).
Another aim was to study the relationship between temporal and regional patterns of neuronal cell death and caspase-3 protein expression in this model. Caspase-3 is a member of the interleukin-1β-converting enzyme family of cysteine proteases, which trigger the execution phase of apoptosis (Yuan et al., 1993; Porter and Janicke, 1999). In various ischemia models, caspase-3 is an important neuronal death effector (Endres et al., 1997; Hara et al., 1997a; Chen et al., 1998; Cheng et al., 1998; Endres et al., 1998; Namura et al., 1998; Ni et al., 1998;Schulz et al., 1998, 1999). Caspase inhibitors are neuroprotective in adult ischemia models (Hara et al., 1997b; Cheng et al., 1998), even when animals are treated 6–9 hr after the onset of ischemia (Endres et al., 1998; Fink et al., 1998). We also examined whether caspase-3 displayed prolonged activation in this model. An extended period of apoptosis and caspase-3 activation in neonatal brain after HI (Li et al., 1998) would support the possibility of a prolonged therapeutic window for intervention (Cheng et al., 1998).
MATERIALS AND METHODS
Unilateral cerebral hypoxia–ischemia model. All experiments were performed in accordance with approved institutional animal care guidelines. Common carotid artery occlusion combined with hypoxia in postnatal day 7 (P7) rat pups was used as described previously (Trescher et al., 1997) to produce hypoxic ischemic brain damage. P7 Sprague Dawley rats from Charles River Laboratories (Wilmington, MA) rat pups were separated from their mothers and placed in a temperature-controlled incubator set to an ambient temperature of 35°C. Under deep ether anesthesia, the right common carotid artery was isolated, double-ligated, and cut between the ligatures. After the surgical procedure, the animals recovered for 2 hr in a temperature-controlled incubator and then were exposed to a 2 hr period of hypoxia. Pups were placed in enclosed, vented chambers that were partially submerged in water (36.5°C). Hypoxia was induced by continuous flow of warmed, humidified gas (8% oxygen, balanced with nitrogen). Nontreated littermates served as controls.
Tissue preparation. Pups were killed at various times after hypoxic ischemic exposure [0, 6, 12, 24, 48, 72, and 168 hr (7 d) after HI]. Four separate sets of animals were subjected to HI. The brains for the first group (Nissl staining and caspase-3 immunocytochemistry) were perfusion-fixed and frozen (n= 2 at 0 and 6 hr; n = 3 at 12 and 168 hr;n = 5 at 24 and 48 hr; n = 4 at 72 hr). Another group of brains [hematoxylin and eosin (H&E), Nissl, and TUNEL staining] were perfusion-fixed and paraffin-embedded (n = 2 at 0, 6, 12, and 168 hr; n = 4 at 24 hr; n = 3 at 48 and 72 hr). A third group of brains were perfusion-fixed and processed for electron microscopy (n = 2 at the 24 and 48 hr survival times). A fourth group of brains (Western blotting) was fresh-frozen (n= 4 for HI brains; n = 1 for nontreated controls at each time point).
Nissl staining. For dark-field microscopy of Nissl-stained sections and caspase-3 immunohistochemistry, rat pups were anesthetized with 300 mg/kg chloral hydrate intraperitoneally and then perfused through the left ventricle with 0.1 m PBS, followed by 4% paraformaldehyde (PAF) in 0.1 mphosphate buffer, pH 7.4 (4°C). Brains were then removed, post-fixed for 6–10 hr in the same fixative, and cryoprotected in 15 and 30% (w/v) sucrose in PBS. They were frozen with powdered dry ice and stored at −70°C. Frozen PAF-fixed brains were sectioned at a thickness of 50 μm on a sliding microtome. For Nissl staining, sections were mounted on gelatin-coated slides and stained with cresyl violet as described previously (Lange et al., 1999).
Immunocytochemistry. Sections adjacent to the Nissl-stained sections were processed for caspase-3 immunocytochemistry. Sections were incubated in PBS with 5% normal goat serum, 0.2% Triton X-100, and 0.2% gelatin solution (PBS-GS) as preblocking step, followed by incubation overnight at 4°C in the caspase-3 antibody (anti-caspase-3/yama/apopain; Upstate Biotechnology, Lake Placid, NY) diluted 1:200 in PBS-GS. The caspase-3 antibody was visualized using the avidin-biotin peroxidase complex method (Blue et al., 1997).
Paraffin embedding. Rat pups were perfused under deep anesthesia with PBS, followed by 4% PAF in 0.1 mPBS. Brains were removed, kept in the same fixative solution for 1 week, and then embedded in paraffin. Paraffin-embedded brains were sectioned at a thickness of 6 μm and processed for H&E or Nissl staining or TUNEL as described previously (Lange et al., 1999).
In situ labeling of 3′-OH DNA ends (ApopTag). To investigate the relationship between DNA fragmentation and cell morphology,in situ end labeling was performed on paraffin-embedded sections to identify DNA fragments from cells dying by apoptosis and necrosis (Yasuda et al., 1995; de Torres et al., 1997). 3′-OH ends of DNA fragments were detected using the ApopTag peroxidase kit from Intergen Company (Purchase, NY). To reduce the background staining and increase the sensitivity, we adopted a combined pretreatment method (Negoescu et al., 1996, 1998; Labat-Moleur et al., 1998). The slides were immersed in 0.01 m citrate buffer, pH 3, heated for two cycles of 5 min microwave irradiation, and incubated in a 20 μg/ml solution of proteinase K. No inhibition of endogenous peroxidase was performed because H2O2 weakens terminal deoxynucleotidyl transferase (TdT) activity (Migheli et al., 1995) and induces DNA breaks (Wijsman et al., 1993). After combined pretreatment, sections were blotted and incubated with a mixture containing TdT enzyme and digoxigenin-tagged dUTP. Negative controls had the TdT omitted. The reaction product was visualized by 1–2 min incubation in 0.05% diaminobenzidine. The TUNEL-labeled slides were counterstained with 1% methyl green (Lange et al., 1999).
Screening of degenerating neurons. Two authors blinded to the experimental protocol evaluated the injury in Nissl-stained sections from seven brain regions (cerebral cortex, hippocampus CA1, dentate gyrus (DG), striatum, globus pallidus, laterodorsal thalamus, and medial habenula). These areas were selected because of their known susceptibility to hypoxic ischemic injury (Volpe, 1995). Each investigator rated the damage using dark-field microscopy. This method identifies cells with clumped chromatin, independent of the type of cell death. The rating scale for injury was as follows: 0, single or no clusters of degenerating neurons; 1, occasional clusters (mild); 2, multiple clusters (moderate); and 3, confluent clusters (severe). For this analysis, only the right hemisphere (ipsilateral to ligation) was evaluated.
Neuropathological analysis of apoptosis and necrosis.Another semiquantitative analysis evaluated, at each time point, in both hemispheres, the degree of apoptotic and necrotic injury in cerebral cortex (frontal, cingulate, parietal, and retrosplenial areas), basal ganglia (striatum and globus pallidus), hippocampus (CA1, CA3, and dentate gyrus), laterodorsal thalamus, subiculum, and medial habenula. Two investigators blinded to treatment examined adjacent H&E-stained, Nissl-stained, and TUNEL-labeled sections. In H&E- and Nissl-stained sections, the identification of apoptotic cells depended on the recognition of round or oval apoptotic bodies (Gavrieli et al., 1992; Wijsman et al., 1993). Because most apoptotic cells have multiple apoptotic bodies (2–20 per cell) (Li et al., 1995b,c, 1998), a cell was defined as apoptotic if it had two or more round, regularly shaped, dark purple chromatin clumps. Necrotic cells in H&E-stained sections were identified by their intense cytoplasmic eosinophilia, dispersed nuclear chromatin, and the loss of nuclear membrane integrity (karyolysis) (Wyllie et al., 1980; Majno and Joris, 1995) or by nuclear changes alone without eosinophilic cytoplasm. Of note, chromatin changes alone could not be used as reliable markers for type of cell death. Karyolysis is indicative of necrosis, but pyknosis (chromatin condensation and nuclear shrinkage) and karyorrhexis (fragmentation of condensed chromatin, also seen with TUNEL) are features of both apoptosis and necrosis.
Statistical analysis. For both semiquantitative analyses, we used the statistical program (SPSS Professional Statistics version 7.5; SPSS Inc., Chicago, IL) to evaluate inter-rater reliability. A split-half reliability analysis was performed to compare the ratings of injury by the two investigators. A multivariate repeated measures ANOVA was also used to study the differences among raters, regions, and survival. For each animal, scores of the two investigators were averaged; reported values represent the mean ± SEM for the animals in each group.
Electron microscopy. Twenty-four and 48 hr after HI, pups (n = 2 at each time point) were anesthetized with chloral hydrate and perfused with 70 ml of PBS, followed by 150 ml of 1% paraformaldehyde and 1.25% glutaraldehyde in 0.1m PBS, pH 7.4. After perfusion, the brains were removed and post-fixed overnight in the same fixative. Samples of cortex, hippocampus, and thalamus from both hemispheres were microdissected from each rat, post-fixed for 2 hr in 1% osmium tetroxide, washed in PBS, dehydrated in ethanol, and embedded in epoxy resin. Semithin (1 μm) sections were stained with cresyl violet and screened. An area of interest was selected, and ultrathin sections were cut and placed on single-hole grids. After staining with uranyl acetate and lead citrate, the sections were examined using a Phillips EM 120 electron microscope.
Western blots. A fourth set of animals was anesthetized with 300 mg/kg chloral hydrate intraperitoneally and killed. The cortex, hippocampus, striatum, and thalamus from both hemispheres were dissected from P7 rat pups at 0, 3, 6, 12, 24, 48, 72, and 168 hr after HI (four animals for HI and one nontreated control at each time point). Tissue was frozen on powdered dry ice and stored at −80°C until use. After thawing in lysis buffer [50 mm Tris-HCl, pH 7.4, containing 2 mm EDTA and one tablet complete protease inhibitor cocktail/10 ml (Boehringer Mannheim, Mannheim, Germany], the tissue was homogenized and centrifuged at 12,000 × g at 4°C for 15 min. Protein was denatured in SDS gel–loading buffer (0.125 m Tris, pH 6.8, 20% glycerol, 10% mercaptoethanol, 4% SDS, and 0.002% bromophenol blue) at 95°C for 2 min and separated on a 10% SDS-PAGE gel using a mini-protean system (Bio-Rad, Hercules, CA) with high-range molecular weight markers. Protein measurements were performed using a Bio-Rad protein assay; equal amounts of total protein (40 μg) were loaded in each lane. A purified caspase-3 standard (nonstimulated A-431 cell lysate; Upstate Biotechnology) was electrophoresed separately to verify that the induced bands migrated to the same location on the gel. After electrophoresis, the protein was transferred to a nitrocellulose membrane. Blots were blocked with 3% nonfat milk in 0.1% TBST (50 mm Tris-HCl, pH 7.4, 150 mmNaCl, and 0.1% Tween 20) and probed with the same caspase-3 antibody used for immunocytochemistry (1:200 dilution in 3% nonfat milk–TBST). Immunoblots were processed with horseradish peroxidase-conjugated anti-rabbit IgG; bound antibodies were detected using a Western blot chemiluminescence reagent kit (Life Science Products, Boston, MA). For a protein loading control, the membrane was reprobed with anti-α-tubulin antibody (1:50,000; Sigma, St. Louis, MO).
RESULTS
General observations
Brains from control rats appeared normal; both cerebral hemispheres had a similar appearance. The mortality rate of pups that underwent HI was ∼10% [group 1, 2 of 26 (8%); group 2, 3 of 21 (14%); group 3, 0 of 4; group 4 = 3 of 31 (10%)]; pups that died were excluded from the study. In the first group (n = 24; perfused and frozen), macroscopic observation of brains before 24 hr after HI revealed no detectable differences between the cerebral hemispheres. At 24 and 48 hr after HI, the hemispheres ipsilateral to the ligated side appeared edematous and slightly larger than the opposite hemispheres. At 72 hr after HI, the infarcted area became cystic. At 7 d, brains exhibited liquefaction with a partial collapse. In a second set of animals (n = 18; paraffin-embedded), the timetable and the severity of damage was different. Swelling in the ipsilateral hemisphere began at 6 hr after HI, and liquefaction was apparent by 48 hr after HI. The variability in the timing and in the degree of damage likely reflected individual difference in the sensitivity to HI. Although the severity of injury varied, at least 70% of rats that were examined at 24 hr after HI (n = 9) had developed infarcts in the ipsilateral cerebral hemisphere. In accordance with a previous report (Towfighi et al., 1995), the contralateral cortex in many of the brains was damaged. Eight of 24 pups (33%) in the first set and 13 of 18 pups (74%) in the second set had mild damage in the contralateral hemisphere.
Regional patterns of neuronal injury
In an adult ischemia model, we showed previously in Nissl-stained sections that the clumped chromatin in neurons undergoing degeneration refracted light under dark-field conditions and appeared as bright objects in a dark background. We found this method useful for detecting the regional patterns of dying neurons in Nissl-stained sections from rats that had undergone hypoxic ischemic injury at postnatal day 7. Using dark-field microscopy, no bright objects were observed 0 hr after hypoxic ischemic injury (Fig.1A,B). Clusters of degenerated cells were detected in the cortex and striatum from 6 to 168 hr (7 d) after HI (Fig. 1C–H). The occasional apoptotic cells in control brains and in the contralateral hemisphere of injured brains were not detected by the dark-field method. In frontal and parietal cortex, degenerated cells formed a columnar pattern (Figs. 1D–H,2A,3A), which matches that reported previously in the same model (Rice et al., 1981). In the striatum, clusters of degenerating cells formed a patchy pattern of bright objects, 24 and 48 hr (Figs. 1C,E,2B) and 7 d (Figs. 1G, 3B) after hypoxic ischemic injury. The striatum had more degenerating cells than the globus pallidus. Under bright field, a patchy pattern of cells with apoptotic and necrotic morphologies was observed (Figs.2B1, 3B1). At the 48 hr time point, we also observed degenerating cells in the lateral and laterodorsal thalamus, the medial habenula, and in CA1, CA3, and the dentate gyrus of the hippocampus (Figs. 1F, 2C). Under dark-field conditions, a ribbon of bright cells was present in right CA1 at 48 hr (Figs. 1F, 2C) and 7 d (Fig. 3C) after HI. Under bright field, the same sections showed a mosaic pattern of normal and degenerating cells in CA1 (Figs.2C1, 3C1). In the cortex, we found many apoptotic cells that possessed two or more round, chromatin clumps in the penumbra (Fig. 2A1). Necrotic cells with small or punctate, irregularly shaped chromatin clumps, predominated in the ischemic core (Fig. 2A2).
Fig. 1.
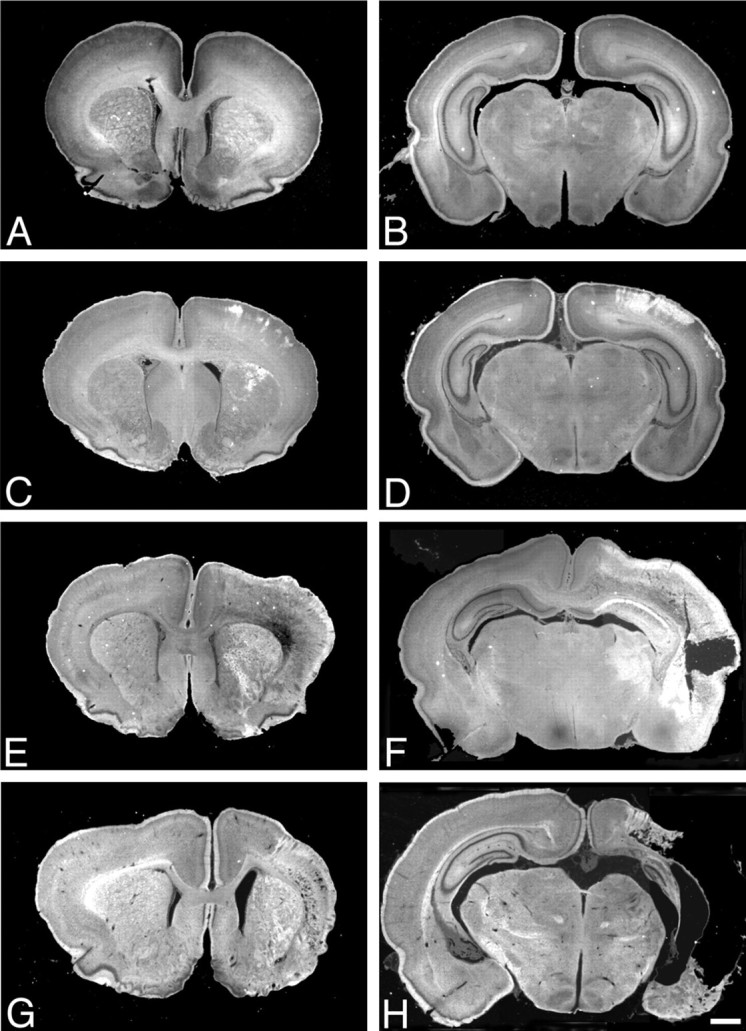
Temporal and regional patterns of damage after neonatal hypoxic ischemic injury. Dark-field photographs of coronal, Nissl-stained sections at rostral (A, C,E, G) and caudal (B,D, F, H) levels from rats killed at 0 (A, B), 24 (C, D), 48 (E,F), and 168 (G,H) hr after HI. Damaged areas appear as bright objects in a dark field. No injury was apparent at the 0 hr time point (A, B). Clusters of degenerated cells were detected in the cortex and striatum from 24 through 168 hr (7 d) after HI (C–H). Forty-eight hours after HI, lateral and laterodorsal thalamus, medial habenula, and CA1 hippocampus showed damage (F). Scale bar, 100 μm.
Fig. 2.
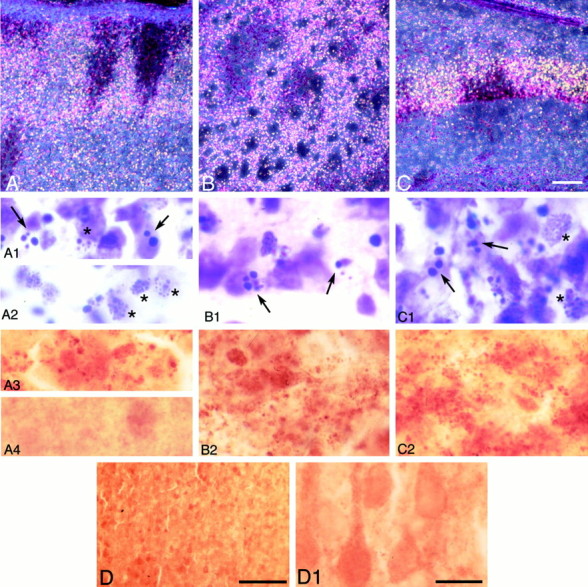
Degenerating neurons and caspase-3 immunostaining 48 hr after HI. Dark-field images from 50 μm, Nissl-stained sections of parietal cortex (A), striatum (B), and CA1 hippocampus (C) on the ligated side. A columnar pattern of degenerating neurons was observed in parietal cortex (A). High-magnification images from the penumbra (A1) and ischemic core (A2) showed apoptotic cells with large, round, chromatin clumps (arrows, A1) and necrotic cells with small, punctate chromatin clumps (asterisks,A1, A2). The striatum showed a patchy pattern of clusters of normal and degenerating neurons (B). At higher magnification, apoptotic cells with large, chromatin clumps (arrows, B1) were observed. In CA1 (C), the bright ribbon of degenerating cells contained apoptotic (arrows,C1) and necrotic (asterisks,C1) cells. Caspase-3 immunoreactivity showed a granular and particulate pattern in damaged areas within cerebral cortex (A3), striatum (B2), and CA1 (C2). Noninjured areas, in right parietal cortex 0 hr after HI, at low (D) and high (A4,D1) magnification, showed only diffuse, nonparticulate background staining. Scale bars: A–D, 100 μm;A1–D1, 10 μm.
Fig. 3.
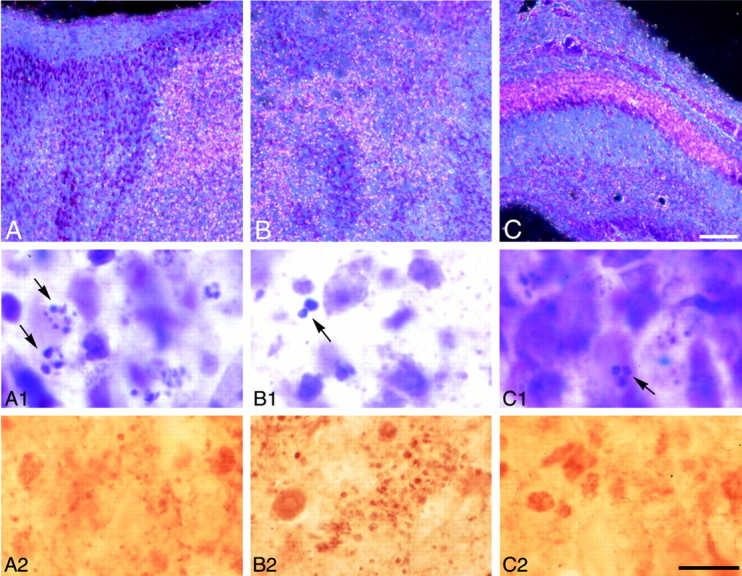
Degenerating neurons and caspase-3 immunostaining 7 d after HI. Dark-field images of parietal cortex (A), striatum (B), and CA1 hippocampus (C) on the ligated side show a pattern similar to that observed 48 hr after HI. High-magnification bright-field views of the fields in A–C show degenerating cells with varied sizes of chromatin clumps (arrows, A1–C1). Punctate caspase-3 immunoreactivity was distributed in cortex (A2) and striatum (B2) and CA1 (C2) in which apoptotic cells were observed, but the density is less in CA1 at this time point than at 48 hr (compare with Fig. 2C2). Scale bars: A–C, 100 μm; A1–C2, 10 μm.
Figure 4 shows the two distinct temporal patterns of degenerating cells observed in different regions after HI. The values represent the semiquantitative assessments of damage at each time point obtained using the dark-field method. Overall, inter-rater correlation was 0.9529, and the scores assigned by two investigators did not differ significantly. Statistical analysis showed significant regional differences and a significant interaction between region and survival time variables (p < 0.001). The density of degenerating cells in dentate gyrus, habenula, and thalamus was highest at 24–48 hr after HI and subsequently declined (Fig.4A). In cortex, striatum, and CA1, damaged cells were detected at 6 hr, and densities remained elevated from 24 hr to 7 d after HI (Figs. 1C–H, Fig. 4B).
Fig. 4.

Semiquantitative analysis of regional and temporal patterns of damage. Using the dark-field detection method, the degree of damage, which includes apoptosis, necrosis, and hybrid forms of cell death, was assessed semiquantitatively on a scale of 0 (little damage) to 3 (severe damage) at each time point by two investigators blinded to treatment (n = 2 at 0 and 6 hr;n = 3 at 12 and 168 hr; n = 5 at 24 and 48 hr; n = 4 at 72 hr). The average of the two scores for each animal was used to calculate the group means ± SEM shown here. In globus pallidus, thalamus, dentate gyrus, and medial habenula, the density of damaged cells peaked between 24 and 48 hr after hypoxic ischemic injury and then subsequently declined (A). In cortex, striatum, and CA1, the density of degenerating cells remained elevated through 7 d after HI (B).
Temporal and regional profiles of apoptotic and necrotic cells
In the second analysis, two raters blinded to treatment, evaluated apoptotic and necrotic injury in adjacent H&E-stained, Nissl-stained, and TUNEL-labeled sections. Overall, the degree of injury was more severe in this group of animals. At 6 hr after HI, we detected many cells with pyknotic nuclei that were TUNEL-positive in frontal, parietal, cingulate, and retrosplenial cortex, striatum, and globus pallidus on the ligated side. Figure 5shows the averaged score of the two raters for apoptotic and necrotic injury for the 12 brain regions examined at each time point. In most regions analyzed, cells dying by necrosis predominated. In frontal and parietal cortex, necrotic cells were distributed predominantly but not exclusively in the ischemic core. By 6 hr after HI, we observed relatively high densities of necrotic cells in the cortex, caudate putamen, hippocampus, and medial habenula. The density of necrotic cells remained elevated in most areas through 7 d after HI, except in the retrosplenial cortex, dentate gyrus, and medial habenula (Fig.5C,I,L). We observed two different temporal patterns of apoptotic injury. In frontal, parietal, and retrosplenial cortex, caudate putamen, globus pallidus, and CA1, the density of apoptotic cells remained high through 7 d after HI (Fig. 5A–F). In cingulate cortex, CA3, DG, subiculum, laterodorsal thalamus, and medial habenula, the density of apoptotic cells peaked between 24 and 72 hr after HI and then declined (Fig. 5G–L). Statistical analysis showed no significant differences between the two raters; overall inter-rater correlation was 0.8955. ANOVA showed significant differences for region (p < 0.02) and injury type (apoptosis vs necrosis, p < 0.001). There were significant interactions for region, injury type, and survival variables (p < 0.05).
Fig. 5.

Semiquantitative analysis of regional and temporal patterns of necrotic and apoptotic forms of cell death. Examining adjacent Nissl-stained, H&E-stained, and TUNEL-labeled sections, two investigators blinded to treatment semiquantitatively (0 to 3; see Fig.4) assessed the degree of apoptotic and necrotic forms of cell death (n = 2 at 0, 6, 12, and 168 hr;n = 4 at 24 hr; n = 3 at 48 and 72 hr). The average of the two scores for each animal was used to calculate the group means ± SEM shown here. In frontal, parietal, and retrosplenial cortex, caudate putamen, globus pallidus, and CA1, the density of apoptotic cells remained high through 7 d after HI (A–F). In cingulate cortex, CA3, dentate gyrus, subiculum, laterodorsal thalamus, and medial habenula (G–L), the density of apoptotic cells peaked between 24 and 72 hr after hypoxic ischemic injury and then subsequently declined.
After HI, TUNEL showed a patchy pattern in striatum (Fig.6A) and a mosaic pattern in CA1 (Fig. 6D). TUNEL-positive cells showed apoptotic and necrotic morphologies (Fig.6B,C,E,F). TUNEL-positive cells were frequently found in the ischemic core. Although TUNEL identified DNA fragmentation in both apoptotic and necrotic cells, the morphology differed between the two types of degenerating cells. The round, darkly stained chromatin of nuclei from apoptotic cells (Fig. 6B,C) contrasted with the diffusely stained chromatin in necrotic cells (Fig.6E,F).
Fig. 6.

TUNEL shows a patchy distribution in the striatum and hippocampus after neonatal hypoxic ischemic injury. Twenty-hour hours after HI, TUNEL-labeled cells form a patchy pattern in the striatum (A). Enlargement of the boxed region in A shows TUNEL-positive cells with round chromatin clumps typical of apoptotic cells (arrows, B); adjacent H&E-stained sections also show neurons with apoptotic features (arrows, C). Seventy-two hours after hypoxic ischemic injury, CA1 hippocampus shows a patchy pattern of TUNEL-positive and counterstained normal cells. Enlargement of theboxed region in D shows TUNEL-positive (E) and H&E-stained cells (F) with apoptotic (arrow,E) and necrotic (asterisks,E, F) morphologies. Scale bars:A, D, 200 μm; B,C, E, F, 10 μm.
By 7 d after HI, karyorrhexis (fragmentation of condensed chromatin) and karyolysis (punctate chromatin fragmentation) were a common feature of dying cells in what remained of the cortex and CA1. In the hemisphere contralateral to the lesion, a few dying cells with apoptotic morphologies were observed between 6 and 72 hr after HI in cortex, striatum, laterodorsal thalamus, and subiculum.
Ultrastructure of degenerating neurons
To confirm the presence of apoptosis in this model, we performed an electron microscopic analysis. This analysis showed degenerating neurons that displayed morphologies that varied along an apoptosis–necrosis continuum. The morphological continuum we observed in this neonatal HI model was similar to that shown previously in the immature brain after excitotoxic injury (Portera-Cailliau et al., 1997). In cortex, we found cells with the regularly shaped, round clumps of condensed chromatin characteristic of apoptotic neurons (Fig.7). Uniformly condensed, regularly shaped, round chromatin clumps were present in cells at relatively early stages of apoptosis; the nucleus and cytoplasm remained relatively intact (Fig. 7B,B1). As apoptosis proceeded, neurons showed nuclear and cytoplasmic shrinkage (Fig. 7C,C1). At an advanced stage, phagocytosed apoptotic bodies and debris were observed (Fig.7D,D1). Apoptotic cells with morphologies that varied from relatively early stages to advanced stages were also observed in the thalamus (Fig.8A,B) and hippocampus (Fig. 8C,D). Both swollen and normal mitochondria were observed in apoptotic cells (Fig.8A1). Cells showing a morphology intermediate between that of classic apoptosis and necrosis, previously referred to as “hybrid” cells (Portera-Cailliau et al., 1997; Martin et al., 1998), also were observed. The nucleus of these cells had large, irregularly shaped chromatin clumps, similar to apoptotic neurons, but the cytoplasm showed changes similar to necrotic neurons (Fig.9A,A1). The majority of necrotic cells had a distinctive morphology. The nucleus of these necrotic cells had relatively small, irregularly shaped, chromatin clumps (Fig.9B,B1) that were distinguishable from the large, round, regularly shaped chromatin clumps typical of apoptotic neurons. These types of necrotic cells were distributed predominantly in the cortex, thalamus, and hippocampus on the ligated side. Necrotic cells with pyknotic nuclei and condensed cytoplasm (Fig.9C,C1), which are typical in adult ischemia models, were only rarely observed.
Fig. 7.
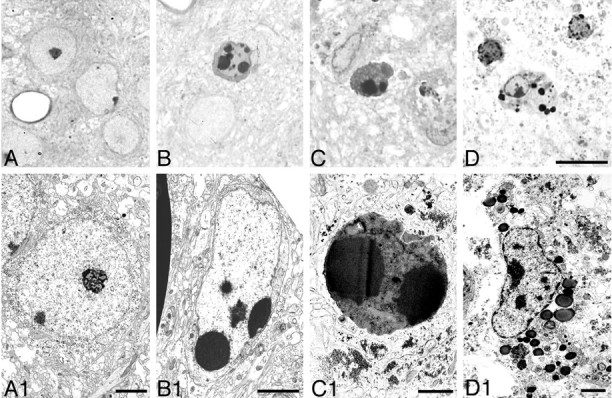
Nuclear changes in apoptotic cells in cortex, 48 hr after HI. Light microscopic photographs of 1 μm, Nissl-stained sections (A–D) and electron micrographs (A1–D1) are shown. Normal neurons are shown inA and A1 for reference. Uniformly condensed, regularly shaped, round chromatin clumps were present in cells at early stages of apoptosis (B,B1). As apoptosis proceeded, neurons showed nuclear and cytoplasmic shrinkage (C, C1). At an advanced stage, phagocytosed apoptotic bodies and debris were observed (D, D1). Scale bars: A–D, 10 μm; A1, B1, D1, 2 μm; C1, 1 μm.
Fig. 8.
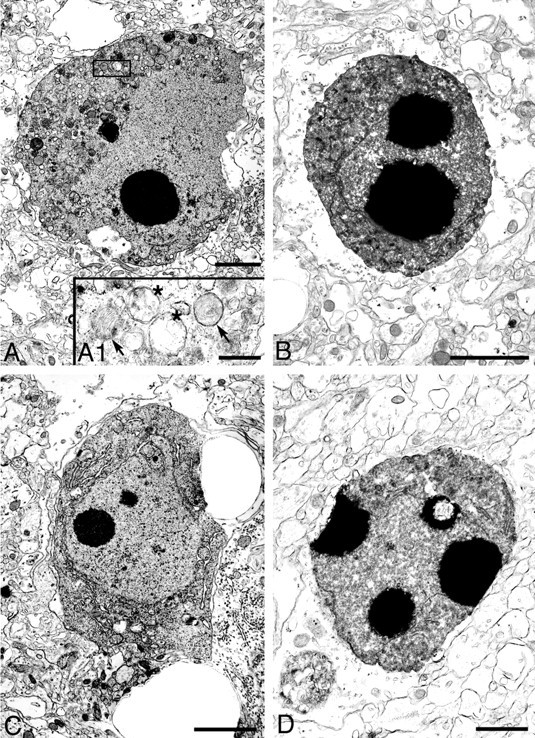
Nuclear changes in apoptotic cells in thalamus and hippocampus, 48 hr after HI. Based on ultrastructural appearance, apoptotic neurons were identified in ligated thalamus (A, B) and hippocampus (C,D). The nucleus of neurons at a middle stage of apoptosis (A, C) showed round chromatin clumps surrounded by an intact nuclear envelope; the cytoplasm was shrunken and condensed but had an intact plasma membrane. Enlargement of the boxed area in A(A1) shows both swollen (asterisks) and normal (arrows) mitochondria, which are typical for cells undergoing apoptotic death. Apoptotic neurons at a more advanced stage (B, D) show more severe nuclear condensation and shrinkage of the cytoplasm. Scale bars:A–C, 2 μm; D, 1 μm;A1, 500 nm.
Fig. 9.
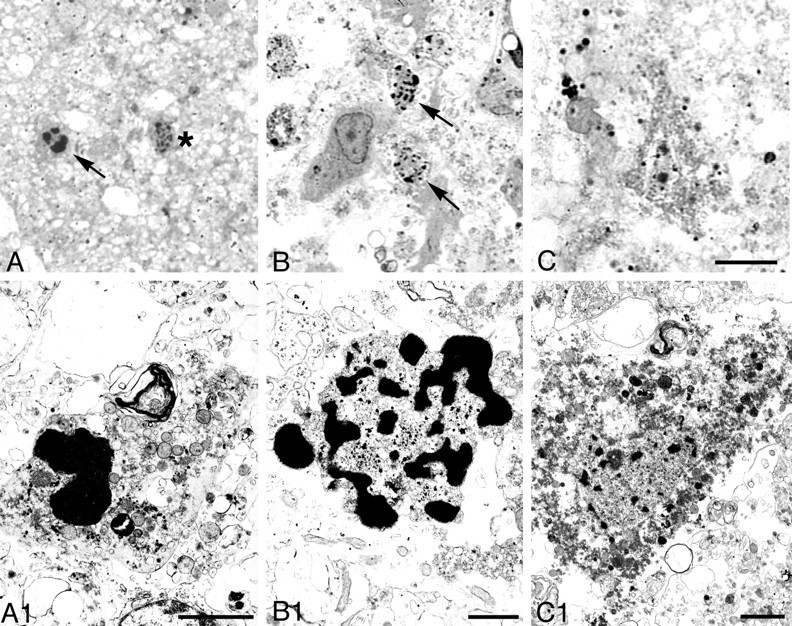
Nuclear changes in degenerating cells in cortex 48 hr after hypoxia–ischemia. Light microscopic photographs of 1 μm, Nissl-stained sections (A–C) and electron micrographs (A1–C1) are shown. We found an intermediate type of degenerating neurons, hybrid cells (arrow, A, A1) with large, chromatin clumps in the nucleus that were similar in size to those found in cells undergoing apoptosis (see Figs. 7, 8) but that were more irregular in shape. Typical necrotic neurons had smaller clumps of irregularly shaped, condensed chromatin (asterisk inA; arrows in B,B1). Necrotic neurons, which had a relatively homogeneous nucleus with a few irregular chromatin clumps and condensed granular cytoplasm (C, C1) and were typically found in adult ischemic models, were rarely identified in this model. Scale bars: A–C, 10 μm;A1, C1, 2 μm; B1, 1 μm.
Caspase-3 immunostaining
Caspase-3 immunostaining was performed on 50 μm sections adjacent to the Nissl-stained sections used for the dark-field analysis. Figure 10 shows the distribution of caspase-3-immunoreactive protein after HI in the P7 rat pups. Caspase-3-immunoreactive protein was expressed in injured cortex, striatum, hippocampus, and thalamus, regions shown previously to be selectively vulnerable to hypoxic ischemic injury in newborn rats. The cerebral cortex in normal control brains and in hemispheres contralateral to the ligation showed very weak caspase-3 immunoreactivity, consistent with observations in adult rat ischemia models (Chen et al., 1998). Another study has shown ubiquitous expression of caspase-3 mRNA in all brain nuclei from P1 to P12, followed by a steep decline so that, in the adult, caspase-3 mRNA is restricted to piriform and entorhinal cortex and to areas in neocortex in which neurogenesis is observed (de Bilbao et al., 1999). Thus, the evenly brown-stained cells that we observed throughout control brains and in the contralateral hemisphere of P7 hypoxic ischemic rats may be caspase-3-positive cells (Fig.2D,D1). In the ischemic core of cortex (Fig. 2A3) and striatum (Fig.2B2), darker, punctate caspase-3 immunoreactivity was present between 24 and 72 hr after HI. Hippocampus CA1 (Fig.2C2) and laterodorsal thalamus exhibited increased caspase-3 immunoreactivity at 48 hr after HI. Caspase-3 staining appeared as dense, fine, or coarse granular deposits (Fig.2A3,B2), making it difficult to know its cellular localization. Although at 7 d after HI severely damaged cortex tissue was often lost during processing, the remaining cortex, including the penumbra, showed increased caspase-3 immunoreactivity (Fig. 3A2). Caspase-3 immunoreactivity was also increased in the striatum (Fig. 3B2) and the thalamus (data not shown).
Fig. 10.

Regional and temporal distribution of increased caspase-3 immunoreactivity after neonatal HI. Distribution of caspase-3-immunoreactive protein at the level of the anterior striatum (top) and at the mid-hippocampal level (bottom). Dots show the general distribution of elevated caspase-3 immunoreactivity but do not reflect quantitative values. Intense, punctate caspase-3 immunoreactivity was enhanced in frontal, parietal, and cingulate cortex and in the striatum 24 hr after hypoxic ischemic injury. The density of caspase-3 immunoreactivity in the striatum was higher dorsally than ventrally and decreased slightly by 7 d after HI. In thalamus, increased caspase-3 immunoreactivity was distributed in lateral, laterodorsal, and reticular nuclei with a peak in density at 48 hr after HI. In hippocampus, intense caspase-3 immunoreactivity was present in CA1 and in the dentate gyrus at 48 hr after insult but had nearly disappeared by 7 d after hypoxic ischemic injury.
Caspase-3 immunoreactivity was enhanced in regions of the ligated hemisphere that displayed neurons undergoing apoptotic and necrotic types of cell death (Figs.2A3,B2,C2,3A1,B1). Overall, the regional and temporal patterns of caspase-3 staining corresponded well to those for degenerating cells. For example, regions such as cortex and striatum in which the densities of degenerating cells remained elevated through 7 d after HI also showed a prolonged increase in caspase-3 immunoreactivity. Likewise, in thalamus and dentate gyrus, regions in which the density of degenerating cells peaked at 48–72 hr and then declined, showed a similar peak in the intensity of caspase-3 staining at 48 hr and diminished immunoreactivity 7 d after HI. CA1 was a notable exception. At 7 d after HI, the density of degenerating neurons in Nissl-stained sections of CA1 remained high (Figs.3C1,B), and apoptotic cells were most abundant at 7 d (Fig. 5F). However, punctate caspase-3 immunoreactivity was diminished compared with its expression at 48 hr (compare Figs. 2C2, 3C2).
Western blots of caspase-3 protein
To investigate the in vivo expression of endogenous caspase-3 after HI, we analyzed the appearance of precursor caspase-3 protein and active subunits on Western blots. This immunoblot method is considered to be more quantitative than immunocytochemistry but less sensitive to spatial changes. Caspase-3 is synthesized as a 32 kDa proform that is cleaved during activation into a large subunit of p20 (20 kDa) or p17 (17 kDa) and a smaller subunit of p12 (12 kDa) (Nicholson et al., 1995; Erhardt and Cooper, 1996; Schlegel et al., 1996). In our study, antibodies to caspase-3 recognized both precursor and the p17 subunit but not the p12 subunit. At all time points, the full-length caspase-3 (p32) was detected in brain tissue homogenates of ligated and contralateral hemispheres and in control brains. No significant time-dependent changes in caspase-3 (p32) expression were detected by densitometric analysis of immunoblots (n = 4 per time point; data not shown). However, the activated p17 subunit showed regional and temporal differences in expression. In the striatum (Fig. 11A), the p17 subunit was present from 24 hr to 7 d after HI, but the levels of precursor protein remained unchanged. In the hippocampus (Fig.11B), a faint band p17 subunit appeared at 12 hr after HI, and the density of the band increased at 24–48 hr and then diminished. In the thalamus (Fig. 11C), the p17 form was present at 72 hr and 7 d after HI. The p17 subunit was not detected in cortex at any time point (Fig. 11D).
Fig. 11.
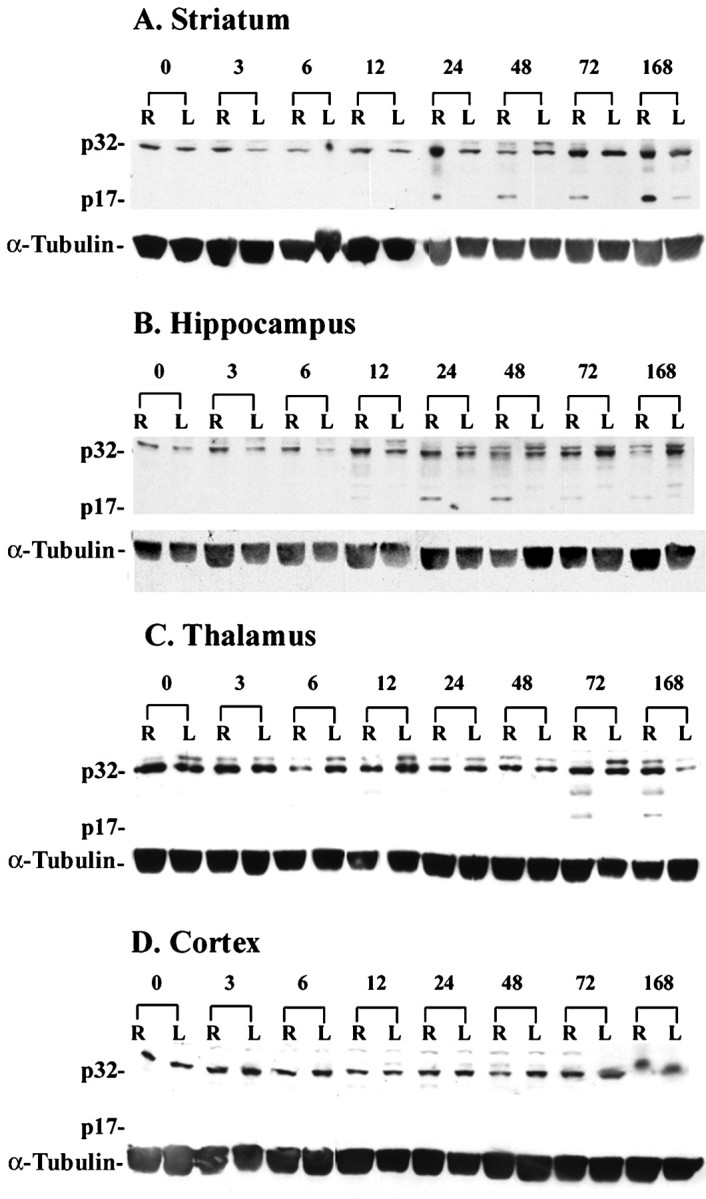
Regional and temporal differences in caspase-3 protein expression after neonatal HI. Brain tissue lysates (40 μg/lane) were subjected in 10% SDS-PAGE. In the striatum (A) on the ligated right side (R), caspase-3 cleavage product (p17) was expressed from 24 hr through 7 d after HI. In the right hippocampus (B), a faint p17 band was present at 12 hr after HI, and the density of the band increased at 24–48 hr but diminished at 72 hr and 7 d after HI. In the thalamus (C), the p17 band was present at the 72 hr and 7 d time points. In cerebral cortex (D), we did not detect the p17 subunit in either hemisphere at any time point. This experiment was performed using five animals at each time point (4 animals for HI and 1 nontreated control); representative blots are shown. α-Tubulin was used as a protein loading control.L, Left side.
DISCUSSION
This study evaluated the regional distribution and the temporal pattern of apoptotic and necrotic degeneration after HI in the newborn rat. In this neonatal HI model, both apoptotic and necrotic forms of cell death contribute to brain damage. By ultrastructural criteria, we confirmed that apoptosis occurs in this model. The temporal pattern of apoptosis varied in each region, but two general patterns were observed. In frontal, parietal, and retrosplenial cortex, striatum, globus pallidus, and CA1, a relatively high density of apoptotic cells were observed from 6 or 12 hr through 7 d after HI, the endpoint of our study. In cingulate cortex, CA3, DG, subiculum, laterodorsal thalamus, and medial habenula, we observed a bell-shaped curve with a peak in density between 24 and 72 hr after HI and a subsequent decline. The timing for the onset of cell death varied between the two groups of animals in which we evaluated temporal patterns of cell death. In the group with moderate to severe damage, apoptotic cells were observed as early as 6 hr after HI, whereas significant densities of apoptotic cells were not observed in the group with mild to moderate brain damage until the 24 hr time point. However, once initiated, both groups showed similar regional and temporal patterns of cell death.
The severity and variability in the injury and the regional pattern of damage in the present study was similar to that reported previously (Rice et al., 1981, Towfighi et al., 1995, 1997; Trescher et al., 1997). The cortex, striatum, CA1 hippocampus, and medial habenula were particularly sensitive to hypoxic ischemic exposure. In the thalamus, the lateral, dorsal, and reticular nuclei were the most severely affected. In our study and in other ischemia models, necrosis predominated in the ischemic core, whereas apoptosis occurred primarily in the penumbra (Li et al., 1995a,b,c, 1997, 1998; Charriaut-Marlangue et al., 1996b; Chopp and Li, 1996). As reported previously (Ito et al., 1975; Vannucci et al., 1993; Bonfoco et al., 1995; Vannucci and Vannucci, 1997), the time course of the injury and amount of neuronal death depended on the severity of the insult.
In the present study, we rarely found eosinophilic, necrotic neurons that are typical in adult ischemia models (Garcia et al., 1995). Instead, necrotic cells with aggregates of irregularly shaped chromatin were common. Apoptotic cells with round, chromatin clumps and intermediate types with large, irregularly shaped, chromatin clumps also were frequently observed. It is possible that the extended period of time over which cells die in this newborn model may account for the heterogeneity in their appearance. The greater severity and the more rapid pace of ischemia in older animals (Towfighi and Mauger, 1998) may not allow for the nuclear and cytoplasmic changes that occur in the newborn model.
Electron microscopy confirmed the apoptosis–necrosis morphological continuum after HI in the neonate. The pattern was similar to that observed in newborn rats after excitotoxic activation of NMDA and non-NMDA glutamate receptors (Portera-Cailliau et al., 1997), suggesting that glutamate plays an important role in neonatal brain injury. The proportions of apoptotic neurons observed in newborn hypoxic ischemic and excitotoxic injury models also may vary between species (e.g., rats vs piglets), perhaps because of differences in the maturity or immaturity of the brains at the time of insult (Martin et al., 2000).
Coexistence of apoptosis and necrosis
Apoptosis and necrosis may not be mutually exclusive forms of cell death (Ankarcrona, 1998; Martin et al., 1998; Roy and Sapolsky, 1999). It also is possible that different subsets of neurons die by apoptosis or necrosis. In our experiments, all regions exhibited both types of cell death, at all the time points examined. Even in the ischemic core, we found cells possessing apoptotic morphological features. Moreover, within a population of neurons (e.g., CA1 pyramidal neurons), the entire continuum of morphological appearance was present. The existence of apoptotic cells adjacent to necrotic cells in both excitotoxic and hypoxic ischemic newborn models (Portera-Cailliau et al., 1997; Martin et al., 1998) suggests that common extracellular microenvironmental signals may be involved.
In an adult middle cerebral artery occlusion ischemia model, at 46 hr after insult, the ratio of apoptotic cells to necrotic cells was 1:9, 1:6, and 1:13 in the ischemic core and inner and outer boundary zones, respectively (Li et al., 1998). Although the majority of ischemic cells appeared necrotic, the findings support the existence of apoptotic cells in the ischemic core. Because of the short half-life of apoptotic cells relative to that of necrotic cells (Wyllie et al., 1980; Bursch et al., 1990; Pittman et al., 1993; Messam and Pittman, 1998), the contribution of apoptosis to the hypoxic ischemic brain damage may have been underestimated (Chopp and Li, 1996; Li et al., 1998). Considering the short time span for apoptosis, apoptosis must continue to be initiated in regions in which apoptotic cells are observed days after hypoxic ischemic injury. Our findings of significant densities of apoptotic figures in cortex, striatum, and CA1 7 d after HI suggest that new cells continue to be triggered to die by apoptosis over an extended period in these regions. In other regions in which apoptotic figures were observed for a shorter period (e.g., habenula, thalamus, and dentate gyrus), there were probably fewer cycles of apoptosis. Our observations of apoptotic cells in cortex, striatum, and CA1 7 d after HI indicate that cell death is an ongoing dynamic process and that therapeutic intervention even 1 week after the onset of hypoxic ischemic injury could be neuroprotective.
In accord with previous studies (MacManus et al., 1994; Yasuda et al., 1995; van Lookeren Campagne and Gill, 1996; de Torres et al., 1997;Charriaut-Marlangue et al., 1998), we found that TUNEL staining primarily detected apoptotic cells with fragmented condensed nuclei. However, the TUNEL method also labeled nonapoptotic DNA fragments and was not always restricted to the nucleus. Some of the TUNEL-positive cells showing cytoplasmic labeling were probably macrophages because of their location in the ischemic core.
Ultrastructural analyses verified our light microscopic observations of apoptotic cells in this model. Our results are consistent with a previous electron microscopic study in the 7-d-old HI model (Pulera et al., 1998). In adult rodent ischemia models, studies provide conflicting evidence for cells dying by apoptosis. Investigators examining 40,000 neurons in CA1 did not detect cells dying by apoptosis 4, 14, and 60 d after insult in an adult gerbil, global ischemia model (Colbourne et al., 1999). Other studies suggest a possible role for apoptosis in the adult (MacManus et al., 1994; Charriaut-Marlangue et al., 1998). However, apoptosis does appear to play a more predominant role in ischemia in the neonate than in the adult (Sidhu et al., 1997; Pulera et al., 1998). The normal occurrence of programmed cell death (Sidhu et al., 1997) and higher caspase-3 expression (Siman et al., 1999) in the developing brain may influence the way cells die after HI in the neonate.
Capsase-3 immunohistochemistry and Western blots
Western blots detected both proenzyme (p32) and activated forms (p17 subunit) of caspase-3. It is likely that both forms of caspase-3 were positively stained in the immunocytochemical experiments, because we used the same antibody for both procedures. Namura et al. (1998)observed immunoreactivity of the caspase-3 precursor throughout normal adult mouse brain within neurons and axonal fibers. In adult rat brain, basal caspase-3 reactivity was found in the cell cytoplasm exclusively and only scattered, shrunken neurons in the cortex and thalamus showed nuclear caspase-3 immunoreactivity (Krajewska et al., 1997; Chen et al., 1998).
In the present study, hypoxic ischemic-induced activation of caspase-3 was demonstrated indirectly by the appearance of the active p17 subunit on Western blots; the active p12 subunit of caspase-3 was not detected. Although we could not show directly that caspase-3 activation caused cell death, the prolonged expression of p17 subunits in the Western blots suggests that caspase-3 remained activated in the ligated hemisphere for an extended period of time. In agreement with our results, caspase-3 is cleaved after transient cerebral ischemia in adult rodent models (Chen et al., 1998; Namura et al., 1998), as well as in cell culture studies (Erhardt and Cooper, 1996; Schlegel et al., 1996). As reported previously (Namura et al., 1998), we did not detect the activated p17 subunit of caspase-3 in injured cortex but did find intense, punctate caspase-3 immunoreactivity in this region.
Our results showed that activation of caspase-3 protein was prolonged and that moderate to high levels of immunoreactive protein remained for at least 7 d after hypoxic ischemic injury. The regional and temporal patterns of caspase-3 immunoreactivity corresponded well with those for apoptosis, lending further support for a prolonged role of apoptosis in hypoxic ischemic injury in the neonatal brain. Our results are consistent with those showing delayed caspase-3 activation and neuroprotection with caspase inhibitors in neonatal HI (Cheng et al., 1998). The protracted period of caspase-3 activation after HI also suggests an extended therapeutic window in which caspase inhibitors may reduce or prevent brain damage after neonatal asphyxia.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS28208 (to M.V.J.) and National Institutes of Health, National Institute on Aging Grant AG16282 (to L.J.M). We thank Karen Smith-Connor, Tae H. Chong, Carol A. Cooke, and Lisa A. Kerrigan for technical assistance.
Correspondence should be addressed to Dr. Michael V. Johnston, Kennedy Krieger Research Institute, 707 N. Broadway, Baltimore, MD 21205. E-mail: johnston@kennedykrieger.org.
REFERENCES
- 1.Ankarcrona M. Glutamate induced cell death: apoptosis or necrosis? Prog Brain Res. 1998;116:265–272. doi: 10.1016/s0079-6123(08)60442-2. [DOI] [PubMed] [Google Scholar]
- 2.Banasiak KJ, Haddad GG. Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 1998;797:295–304. doi: 10.1016/s0006-8993(98)00286-8. [DOI] [PubMed] [Google Scholar]
- 3.Blue ME, Martin LJ, Brennan EM, Johnston MV. Ontogeny of non-NMDA glutamate receptors in rat barrel field cortex. I. Metabotropic receptors. J Comp Neurol. 1997;386:16–28. [PubMed] [Google Scholar]
- 4.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bursch W, Paffe S, Putz B, Barthel G, Schulte-Hermann R. Determination of the length of the histological stages of apoptosis in normal liver and in altered hepatic foci of rats. Carcinogenesis. 1990;11:847–853. doi: 10.1093/carcin/11.5.847. [DOI] [PubMed] [Google Scholar]
- 6.Charriaut-Marlangue C, Aggoun-Zouaoui D, Represa A, Ben-Ari Y. Apoptotic features of selective neuronal death in ischemia, epilepsy and gp 120 toxicity. Trends Neurosci. 1996a;19:109–114. doi: 10.1016/s0166-2236(96)80039-7. [DOI] [PubMed] [Google Scholar]
- 7.Charriaut-Marlangue C, Margaill I, Represa A, Popovici T, Plotkine M, Ben-Ari Y. Apoptosis and necrosis after reversible focal ischemia: an in situ DNA fragmentation analysis. J Cereb Blood Flow Metab. 1996b;16:186–194. doi: 10.1097/00004647-199603000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Charriaut-Marlangue C, Remolleau S, Aggoun-Zouaoui D, Ben-Ari Y. Apoptosis and programmed cell death: a role in cerebral ischemia. Biomed Pharmacother. 1998;52:264–269. doi: 10.1016/S0753-3322(98)80012-7. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng Y, Deshmukh M, D, Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chopp M, Li Y. Apoptosis in focal cerebral ischemia. Acta Neurochir Suppl (Wien) 1996;66:21–26. doi: 10.1007/978-3-7091-9465-2_4. [DOI] [PubMed] [Google Scholar]
- 12.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Bilbao F, Guarin E, Nef P, Vallet P, Giannakopoulos P, Dubois-Dauphin M. Postnatal distribution of cpp32/caspase 3 mRNA in the mouse central nervous system: an in situ hybridization study. J Comp Neurol. 1999;409:339–357. doi: 10.1002/(sici)1096-9861(19990705)409:3<339::aid-cne1>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 14.de Torres C, Munell F, Ferrer I, Reventos J, Macaya A. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci Lett. 1997;230:1–4. doi: 10.1016/s0304-3940(97)00445-x. [DOI] [PubMed] [Google Scholar]
- 15.Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Erhardt P, Cooper GM. Activation of the CPP32 apoptotic protease by distinct signaling pathways with differential sensitivity to Bcl-xL. J Biol Chem. 1996;271:17601–17604. doi: 10.1074/jbc.271.30.17601. [DOI] [PubMed] [Google Scholar]
- 18.Fink K, Zhu J, Namura S, Shimizu-Sasamata M, Endres M, Ma J, Dalkara T, Yuan J, Moskowitz MA. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J Cereb Blood Flow Metab. 1998;18:1071–1076. doi: 10.1097/00004647-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JH, Liu KF, Ho KL. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke. 1995;26:636–642. doi: 10.1161/01.str.26.4.636. [DOI] [PubMed] [Google Scholar]
- 20.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hara H, Fink K, Endres M, Friedlander RM, Gagliardini V, Yuan J, Moskowitz MA. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997a;17:370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin-1β-converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA. 1997b;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito U, Spatz M, Walker JT, Jr, Klatzo I. Experimental cerebral ischemia in mongolian gerbils. I. Light microscopic observations. Acta Neuropathol (Berl) 1975;32:209–223. doi: 10.1007/BF00696570. [DOI] [PubMed] [Google Scholar]
- 24.Johnston MV. Neurotransmitter alterations in a model of perinatal hypoxic-ischemic brain injury. Ann Neurol. 1983;13:511–518. doi: 10.1002/ana.410130507. [DOI] [PubMed] [Google Scholar]
- 25.Johnston MV. Hypoxic and ischemic disorders of infants and children. Lecture for 38th meeting of Japanese Society of Child Neurology, Tokyo, Japan, July 1996. Brain Dev. 1997;19:235–239. doi: 10.1016/s0387-7604(96)00561-x. [DOI] [PubMed] [Google Scholar]
- 26.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krajewska M, Wang HG, Krajewski S, Zapata JM, Shabaik A, Gascoyne R, Reed JC. Immunohistochemical analysis of in vivo patterns of expression of CPP32 (Caspase-3), a cell death protease. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- 28.Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, Negoescu A. TUNEL apoptotic cell detection in tissue sections: critical evaluation and improvement critical evaluation and improvement. J Histochem Cytochem. 1998;46:327–334. doi: 10.1177/002215549804600306. [DOI] [PubMed] [Google Scholar]
- 29.Lange MS, Johnston MV, Tseng EE, Baumgartner WA, Blue ME. Apoptosis detection in brain using low-magnification dark-field microscopy. Exp Neurol. 1999;158:254–260. doi: 10.1006/exnr.1999.7097. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C. Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke. 1995a;26:1252–1257. doi: 10.1161/01.str.26.7.1252. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995b;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M. Ultrastructural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol. 1995c;146:1045–1051. [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Chopp M, Powers C, Jiang N. Apoptosis and protein expression after focal cerebral ischemia in rat. Brain Res. 1997;765:301–312. doi: 10.1016/s0006-8993(97)00524-6. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Powers C, Jiang N, Chopp M. Intact, injured, necrotic and apoptotic cells after focal cerebral ischemia in the rat. J Neurol Sci. 1998;156:119–132. doi: 10.1016/s0022-510x(98)00036-7. [DOI] [PubMed] [Google Scholar]
- 35.Macaya A. Apoptosis in the nervous system. Rev Neurol. 1996;24:1356–1360. [PubMed] [Google Scholar]
- 36.MacManus JP, Hill IE, Huang ZG, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal ischaemic neocortex. NeuroReport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 37.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 38.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 39.Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, Traystman RJ. Neonatal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis. 2000;7:169–191. doi: 10.1006/nbdi.2000.0282. [DOI] [PubMed] [Google Scholar]
- 40.McDonald JW, Behrens MI, Chung C, Bhattacharyya T, Choi DW. Susceptibility to apoptosis is enhanced in immature cortical neurons. Brain Res. 1997;759:228–232. doi: 10.1016/s0006-8993(97)00248-5. [DOI] [PubMed] [Google Scholar]
- 41.Mehmet H, Yue X, Squier MV, Lorek A, Cady E, Penrice J, Sarraf C, Wylezinska M, Kirkbride V, Cooper C, Brown GC, Wyatt JS, Reynolds EOR, Edwards AD. Increased apoptosis in the cingulate sulcus of newborn piglets following transient hypoxia-ischaemia is related to the degree of high energy phosphate depletion during the insult. Neurosci Lett. 1994;181:121–125. doi: 10.1016/0304-3940(94)90574-6. [DOI] [PubMed] [Google Scholar]
- 42.Messam CA, Pittman RN. Asynchrony and commitment to die during apoptosis. Exp Cell Res. 1998;238:389–398. doi: 10.1006/excr.1997.3845. [DOI] [PubMed] [Google Scholar]
- 43.Migheli A, Attanasio A, Schiffer D. Ultrastructural detection of DNA strand breaks in apoptotic neural cells by in situ end-labelling techniques. J Pathol. 1995;176:27–35. doi: 10.1002/path.1711760106. [DOI] [PubMed] [Google Scholar]
- 44.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Negoescu A, Lorimier P, Labat-Moleur F, Drouet C, Robert C, Guillermet C, Brambilla C, Brambilla E. In situ apoptotic cell labeling by the TUNEL method: improvement and evaluation on cell preparations. J Histochem Cytochem. 1996;44:959–968. doi: 10.1177/44.9.8773561. [DOI] [PubMed] [Google Scholar]
- 46.Negoescu A, Guillermet C, Lorimier P, Brambilla E, Labat-Moleur F. Importance of DNA fragmentation in apoptosis with regard to TUNEL specificity. Biomed Pharmacother. 1998;52:252–258. doi: 10.1016/S0753-3322(98)80010-3. [DOI] [PubMed] [Google Scholar]
- 47.Ni B, Wu X, Su Y, Stephenson D, Smalstig EB, Clemens J, Paul SM. Transient global forebrain ischemia induces a prolonged expression of the caspase-3 mRNA in rat hippocampal CA1 pyramidal neurons. J Cereb Blood Flow Metab. 1998;18:248–256. doi: 10.1097/00004647-199803000-00003. [DOI] [PubMed] [Google Scholar]
- 48.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 49.Pittman RN, Wang S, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 51.Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997;378:70–87. [PubMed] [Google Scholar]
- 52.Pulera MR, Adams LM, Liu H, Santos DG, Nishimura RN, Yang F, Cole GM, Wasterlain CG. Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke. 1998;29:2622–2630. doi: 10.1161/01.str.29.12.2622. [DOI] [PubMed] [Google Scholar]
- 53.Renolleau S, Aggoun-Zouaoui D, Ben-Ari Y, Charriaut-Marlangue C. A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat: morphological changes indicative of apoptosis. Stroke. 1998;29:1454–1460. doi: 10.1161/01.str.29.7.1454. [DOI] [PubMed] [Google Scholar]
- 54.Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 55.Roy M, Sapolsky R. Neuronal apoptosis in acute necrotic insults: why is this subject such a mess? Trends Neurosci. 1999;22:419–422. doi: 10.1016/s0166-2236(99)01435-6. [DOI] [PubMed] [Google Scholar]
- 56.Schlegel J, Peters I, Orrenius S, Miller DK, Thornberry NA, Yamin TT, Nicholson DW. CPP32/apopain is a key interleukin-1β-converting enzyme-like protease involved in Fas-mediated apoptosis. J Biol Chem. 1996;271:1841–1844. doi: 10.1074/jbc.271.4.1841. [DOI] [PubMed] [Google Scholar]
- 57.Schulz JB, Weller M, Matthews RT, Heneka MT, Groscurth P, Martinou JC, Lommatzsch J, von Coelln R, Wullner U, Loschmann PA, Beal MF, Dichgans J, Klockgether T. Extended therapeutic window for caspase inhibition and synergy with MK-801 in the treatment of cerebral histotoxic hypoxia. Cell Death Differ. 1998;5:847–857. doi: 10.1038/sj.cdd.4400420. [DOI] [PubMed] [Google Scholar]
- 58.Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 59.Scott RJ, Hegyi L. Cell death in perinatal hypoxic-ischaemic brain injury. Neuropathol Appl Neurobiol. 1997;23:307–314. [PubMed] [Google Scholar]
- 60.Sidhu RS, Tuor UI, Del Bigio MR. Nuclear condensation and fragmentation following cerebral hypoxia-ischemia occurs more frequently in immature than older rats. Neurosci Lett. 1997;223:129–132. doi: 10.1016/s0304-3940(97)13426-7. [DOI] [PubMed] [Google Scholar]
- 61.Siman R, Bozyczko-Coyne D, Meyer SL, Bhat RV. Immunolocalization of caspase proteolysis in situ: evidence for widespread caspase-mediated apoptosis of neurons and glia in the postnatal rat brain. Neuroscience. 1999;92:1425–1442. doi: 10.1016/s0306-4522(99)00034-2. [DOI] [PubMed] [Google Scholar]
- 62.Stroemer RP, Rothwell NJ. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1β in the rat. J Cereb Blood Flow Metab. 1998;18:833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Towfighi J, Mauger D. Temporal evolution of neuronal changes in cerebral hypoxia-ischemia in developing rats: a quantitative light microscopic study. Brain Res Dev Brain Res. 1998;109:169–177. doi: 10.1016/s0165-3806(98)00077-7. [DOI] [PubMed] [Google Scholar]
- 64.Towfighi J, Zec N, Yager J, Housman C, Vannucci RC. Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta Neuropathol (Berl) 1995;90:375–386. doi: 10.1007/BF00315011. [DOI] [PubMed] [Google Scholar]
- 65.Towfighi J, Mauger D, Vannucci RC, Vannucci SJ. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Res Dev Brain Res. 1997;100:149–160. doi: 10.1016/s0165-3806(97)00036-9. [DOI] [PubMed] [Google Scholar]
- 66.Trescher WH, Ishiwa S, Johnston MV. Brief post-hypoxic-ischemic hypothermia markedly delays neonatal brain injury. Brain Dev. 1997;19:326–338. doi: 10.1016/s0387-7604(97)00027-2. [DOI] [PubMed] [Google Scholar]
- 67.van Lookeren Campagne M, Gill R. Ultrastructural morphological changes are not characteristic of apoptotic cell death following focal cerebral ischaemia in the rat. Neurosci Lett. 1996;213:111–114. doi: 10.1016/0304-3940(96)12839-1. [DOI] [PubMed] [Google Scholar]
- 68.Vannucci RC. Experimental biology of cerebral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res. 1990;27:317–326. doi: 10.1203/00006450-199004000-00001. [DOI] [PubMed] [Google Scholar]
- 69.Vannucci RC, Vannucci SJ. A model of perinatal hypoxic-ischemic brain damage. Ann NY Acad Sci. 1997;835:234–249. doi: 10.1111/j.1749-6632.1997.tb48634.x. [DOI] [PubMed] [Google Scholar]
- 70.Vannucci RC, Christensen MA, Yager JY. Nature, time-course, and extent of cerebral edema in perinatal hypoxic-ischemic brain damage. Pediatr Neurol. 1993;9:29–34. doi: 10.1016/0887-8994(93)90006-x. [DOI] [PubMed] [Google Scholar]
- 71.Volpe J. Neurology of the newborn, Ed 3, pp 214–217. Sanders; Philadelphia: 1995. [Google Scholar]
- 72.Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH. A new method to detect apoptosis in paraffin sections: in situ end-labeling of fragmented DNA. J Histochem Cytochem. 1993;41:7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]
- 73.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 74.Yasuda M, Umemura S, Osamura RY, Kenjo T, Tsutsumi Y. Apoptotic cells in the human endometrium and placental villi: pitfalls in applying the TUNEL method. Arch Histol Cytol. 1995;58:185–190. doi: 10.1679/aohc.58.185. [DOI] [PubMed] [Google Scholar]
- 75.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 76.Yue X, Mehmet H, Penrice J, Cooper C, Cady E, Wyatt JS, Reynolds EO, Edwards AD, Squier MV. Apoptosis and necrosis in the newborn piglet brain following transient cerebral hypoxia-ischaemia. Neuropathol Appl Neurobiol. 1997;23:16–25. [PubMed] [Google Scholar]