

Abstract
The amyloid precursor protein (APP) involved in Alzheimer's disease is a member of a larger gene family including amyloid precursor-like proteins APLP1 and APLP2. We generated and examined the phenotypes of mice lacking individual or all possible combinations of APP family members to assess potential functional redundancies within the gene family. Mice deficient for the nervous system-specific APLP1 protein showed a postnatal growth deficit as the only obvious abnormality. In contrast to this minor phenotype, APLP2−/−/APLP1−/−and APLP2−/−/APP−/−mice proved lethal early postnatally. Surprisingly, APLP1−/−/APP−/−mice were viable, apparently normal, and showed no compensatory upregulation of APLP2 expression. These data indicate redundancy between APLP2 and both other family members and corroborate a key physiological role for APLP2. This view gains further support by the observation that APLP1−/−/APP−/−/APLP2+/−mice display postnatal lethality. In addition, they provide genetic evidence for at least some distinct physiological roles of APP and APLP2 by demonstrating that combinations of single knock-outs with the APLP1 mutation resulted in double mutants of clearly different phenotypes, being either lethal, or viable. None of the lethal double mutants displayed, however, obvious histopathological abnormalities in the brain or any other organ examined. Moreover, cortical neurons from single or combined mutant mice showed unaltered survival rates under basal culture conditions and unaltered susceptibility to glutamate excitotoxicity in vitro.
Keywords: amyloid precursor protein, amyloid precursor-like protein, knock-out mice, functional redundancy, excitotoxicity, cortical neurons, Alzheimer's disease
Neurofibrillary tangles and senile neuritic plaques are the major pathological features of Alzheimer's disease. The predominant constituent of neuritic plaques is the β-amyloid peptide (βA4), proteolytically derived from the larger β-amyloid precursor protein (APP). APP is a member of a larger gene family including the two amyloid precursor-like proteins APLP1 and APLP2 from mammals (Wasco et al., 1992, 1993; Sprecher et al., 1993;Sandbrink et al., 1994; Slunt et al., 1994). Both APLPs are highly homologous to APP and are proteolytically processed in a similar way, leading to the secretion of the large ectodomains (sAPP and sAPLP) (Slunt et al., 1994; Paliga et al., 1997). Using in situhybridization and RT-PCR analysis, we and others have demonstrated that APP and APLP2 are expressed ubiquitously in largely overlapping patterns during embryonic development and in adult tissue (Slunt et al., 1994; Lorent et al., 1995). In contrast, APLP1 is found primarily in the nervous system (Lorent et al., 1995; Thinakaran et al., 1995c). APP is axonally transported, has been localized to synapses, and undergoes retrograde and transcytotic transport in neurons (Simons et al., 1995; Yamazaki et al., 1995). APLP2 has been detected in postsynaptic compartments throughout the cortex and in axonal termini of olfactory sensory neurons (Thinakaran et al., 1995a,b). In summary, APP/APLP proteins are highly related, are similarly processed, share overlapping domains of expression, and may therefore also be functionally conserved.
Multiple functions have been proposed for APP, mainly based on in vitro experiments (for review, see Mattson, 1997). To address the physiological functions of APP directly, we and others have generated mice carrying a hypomorphic mutation of APP (APPΔ; Müller et al., 1994) and APP-deficient null mutants (Zheng et al., 1995; Li et al., 1996). The phenotypes of these mutants suggested that APP may play a role in neurite outgrowth and the formation of forebrain commissures, postnatal somatic growth and neurobehavioral development, locomotor activity and grip strength, copper homeostasis, and the susceptibility to epileptic seizures and excitotoxic agents (Zheng et al., 1995; Li et al., 1996; Perez et al., 1997; Steinbach et al., 1998; Tremml et al., 1998; Magara et al., 1999; White et al., 1999a,b). APLP2−/−mice showed no apparent abnormalities, but double mutants obtained by crossing APP−/− mice (obtained from H. Zheng, H. Chen, M. Trumbauer, and L. H. T. van der Ploeg) to APLP2−/−mice were perinatally lethal, suggesting functional redundancy (von Koch et al., 1997).
This study investigates the phenotype of APLP1−/−mice and addresses the question of functional complementation within the gene family by analyzing all possible combinations of double mutants. We show that APP family members serve essential, at least partially redundant functions by demonstrating early postnatal lethality for both APLP2−/−/APLP1−/−and APLP2−/−/APP−/−double mutants. In addition, we provide genetic evidence for distinct physiological roles of APP and APLP2, by showing that crosses of the respective single knock-outs with APLP1 knock-out mice result in double mutants of clearly different phenotypes, being either lethal (APLP2−/−/APLP1−/−), or viable (APP−/−/APLP1−/−). Moreover, we investigated the presumed neuroprotective role of endogenous APP family members.
MATERIALS AND METHODS
Generation of APLP1−/−embryonic stem cells
Using as a probe a 0.4 kb PstI fragment derived from the 5′-end of the APLP1 cDNA (Wasco et al., 1992) two overlapping λ clones encompassing ∼19 kb of genomic sequence were isolated from an isogenic λGem11 genomic library constructed from AB-1 [129/Sv(ev)] embryonic stem (ES) cells. As judged from hybridization experiments with various oligonucleotides derived from different regions of the APLP1 cDNA and partial sequencing the genomic clones contained the whole coding region including the putative first exon (cDNA positions 9–229) harboring the ATG start codon. Apart from exon1 and exon2 the precise location of the other exons was not mapped. The targeting vector pAPLP1targ was constructed by inserting a blunted 0.9 kb SacI/XhoI fragment lying 2.5 kb upstream of putative exon1 into the NotI site of pTKNEOUMSPSA, a slightly modified vector (containing additional restriction sites) derived from the previously described plasmid pTKNEOUMS (Ruffner et al., 1993). Subsequently a blunted 4.7 kbXhoI/KpnI fragment was inserted as the “long arm” of homology into the unique ClaI site of the vector. In this construct the neoR gene under the control of the phosphoglycerate kinase (PGK) promoter was in antisense orientation to the transcriptional direction of the APLP1 gene and was followed by the 360 bp UMS sequence reported to mediate transcription termination. A herpes simplex virus thymidine kinase (HSV-TK) cassette was added at the 3′ end to allow for counterselection. Homologous recombination leads to a ∼8 kb genomic deletion containing ∼2.5 kb of the promoter. To determine how much of the coding region would be deleted by gene targeting, hybridization experiments were performed showing that the deleted region contained coding sequences up to approximately position 1000 of the 2.36 kb cDNA, as judged by hybridization to oligo UM14 (cDNA position 1014–1034).
The targeting vector was subsequently linearized with SacII at the 5′ border of the APLP1 genomic region, electroporated into GS1 ES cells [established by Gerlinde Stark from 129 Sv(ev) mice] grown on irradiated mouse embryonic fibroblasts in DMEM supplemented with 20% fetal calf serum (D20). Colonies resistant to neomycin and 1-(2-deoxy-2-flouro-β-d-arabinosifuranosyl)-5-ioduracil (FIAU) were selected in D20 containing 400 μg of G-418 and 0.2 μm FIAU and screened as described (Müller et al., 1994) by PCR using a primer derived from the 3′ end of the neo-cassette (P3: 5′-ATTCGCAGCGCATCGCCTTCTATCGCC-3′) and a primer corresponding to a genomic sequence 5′ from the targeting vector (UM20: 5′-GGATTTCAGCCCTGGTTCCCATTCTAACCC-3′). The frequency of homologous recombination was ∼1 in 500 G-418/FIAU-resistant colonies.
Generation of APLP1−/− mice
ES cells heterozygous for the mutant APLP1 gene were injected into 3.5-d-old C57BL/6 blastocysts that were transferred into the uteri of pseudopregnant NMRI foster mothers. One of four clones investigated (clone GS-34.4) gave rise to a chimeric male that was mated to C57BL/6 females [and 129 Sv(ev) females for pure genetic background, respectively] and transmitted the mutant allele in the germline as revealed by PCR and Southern blot analysis. Heterozygous offspring were intercrossed and generated 20% homozygous APLP1−/− animals (65 animals analyzed).
Animals and PCR genotyping
APLP1 knock-out mice [of either pure 129Sv(ev) or mixed 129 Sv(ev)xC57BL/6 genetic background] were initially screened with primers UM20/P3 as described for ES cells. Later, a simpler 3-primer PCR was used amplifying for the wild-type (wt) allele a 600 bp fragment with primers UM30 (5′-GCTTTCTGCCTTCATGCCTATCTCTAG-3′) and UM31 (5′-ACTTTGGCTGAACTGAGTGTACACC-3′) derived from the short arm of the targeting vector or from the region replaced by neo, respectively. For the mutant allele a 450 bp product was obtained with primers UM30 and P4-neo (5′-ATGCGGTGGGCTCTATGGCTTCTGA-3′) derived from the PGK-Neo-cassette.
APLP2 knock-out mice [129Sv(ev)xC57BL/6 genetic background] were generated and genotyped as described (von Koch et al., 1997).
APP knock-out animals (129OLAxC57BL/6 genetic background) harbor a 200 kb genomic deletion within the APP locus encompassing exon 2–17 of the APP gene and were generated as described (Li et al., 1996; Magara et al., 1999). For genotyping, a 3-primer PCR was set up amplifying for the wt allele a 650 bp fragment with primers UM44 (5′-GAGACGAGG ACGCTCAGTCCTAGGG-3) and UM42 (5′-ATCACCT-GGTTCTAATCAGAGGCCC-3′) flanking exon 17 and for the mutant allele a 430 bp fragment with primers UM42 located in intron 17 and P3-hygro (5′-CGAGATCAGCAGCCTCTGTTCCACA-3′) derived from the PGK-Hygro-cassette, respectively. PCR was done on DNA obtained from tail biopsies as previously described (Müller et al., 1994).
Animals were housed in a special pathogen-free unit kept under optimal hygiene conditions. Timed matings were set up in the late afternoon followed by plug check on the next morning. The time point of a detected plug was considered as embryonic day 0.5 (E0.5).
Generation of combined mutants
Double mutants were generated by three consecutive crosses. Single mutants of APP family members (e.g., APLP1−/− × APLP2−/−) were intercrossed, and animals heterozygous for both loci were backcrossed to single mutants (e.g., APLP1+/−APLP2+/−× APLP2−/−) to obtain 25% offspring homozygous knock-out for one gene and heterozygous for the other (e.g., APLP1+/−APLP2−/−). These animals were further intercrossed (APLP1+/−APLP2−/− × APLP1+/−APLP2−/−) to obtain double knock-outs in the next generation (e.g., 25% APLP1−/−APLP2−/−, 25% APLP1+/+APLP2−/−, and 50% APLP1+/−APLP2−/−). Accordingly, to obtain APP/APLP2 and APP/APLP mutants the respective analogous crosses were set up, and double knock-outs were generated in the third round of crossing from the following matings: (APP+/−APLP2−/−× APP+/−APLP2−/−) and (APP+/−APLP1−/−× APP+/−APLP1−/−). APP−/−/APLP1−/−/APLP2+/−mice were generated by crossing APP−/−/APLP1−/−mice with APP−/−/APLP1+/−/APLP2+/−mice.
Southern and Northern blot analysis of APLP−/−mice
Southern blots were prepared from APLP1 ES cell or tail DNA digested with HindIII or EcoRI, cutting outside of the targeting vector and separated by conventional or pulsed-field gel electrophoresis [1% agarose in 45 mm Tris-borate, 1 mm EDTA run for 15 hr at 6 V/cm, 120° angle, switch time being from 0.5–2.0 sec in a Bio-Rad (Hercules, CA) DRIII apparatus]. Hybridization was performed with a random primer32P-labeled (PrimeIt; Stratagene, La Jolla, CA) genomic 0.5 kb PstI fragment (Fig.1, probe B) and a 0.6 kbPstI/XbaI-fragment of pGKneo.
Fig. 1.

Disruption of the APLP1 gene by gene targeting in ES cells. Our targeting strategy was aimed at abolishing transcription and translation by generating an ∼8 kb genomic deletion comprising 2.5 kb of the putative promoter, the first exon containing the ATG translational start codon and genomic sequences containing ∼50% of the APLP1 coding region. Top, Genomic segment containing the APLP1 locus. Apart from the first and second exon the precise location of the other exons (Exn) was not mapped. The stippled box represents an arbitrary positioned exon corresponding to coding sequences around cDNA position 1000. Parentheses indicate a restriction site derived from one of two overlapping λ phages. Middle, Targeting vector pAPLP1-targ. Horizontal arrows indicate the direction of transcription. The hatched boxrepresents a HSV-TK gene. Bottom, The disrupted allele after homologous recombination. E,EcoRI; H, HindIII;K, KpnI; P,PstI; S, SacI;X, XhoI; neo-probe, 0.6 kbPstI/XbaI fragment of pGKneo; probe B, genomic 0.5 kb PstI fragment. Bracketsrepresent restriction sites that were destroyed during cloning.
For Northern blots, total RNA (prepared as described in Müller et al., 1994) or polyA-RNA [prepared using Oligotex columns from Qiagen (Hilden, Germany)] was separated on 1% denaturing agarose gels and transferred by capillarity to HybondN+ (Amersham, Arlington Heights, IL) membrane. A 32P-labeled RNA probe containing 570 nt from the 3′ end of the APLP1 cDNA was prepared byin vitro transcription with T7 polymerase usingEcoRV linearized pBSKAPLP1 as the template (Wasco et al., 1992). To check for APP expression, a genomic 0.7 kbEcoRI/ScaI fragment subcloned into pBluescript and comprising exon 2 was used as the template for in vitrotranscription.
Northern blots were hybridized with riboprobes overnight at 65°C in 50% formamide solution containing 5× SSC, 5× Denhardt's solution, 1% SDS, 0.1% Na Pyrrophosphate, 5 mm EDTA, and 300 μg/ml yeast tRNA. Washing of blots was at 65°C for 20 min in 0.2× SSC and 0.5% SDS. After autoradiography blots were stripped by boiling for 10 min in 20 mm Tris-HCl, pH 8.0, 2 mm EDTA, and 0.5% SDS and rehybridized with a random primer 32P-labeled 490 bp XhoII fragment of the rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA.
Western blot analysis
Mouse organs were homogenized in ∼10 vol of homogenization buffer (0.5% SDS and 50 mm Tris-HCl, pH 6.8, 5 mm EDTA, 50 μg/ml pepstatin, and 0.25 mmPMSF) per gram of tissue, using a Ultra-Turrax T25 (IKA Labortechnik) blender. The homogenates were boiled for 10 min, centrifuged for 10 min at 12,000 × g, and the supernatants were recovered. Protein concentrations were determined by the BCA method (Pierce, Rockford, IL). Proteins were separated on Standard (Lämmli) 12% SDS-polyacrylamide gels using the protean II system (Bio-Rad) and electrotransferred to nitrocellulose membrane in low glycine buffer at 10 mA for 17 hr with wet transfer equipment (Bio-Rad). Filters were blocked for 1 hr in TBST (20 mm Tris-HCl, pH 7.6, 137 mm NaCl, and 0.1% Tween 20) containing 5% nonfat dry milk. The blot was incubated overnight with the primary antibody (e.g., rabbit antiserum CT-11 at a dilution of 1:2000 in TBST and 1% nonfat dry milk) followed by an 1 hr incubation with HRP-coupled secondary antibody [swine anti-rabbit IgG from Dako (Carpinteria, CA) at a dilution of 1:2500 in TBST and 1% nonfat dry milk] and developed by the ECL reaction (Pierce).
Antibodies used for Western blotting: CT-11 is a rabbit polyclonal serum raised against a synthetic peptide comprising the 11 C-terminal amino acids of murine APLP1 and is not cross-reacting with either APP or APLP2 (Thinakaran et al., 1995c). APLP2 was specifically detected with rabbit serum D2II (dilution 1:2000) as described (Thinakaran and Sisodia, 1994), and APP-specific staining was obtained with antibody 22C11 (dilution 1:1000; Chemicon). anti-Actin staining was performed with antibody C4 (dilution 1:10,000; ICN Biomedicals, Cleveland, OH).
Anatomical and histological analysis
Immunohistochemistry. Immunohistochemistry was done as described previously (Steinbach et al., 1998). Formaldehyde-fixed, paraffin-embedded, rehydrated, 2-μm-thick sections were incubated with monoclonal mouse and polyclonal rabbit antibodies (MAP II; 1:400, Boehringer, Mannheim, Germany), anti-synaptophysin (1:50, Dako), glial fibrillary acid protein (GFAP; 1:50, Dako), and β-III tubulin (1:2000, Promega) diluted in PBS for 2 hr at room temperature. This was followed by incubation with the secondary antibody (rabbit-anti mouse IgG, Dako) diluted 1:50 in PBS for 45 min at room temperature. Bound secondary antibody was detected by using the alkaline phosphatase-anti-alkaline phosphatase complex (APAAP; mouse monoclonal, Dako) diluted 1:40 in PBS and incubated for 45 min at room temperature. The alkaline phosphatase activity was visualized by using Astranenfuchsin (Aldrich, Milwaukee, WI). The sections were counterstained with hemalaun. The immunohistochemical methods used have previously been validated by us as a sensitive assay to detect lesions induced by systemic injection of sublethal, low doses of kainate (Steinbach et al., 1998).
In situ end-labeling assay. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays were performed on formalin-fixed sections: staining was performed incubating sections under coverslips with 50 μl of labeling mix [25 U/μl terminal deoxynucleotidyl transferase (TdT) (Boehringer), Dig-DNA labeling mix 10× concentration (Boehringer), and 2 mm CaCl2 in reaction buffer for terminal transferase (Boehringer) containing 0.2 mpotassium cacodylate and 25 mm Tris-HCl, pH 7.5] for 60 min at 37°C. After rinsing in Tris-buffered saline (TBS), sections were blocked with 10% fetal calf serum (FCS) (Seromed). Sections were then treated for 60 min with alkaline phosphatase-labeled anti-dioxigenin antibody Fab-fragment (Boehringer) at a dilution of 1:250 in 10% FCS. After washing in TBS, the color reaction was visualized by incubating the sections in reaction buffer (in mm: Tris 100, NaCl 100, and MgCl2 50, pH 9.5) containing 100 mg/ml 4-nitroblue tetrazolium salt and 50 mg/ml 5-bromo-4-chloro-3-indolyl phosphate (Boehringer) for 5–15 min. The reaction was stopped with TBS, and sections were counterstained with nuclear fast red (Merck).
Electron microscopy
Brains from newborn mice obtained from wt matings or by intercrosses of ALPL1+/−APLP2−/−mice, or APP+/−APLP2−/−mice, respectively, were dissected and fixed by immersion in 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, containing 2% polyvinyl-pyrrolidone (PVP) for 2 hr. After several rinses in cacodylate buffer with 2% PVP, the brainstems were post-fixed in 1% OsO4 in cacodylate buffer followed by dehydration in ethanol and embedding in Epon 812 substitute (Fluka, Neu-Ulm, Germany). Ultrathin 75 nm sections were cut with an ultramicrotome (Reichert Ultracut E). Sections were stained with uranyl acetate/lead citrate and examined under the electron microscope (ZeissEM 10C). Images were recorded with a BioScan Camera (Gatan).
Primary cultures from mouse cortex
Cultures of cortical neurons were prepared with some minor modifications in analogy to the procedure described by Banker and Goslin (1998) for hippocampal neurons. Embryos for cortical cultures were obtained from intercrosses of APLP1+/−APLP2−/−mice or APP+/−APLP2−/−mice, respectively. Genotyping was done by PCR on tail tissue. Wild-type control mice were obtained from 129 Sv(ev) × C57B6 matings and processed in parallel. Cortices from single E14.5 mouse embryos were collected in Ca 2+- and Mg2+-free HBSS/10 mmHEPES, pH 7.2 (Life Technologies, Gaithersburg, MD). After addition of trypsin (final concentration 0.05%) neuronal tissue was incubated for 8 min at 37°C. Trypsin was removed, the suspension was washed with 15 ml of HBSS/HEPES, and cortices were triturated with 5 ml glass pipettes in plating medium (serum-free Neurobasal medium supplemented with B27, 0.5 mm glutamine, and 50 U of penicillin–streptomycin; Life Technologies). Cells were seeded in 500 μl of plating medium in 24-well plastic dishes (coated overnight with 10 μg/ml poly-l-lysine hydrobromide in 0.1 mborate buffer, pH 8.5) at a density of 130,000 cells/well (∼73,000 cells/cm2) and maintained at 37°C in 5% CO2. Neuronal cultures were treated with cytosine-β-d-arabinofuranoside (Ara-C; final concentration 3 μm, Sigma) dissolved in 250 μl of plating medium on day 3 in vitro (DIV 3) to prevent glial proliferation. Neuronal purity was >95% as determined by immunohistochemical staining against GFAP, performed on DIV 10. Cultures were kept without further medium change until DIV 15 when survival experiments were done.
Survival assays and glutamate treatment
To assess spontaneous survival rates, neurons from individual embryos were cultured on gridded cellocate coverslips (Eppendorf, Hamburg). Live neurons, as judged by their morphological integrity (with an extended neurite network, smooth membrane appearance, and noncondensed soma) were counted within the same area (six nonoverlapping gridded fields, each covering a total area of 350 × 350 μm, containing ∼40–60 neurons per field, on average 240–360 neurons per sample) once on DIV 1 and again on DIV 7. Neuron counts were performed for two or three independent embryos for each genotype. Values represent averages ± SEM. The rate of spontaneous survival on DIV 7 was calculated relative to neuron counts obtained on DIV 1.
Cell survival after glutamate treatment was monitored by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium-bromide (MTT) assays: yellow MTT (Sigma) dissolved in PBS was added to the neuronal cultures to a final concentration of 0.5 mg/ml and incubated for 1 hr at 37°C. A violet formazan product is produced by viable cells because of the metabolic activity of mitochondrial enzymes. The formazan product is dissolved in 250 μl of DMSO and detected by its optical density (OD)Δ540–690. The ODΔ540–690 is taken as a measure of the number of living cells (≥95% neurons) in the culture (Mosmann, 1983).
To investigate cell survival in response to glutamate, neurons were prepared from individual embryos (8–12 processed in parallel), and cell suspensions of two cultures were counted before seeding. To correct for small differences in initial plating densities between individual cultures, cell survival was always expressed as relative values of glutamate-treated cultures normalized to values obtained from untreated cultures derived from the same embryo. For dose–response experiments different concentrations of glutamate (10, 25, 50, 100 μm) dissolved in plating medium were added to cortical neurons for 24 hr followed by MTT assays. Average values from triplicate cultures were calculated relative to values from nontreated cultures for which the mean was set as 100% survival. For each experiment two or three independent embryos were used, and the experiment was replicated at least three times. In addition, cortical neurons were treated with 50 μm glutamate for 1 or 3 hr. For each time point, a triplicate of 24-well cultures were processed in parallel, and mean values were determined. In addition, triplicate sets of nontreated cultures were analyzed for each time point. The mean MTT value obtained from these nontreated cultures (after 0, 1, and 3 hr of incubation) was set as 100% cell survival for each embryo. Percentage of survival was calculated relative to these mean values.
RESULTS
Generation and characterization of APLP1 knock-out mice
Inactivation of the murine APLP1 gene was achieved by gene targeting with a strategy depicted in Figure 1. By homologous recombination in ES cells we generated an ∼8 kb genomic deletion comprising 2.5 kb of the putative promoter, the first exon containing the ATG translational start codon and genomic sequences containing ∼50% of the APLP1 coding region. Deletion of the transcription and translation start sites should completely abolish APLP1 expression. Mutant ES cells were injected into blastocysts and gave rise to one chimera that transmitted the mutant allele in the germ line. Correct homologous recombination was confirmed by Southern blot analysis of tail DNA that showed the expected pattern for an interrupted APLP1 locus (Fig. 2A,B). Heterozygous animals were intercrossed and yielded homozygous APLP1 knock-out animals at a normal Mendelian frequency. To determine whether the APLP1 gene was also functionally inactive, Northern and Southern blot analysis was performed on brain tissue. An antisense RNA probe (cDNA position 1788–2360) lying downstream of the targeted deletion revealed a band of ∼2.6 kb on brain of wt animals, whereas no transcript was detected in APLP1−/−mutants (Fig. 2D). Western blot analysis with the antibody CT-11, which is directed against the 11 C-terminal amino acids of APLP1 and discriminates against the related APP and APLP2 proteins, gave rise to a set of APLP-1-specific bands of 85–100 kDa for wt brain, but not for mutants, indicating that the gene had been completely inactivated. Expression of the other family members APP and APLP2 was unaltered, as judged from Northern (data not shown) and Western blot analysis (Fig. 3A, lane 1, lane 2 ).
Fig. 2.

Molecular validation of the APLP1 knock-out. Functional inactivation of the APLP1 gene was confirmed by a combination of Southern (A, B), Western (C), and Northern (D) blot analysis. Genomic tail DNA from F1 offspring of the germline chimera was digested with HindIII and hybridized with genomic probe B, as depicted in Figure 1. Note that wt animals show a single band of 18 kb, whereas in heterozygous APLP1 mutants an additional band of the expected size of 14 kb was detected (A). Intercrossing of these heterozygous animals lead to homozygous mutants (B), as judged by analysis of DNA from mouse embryonic fibroblasts (MEF). The respective genotype is depicted above the blots. Western blots (C) of wt and APLP1−/− mice were prepared as described in Materials and Methods. Forty micrograms of brain extracts from two APLP1−/− mice (lanes 1, 2), a wt APLP1+/+littermate (lane 3), and a 129 Sv(ev) wt mouse (lane 4) were separated on a 12% standard Lämmli gel. Incubation with APLP1-specific antiserum CT11 followed by anti-rabbit horseradish peroxidase-linked secondary antibody showed a set of APLP-1-specific bands of 85–100 kDa for wt brain homogenates. In contrast, brain homogenates from APLP1−/− animals showed neither APLP1-specific bands of wt size nor any shorter polypeptides. The blot was developed with chemiluminescence reagents (ECL; Amersham). Marker proteins of the indicated sizes were from Bio-Rad (broad range rainbow markers). Total RNA was isolated from brain of either mutant or wt animals, and poly(A+) RNA was prepared from 120 μg of total RNA. Northern blot analysis (D) with an antisense RNA probe (cDNA position 1788–2360) lying downstream of the targeted deletion revealed a band of 2.4 kb on brain of wt animals, whereas no transcript was detected in organs from APLP1−/− mutant animals. After autoradiography, filters were stripped and rehybridized with a GAPDH probe to monitor loading (bottom panel).
Fig. 3.
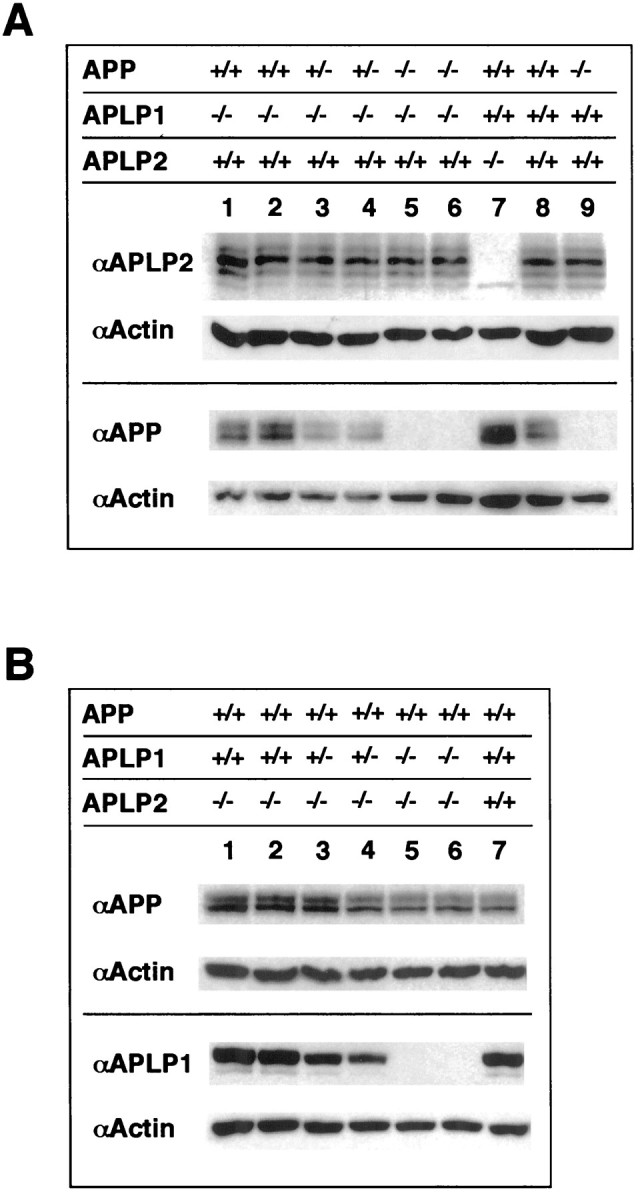
Western blot analysis of APP/APLP expression in single and double mutants. A, Total brains of newborn APP−/− (lane 9), APLP2−/− (lane 7), and wt (lane 8) mice and in addition brains of newborn mice generated by intercrossing heterozygous APP+/−/APLP1−/−mice (lanes 1–6) were homogenized, and equal amounts of protein (20 μg/lane) were resolved on 8% PAA gels. From each blot the bottom half was cut off and probed with an Actin-specific antibody to monitor loading (second row of each panel).Top, Note that probing with an antiserum specific for APLP2 (D2II) showed no major alterations of APLP2 expression in animals of different APP/APLP1 genotype compared to wt levels (lane 8). Bottom, Probing with an APP-specific antibody (22C11) showed comparable amounts of total APP expression in APLP1−/− (lanes 1, 2) and wt (lane 8) mice. The seemingly higher expression of APP in APLP2−/−mice (lane 7) is attributable to unequal loading as evidenced by more intense actin staining. As expected, APP expression was abolished in APP−/− single mutants (lane 9) and APP−/−/APLP1−/−double mutants (lanes 5, 6). Note that in heterozygous APP+/−/APLP1−/−mice APP expression is reduced to ∼50%, arguing against a compensatory upregulation of APP expression. B, Total brains of newborn pups obtained from heterozygous APLP2+/−/APLP1−/−intercrosses were homogenized, and equal amounts of protein (20 μg/lane) were separated on a 8% PAA gel. Top, Probing with an APP-specific antibody (22C11) showed similar amounts of total APP expression in both viable APLP1+/−/APLP2−/−heterozygotes (lanes 3, 4) or in lethal APLP1−/−/APLP2−/−double knock-outs (lanes 5, 6), compared to the amount of APP expression in APLP2−/− single mutants (lanes 1,2) or in a wt control (lane 7). Note that no significant compensatory upregulation of APP expression was found. Bottom, Probing with an antiserum specific for APLP1 (CT11) showed similar APLP1 expression levels in APLP2−/−mice (lanes 1,2) compared to the wt control. Note that in heterozygous APLP1+/−/APLP2−/−animals (lanes 3, 4) expression is reduced to ∼50% and abolished in APLP1−/−/APLP2−/−double mutants (lanes 5, 6). Genotypes of animals analyzed are as indicated above blot panels.
APLP1−/−-animals were viable, fertile, and showed, apart from a significant body weight deficit of ∼10% (mutant males, 25.37 ± 0.70 gm; wt males, 27.72 ± 0.76 gm; mutant females, 20.01 ± 0.47 gm; wt females, 22.12 ± 0.55 gm; average age, 9 weeks; n= 59; p < 0.001 by two-way ANOVA), emerging in the second week postnatally (data not shown), no overt abnormalities until at least 18 months of age. In contrast to APP-deficient mice that exhibit reduced locomotor activity and reduced forelimb grip strength (Müller et al., 1994; Zheng et al., 1995; Tremml et al., 1997), APLP1−/−mice showed, compared to wt controls, normal locomotor activity, and in adult animals grip strength was unchanged (data not shown). A subtle retardation of neurobehavioral development [as assessed by tests of the Fox (1965) battery] was observed early postnatally (Tremml et al., 1997; data not shown). Histopathological analysis of brain sections (see Materials and Methods) showed no morphological alterations; in particular no agenesis of the corpus callosum was observed [even on a pure 129Sv(ev) genetic background], as opposed to APP-deficient mice exhibiting commissure defects and a high incidence of callosal agenesis on a 129-specific background (Magara et al., 1999). Spatial learning was analyzed by the Morris water maze test, which revealed similar cognitive abilities of APLP1−/−animals as wt controls (as assessed by their normal performance in reversal learning). Surprisingly, however, APLP1-deficient mice showed improved acquisition learning, which could be attributed to altered noncognitive components of the behavior, in particular reduced thigmotaxis (Tremml et al., 1997; P. Tremml, U. Müller, H.-P. Lipp, and D. Wolfer, manuscript in preparation). In summary, ablation of the neuron-specific APLP1 gene results in a rather minor phenotype that is clearly distinct from that of APP-deficient mice, suggesting at least some different functions of both proteins.
Generation of mutants with combined gene deficiencies
To address the question of whether the minor phenotype of single mutants is caused by functional compensation by other APP family members, we set out to generate all three possible combinations of double mutants (APP−/−/APLP2−/−, APLP1−/−/APLP2−/−, and APP−/−/APLP1−/−) by crossing the respective single mutants (see Materials and Methods for details). To obtain APP−/−/APLP2−/−mice, animals deficient for APLP2 and heterozygous for APP (APLP2−/−/APP+/−) were intercrossed and expected to yield 25% offspring deficient for both proteins. When we genotyped the offspring at weaning (∼4 weeks of age) initially none and, after analysis of a larger sample of 355 mice, only a single surviving double mutant was found (Table 1A), suggesting that a combined deficiency of APP/APLP2 is lethal either during development or within the first weeks after birth. Analysis of the genotype distribution of offspring at E19 and shortly after birth (P0) showed that almost the theoretically expected number of double mutants (21% of 131 animals screened; Table 1A) survived until this time point. A smaller sample of 26 mice was analyzed at postnatal day 1 (P1), however, only two APP−/−/APLP1−/−mice were found (one of which died at P2 and the other after 4 weeks) indicating postnatal lethality, predominantly within the first day after birth.
Table 1A.
Genotype analysis of offspring obtained from APP/APLP2 matings

Survival rates of APP/APLP2 double mutants reveal postnatal lethality. At E19, or shortly after birth (at P0), a total of 131 animals obtained from parents of the indicated genotypes were examined. At this time point double knock-out animals were obtained at about the expected frequency (21%). Of 26 mice analyzed at P1 only two APP−/−/APLP1−/− mice were found, one of which died at P2 and the other at the age of 4 weeks. At weaning only one APP−/−/APLP2−/− double knock-out mouse of 355 animals examined was found that has so far survived for 13 months.
Table 1B.
Genotype analysis of offspring obtained from APLP1/APLP2 matings

Survival rates of APLP1/APLP2 double mutants reveal postnatal lethality. Shortly after birth (at P0) a total of 171 newborn animals obtained from parents of the indicated genotypes were examined. At this early time point double knock-out animals were obtained at a normal frequency (29%), indicating that APLP1/APLP2s are not essential for embryonic development. Note that no APLP2−/−/APLP1−/− double knock-out mice were found among 28 animals examined at P1 or among 326 animals analyzed at weaning.
Analogously, APLP1+/−/APLP2−/−mice were intercrossed, and the offspring were genotyped at weaning. As seen for APP−/−/APLP2−/−mice, a combined APLP1/APLP2 deficiency proved lethal, in this case with 100% penetrance. Among 326 animals analyzed at weaning, not a single surviving double mutant was found (Table 1B). A smaller sample of pups was screened shortly after birth (P0), and another set on P1 (Table 1B). Whereas APLP1−/−/APLP2−/−mice were born at normal Mendelian frequency (29% of 171 animals), no surviving double mutants were found at P1, indicating that a combined APLP1/APLP2 deficiency results in lethality within the first day after birth. These results demonstrate that APP family members serve essential but at least partially redundant functions in vivoand corroborate a key physiological role for APLP2.
To our surprise, APP−/−/APLP1−/−mice generated in a similar manner were viable, fertile, and showed no apparent abnormalities, apart from a body weight deficit comparable to that of single mutants, until at least 18 months of age. This unexpected finding that not all three possible combinations of the single mutants results in a lethal phenotype has crucial implications for the specific and redundant functions exerted by APP family members (see Discussion).
The key physiological function or functions of APLP2 are further supported by recent preliminary data demonstrating postnatal lethality for APP−/−/APLP1−/−/APLP2+/−mice, which suggests haploinsufficiency for the remaining single APLP2 allele. In an attempt to ultimately generate triple knock-outs, which should theoretically be feasible by intercrossing APP−/−/APLP1−/−/APLP2+/−mice, we set up appropriate matings (see Material and Methods) that were expected to yield 25% offspring of the respective (APP−/−/APLP1−/−/APLP2+/−) genotype. Among 350 animals screened at weaning, we found only four APP−/−/APLP1−/−/ APLP2+/−mice (1% instead of the expected 25%). As observed for double mutants (see below) APP−/−/APLP1−/−/APLP2+/−mice showed (when analyzed on P0) no apparent histopathological abnormalities (data not shown). The surviving animals were severely impaired in breeding, and we have so far been unable to obtain any litters from these mice. Again, lethality occurred postnatally, because an APP−/−/APLP1−/−/APLP2+/−allele frequency of 27% was found in a smaller sample of mice, analyzed shortly after birth (data not shown). We conclude from these data that the presence of a single APLP2 allele in the absence of other APP family members is not sufficient for survival.
Analysis of APP/APLP expression in double mutants
We then asked whether the viable phenotype of APP−/−/APLP1−/−mice may be attributed to a compensatory upregulation of APLP2 (Fig.3). Western blot analysis of brain homogenates prepared from newborn mice showed APLP2 levels comparable to those found in single mutants or wt animals (Fig. 3A, top) suggesting that basal wt-APLP2 levels are not limiting for survival. More subtle or region-specific alterations in APLP2 expression cannot be ruled out, however. Likewise, APP protein levels were not upregulated in heterozygous, viable APP+/−/APLP1−/−littermates, showing an ∼50% reduction of APP as compared to APP+/+/APLP1−/−single knock-outs (Fig. 3A, bottom). We then determined whether in the lethal APLP2−/−/APLP1−/−double knock-out, expression of the remaining third family member, APP, is altered. Both Northern (data not shown) and Western blot analysis failed to reveal changes in the expression level of APP in the brains of both newborn viable APLP2−/−/APLP1+/−pups and of APLP2−/−/APLP1−/−littermates that would have died within the next hour (Fig. 3B, top). However, APLP1 expression was found to be reduced to ∼50% of wt level in viable APLP2−/−/APLP1+/−heterozygous newborns (Fig. 3B, bottom). Thus, a loss of APP/APLP family members does not cause compensatory upregulation of related family members in double knock-outs.
Gross and histopathological analysis of double mutants
Newborn double mutants were initially (for several hours) indistinguishable from their littermates and were able to suckle. Between ∼5 and 15 hr after birth, however, they had no or less milk in their stomachs, appeared weaker, became pale, and died. From the observation of several litters of mice of both types of lethal double mutants, we have no evidence for motor paralysis or overt seizure activity. To address the cause of the early postnatal lethality observed in APLP2−/−/APLP1−/−and APLP2−/−/APP−/−double knock-outs, we used various histological approaches to examine newborn double mutants, as compared to APLP2−/−single mutants and wt controls. No obvious abnormalities were observed after macroscopic examination, revealing normal sizes of the head, face, and body postnatally. Hematoxylin–eosin-stained sections of internal organs also did not show any abnormalities. In particular, no malformations of the palate, esophagus, stomach, intestine, colon, and rectum were found that could result in impaired feeding behavior. Moreover, meconium was present in the intestine of all mutants. Furthermore, cranial nerves implicated in feeding, including trigeminal ganglion and facial motor nucleus, were present and appeared of normal size. The heart and lungs appeared normal in maturity and size, which argues against an impairment of cardiopulmonary function as the cause of lethality, however alterations in the efferent autonomic innervation of the heart cannot be excluded. None of the organs examined (thyroid, thymus, spleen, pancreas, liver, kidneys, bladder, testis, and ovaries) showed differences in size or histopathological abnormalities in both types of double mutants, or single mutants, compared to wt controls (data not shown).
In the brain no signs of increased cell death were found, as evidenced by pyknotic nuclei in Nissl-stained sections or by TUNEL staining (Fig.4). The neocortex showed the normal layered structure, and cortical neurons were of normal appearance. CA1 through CA3 regions and the dentate gyrus of the hippocampus appeared normal, based on Nissl staining pattern and MAP-II and synaptophysin immunohistochemistry, indicating that lack of APLP2 either in combination with a deficiency for APP, or APLP1, respectively, did not affect hippocampal architecture and cell/neurite density (Fig. 4). GFAP immunohistochemistry revealed no signs of gliosis (data not shown).
Fig. 4.
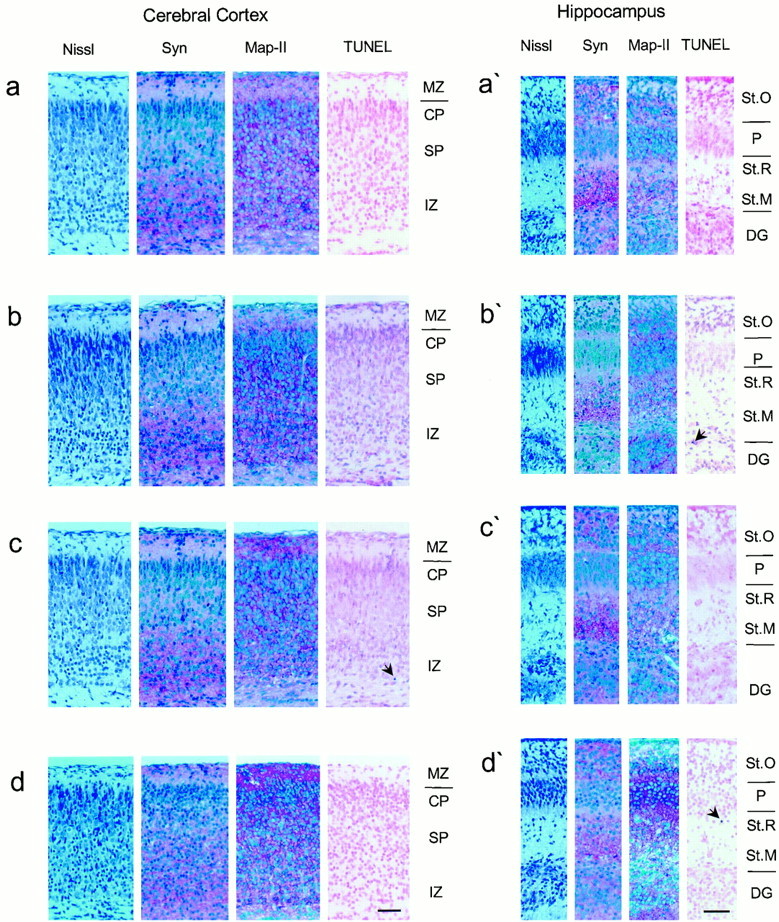
Characterization of mutants by immunocytochemistry on brain sections. Histological analysis of the cortex and hippocampus from age-matched newborn wild-type, APLP2−/−, and double knock-out mice lacking either APLP2/APLP1 or APLP2/APP revealed no apparent anomalies in any of the mutants examined. Pictures show frontal sections of parietal cortex (a–d) and hippocampus (a′–d′) from wild-type (a), APLP2−/−(b), APP−/−/APLP2−/−(c), and APLP1−/−/APLP2−/−(d) knock-out mice. Shown are Nissl stains and immunohistochemistry with antibodies directed against synaptophysin (Syn) and MAP-II, as well as TUNEL stains. Scale bars: d, d′, 50 μm (applies to all panels). The cortical layers are indicated as:MZ, marginal zone; CP, cortical plate;SP, subplate; IZ, intermediate zone. The hippocampal structures are indicated as follows: St.O, stratum oriens; P, CA1 pyramidal cells;St.R, stratum radiatum; St.M, stratum moleculare; DG, dentate gyrus granule cells. Thearrows mark apoptotic nuclei.
Because APLP2 has been suggested to play a role in axon outgrowth in the olfactory system, where it is highly expressed in olfactory sensory neurons and axon terminal glomeruli (Thinakaran et al., 1995a), we analyzed the olfactory system of both types of double mutants. However, the distribution and density of neurons in the olfactory bulb, as well as the number and thickness of neuron-specific tubulin-positive axon bundles, were not found to be different between the lines studied (data not shown). No differences were observed in size and nerve cell density of brainstem nuclei (data not shown).
APP undergoes fast axonal trafficking to presynaptic terminals (Tienari et al., 1996) and has been detected in rab5-containing (Ikin et al., 1996) and clathrin-coated vesicles (Nordstedt et al., 1993), organelles that are likely to be involved in endocytosis. To assess whether a lack of APP family members affected synaptic architecture, we examined by electron microscopy coronal sections of the cortex and the brainstem from newborn double mutants and controls (see Fig.5 for representative images). Inspection of many electron micrographs revealed in both types of lethal double mutants (Fig. 5B,C) mature synapses in the brainstem, at densities similar to that found in wt controls (Fig. 5A). In contrast, predominantly immature synapses were detectable in the developing cortices of these mice (data not shown). Again, no apparent differences in synapse densities could be detected between sections from wt and the lethal double mutants (data not shown). Also, no obvious changes were found in the ultrastructure of the nerve terminals (Fig. 5). We therefore conclude that, at least with regard to these morphological aspects, synapse formation proceeds apparently normal in the brainstem of mutant animals. We also consider neuromuscular deficits as an unlikely cause of lethality in double mutants because some of us have addressed this question in a previous study investigating APP/APLP2 double knock-outs (von Koch et al., 1997).
Fig. 5.

Ultrastructure of brainstem synapses from single or combined mutants. Comparison of the morphology of synapses in brainstem ultrathin sections from newborn mice revealed no obvious changes in the ultrastructure of the nerve terminals in mutant compared to wt mice. Shown are electron micrographs of representative active zones obtained from wt (A), APLP2−/−/APP−/−(B), and APLP2−/−/APLP1−/−(C) samples processed as described in Materials and Methods. At the presynaptic site mutants and control mice showed comparably sized vesicle clouds, including docked vesicles in close proximity to the membrane. Electron dense postsynaptic specializations were clearly detectable in both double mutants.
Viability of primary neuronal cultures derived from single and combined mutants
Viability of cortical neurons derived either from APLP2−/−single mutants or from both types of lethal double mutants (APP−/−/APLP2−/−and APLP1−/−/APLP2−/−) was investigated by counting viable neurons on gridded coverslips after plating (DIV 1) and again after 7 d in culture (DIV 7; Fig.6). No significant differences relative to wt neurons were detectable for any of the mutants analyzed, indicating that under the culture conditions used, loss of endogenous APLP2 alone, or in combination with APLP1 (see Fig.6A for APLP1−/−/APLP2−/−) or with APP (see Fig. 6B for APP−/−/APLP2−/−), respectively, does not affect neuronal survival. Moreover, we saw no significant differences in plating efficiency of viable neurons between wt and mutant cultures on DIV 1 (data not shown), indicating that lack of APP family members did not affect the viability of major neuronal subpopulations in these cultures.
Fig. 6.

Viability of cortical neurons from single or combined mutants. Neuronal viability was similar to that of wt neurons in APLP2−/− single mutants (A, B) or both types of double mutants.A,APLP2−/−/APLP1−/−double mutants. B,APLP2−/−/APP−/−double mutants. Cortical neurons were obtained from individual embryos, as described in Materials and Methods. Viable neurons were counted using gridded coverslips once at DIV 1 (set as 100%) and again on DIV 7. For each embryo ∼240–360 neurons were counted, and two or three embryos were analyzed for each genotype. Values represent mean neuron counts obtained for mice of the indicated genotypes ± SEM, normalized to initial values on DIV 1.
Survival of neuronal cultures derived from single or combined mutants against excitotoxic stress
Studies by Mattson et al. (1993) and Mattson (1994) have demonstrated that primary neuronal cultures can be protected from glutamate-induced excitotoxicity by addition of exogenous secreted forms of human APP (sAPP) to the culture medium. We have previously shown that APP-deficient mice are hypersensitive to seizures induced by the glutamate receptor agonist kainate, although the amount of tissue damage was similar to that observed for wt animals (Steinbach et al., 1998). However, using primary neuronal cultures derived from APP mutant mice, we and others (Steinbach et al., 1998; White et al., 1998) have been unable to show differences in the glutamate sensitivity of APP-deficient neurons. To unravel whether endogenous APP plays any role in the protection of neurons against glutamate excitotoxicity and whether the related APLPs may compensate for such a protection by APP, we compared the survival of neurons derived from single APP family mutants and from lethal mutants deficient for a combination of APLP2/APP, or APLP2/APLP1, respectively. Two sets of experiments were performed. We first investigated the response of neurons prepared from single embryos obtained by intercrossing heterozygous APP+/−/APLP2−/−mice and as controls, neurons prepared from wt embryos of 129Sv(ev)xC57BL/6 matings (Fig.7A,C). Second, we analyzed neurons of embryos derived from intercrosses of APLP1+/−/APLP2−/−mice in comparison to wt neurons of 129Sv(ev)xC57BL/6 matings (Fig.7B,D). Cortical neurons were used because sufficient numbers of cells can be obtained from individual cortices and moreover, this cell type has previously been shown to be protected from glutamate excitotoxicity by (hu)sAPP (Mattson et al., 1993). Cortical neurons cultured under serum-free conditions for 15 d in vitrowere treated with increasing amounts of glutamate (10–100 μm) for 24 hr, and their survival was assessed by MTT assays. This resulted in a glutamate concentration-dependent reduction in cell survival for all cultures examined. Neither APLP2-deficient neurons nor APP−/−/APLP2−/−double knock-out cultures differed, however, significantly in their survival rates compared to wt control neurons at any glutamate concentration used (Fig. 7B). Likewise, APLP1−/−/APLP2−/−double knock-out cultures showed similar survival rates as APLP2−/−single knock-outs or wt control cultures (Fig. 7A). To test for possible differences in the response kinetics of the various mutants we investigated cell survival at 1, 3, and 24 hr after addition of a constant amount of 50 μm glutamate (Fig. 7C,D for 1 and 3 hr, Fig. 7A,B for 24 hr time point). Again, no significant differences for both sets of mutants compared to wt neurons were found at any time point investigated. As expected from these results, we were also unable to detect significant differences when we tested the whole panel of single mutants APP−/−, APLP2−/−, and APLP1−/−relative to wt controls (data not shown).
Fig. 7.

Susceptibility of neurons from single or combined mutants to glutamate excitotoxicity. Cortical neurons from single mutants, heterozygotes, or double mutants showed no difference in their resistance to glutamate toxicity compared to wt neurons. Primary cortical neurons of the indicated genotypes were cultivated for 15 d before they were treated with glutamate, and their survival was analyzed by MTT assays. A, B, Cultures were exposed to different glutamate concentrations (10, 25, 50, 100 μm) for 24 hr. C, D,Glutamate (50 μm) was applied for 1 and 3 hr. MTT measurements were performed immediately after the glutamate exposures. Neurons from embryos of the respective genotypes were generated as described in Materials and Methods.
We have conducted MTT assays to monitor cell death in response to glutamate challenge. Because no difference was found between mutants and wt neurons either at different concentrations or after different length of exposure to glutamate, we did not extensively investigate the actual mode of cell death. Apoptotic cell death has been reported in cultures of cortical neurons when assayed late (18–24 hr) after exposure to excitotoxins (Tenneti and Lipton, 2000). After incubation with 50 μm glutamate for 24 hr we detected predominantly apoptotic cell death to a similar extent in both wt and mutant cortical cultures, as evidenced by staining with fluorescent chromatin dyes (data not shown). It is conceivable that a mutation could have a differential effect on the type of cell death, altering either necrotic or apoptotic responses that may contribute to overall amounts of cell death depending on the specific dose and duration of the insult (Nicotera et al., 1999). Because we saw no shift in the kinetics of overall cell death (as measured by MTT, see Fig. 7) we consider that as unlikely. Moreover, for none of the groups we detected any significant differences in the endpoint of maximal values of cell death. In summary, these findings indicate that cortical neurons deficient in either individual APP family members or doubly deficient for APP/APLP2 or APLP1/APLP2, respectively, do not differ significantly in their ability to survive glutamate-induced excitotoxic insults.
DISCUSSION
This paper investigates the phenotype of APLP1 knock-out mice and a complete set of double mutants comprising all combinations of known APP family members. A summary of our findings is given in Table 2. To analyze the physiological role of the nervous system-specific APLP1 protein, we have generated APLP1−/−mice that were viable and fertile, but revealed a body weight deficit, similar to that observed for APP knock-out mice (Zheng et al., 1995;Tremml et al., 1997; Magara et al., 1999). Thus, both APP and APLP1 appear to play a role in postnatal somatic growth. At least for APLP1-deficient mice, this phenotype might reflect a neural deficit, because APLP1 expression is restricted to the nervous system. Other abnormalities, including those observed for APP−/−mice, were absent in APLP1-deficient mice, suggesting either functional compensation by other family members or distinct functions for APLP1 and APP.
Table 2.
Phenotypes of APP family knock-outs
| Genotype | Phenotype | Reference |
|---|---|---|
| Single mutants | ||
| APP−/− | Viable and fertile | Zheng et al. |
| Reduced body weight | Müller et al. | |
| Reduced grip strength | Li et al. | |
| Reduced locomotor activity | ||
| Alterations in sensorimotor development | Tremml et al. | |
| Commissure defects | Magara et al. | |
| Hypersensitivity to epileptic seizures | Steinbach et al. | |
| APLP1−/− | Viable and fertile | This paper |
| Reduced body weight | ||
| APLP2−/− | Viable and fertile | von Koch et al. |
| No apparent abnormalities | ||
| Double mutants | ||
| APLP2−/−, APP−/− | Perinatally lethal | This paper |
| No histopathological anomalies | von Koch et al. | |
| APLP2−/−, APLP1−/− | Perinatally lethal | This paper |
| No histopathological anomalies | ||
| APLP1−/−, APP−/− | Viable and fertile | This paper |
| Reduced body weight | ||
| APLP1−/−, APP−/−, APLP2+/− | Perinatally lethal | This paper |
| No histopathological anomalies |
Postnatal lethality of double mutants
In contrast to the minor phenotype of mice singly deficient for individual APP family members, two of three double mutants generated in this study (APP−/−/APLP2−/−and APLP1−/−/APLP2−/−) proved perinatally lethal, indicating essential but partially redundant functions within the gene family (Table 2). Our findings that APP−/−/APLP2−/−mice die shortly after birth are in agreement with previous results from von Koch et al. (1997), who had crossed the same line of APLP2−/−mice used here to the APP−/−line generated by Zheng et al. (1995). Interestingly, we observed an almost complete penetrance of early postnatal lethality for our strain of APP−/−/APLP2−/−mice, whereas 26% of double mutants generated by von Koch et al. (1997) survived well into adulthood. This difference in penetrance is likely attributable to the respective APP knock-out strains, which differ in the targeted deletion introduced into the APP locus and in the composition of genetic background alleles. Because the phenotypes of both APP single knock-out lines are largely congruent, it appears more likely that differences in the genetic backgrounds affected penetrance (Müller, 1999).
Given the similarities of the phenotypes of the two lethal double mutants (APP−/−/APLP2−/−and APLP1−/−/APLP2−/−), i.e., death within the first day or days in the absence of gross morphological anomalies, a common underlying mechanism seems likely, although the actual cause is presently unknown. Because APLP1 is, unlike the ubiquitously expressed APP and APLP2 proteins, restricted to the nervous system, it is conceivable that the cause of lethality resides, at least for APLP1−/−/APLP2−/−mice, in the nervous system. This hypothesis gains further support from recent studies on the Caenorhabditis elegans homolog APL-1 by Li et al. (Daigle and Li, 1993; Hornsten et al., 1999). Most interestingly, a knock-out of the ubiquitously expressed apl-1 gene proved lethal in C. elegans, but could be rescued by transgenic expression of APL-1 in neurons, indicating lethal neuronal deficits in the absence of APL-1 (Hornsten et al., 1999).
APP family members could be compensating for each other by binding to and stimulating a common, as yet unidentified receptor. Alternatively, APP/APLPs might act themselves as receptors for a common ligand. Several proteins containing phosphotyrosine-binding (PTB) domains (e.g., Fe65- and X11-family proteins) interact with the intracellular C-terminal domain of APP/APLPs, including the cytoplasmic adapter protein Dab1 (Homayouni et al., 1999). Dab1 is involved in the reelin (Reln) signal transduction pathway, which directs cell positioning in the developing CNS (Rice and Curran, 1999). Our histopathological analysis of single or combined APP/APLP mutants failed to reveal, however, alterations in the formation of laminated brain structures, typically seen in Dab1−/− and Reln−/−mice (D. Rice, R. Homayouni, M. Sheldon, and T. Curran, personal communication). Interestingly, mice lacking cyclin-dependent kinase 5 (cdk5), a kinase known to phosphorylate tau protein implicated in Alzheimer's disease, also display late embryonic or perinatal lethality. In contrast to APP/APLP mutants they exhibit, however, lamination defects similar to Dab1−/− and Reln−/−mice (Ohshima et al., 1996). Recently, APP has been shown to have antiapoptotic effects in a neuroblastoma (B103) cell line challenged with apoptosis-inducing insults (Xu et al., 1999). It will therefore be interesting to see whether neurons derived from APP/APLP knock-out mice are, accordingly, more sensitive to apoptotic challenge. In this study, however, no increase in the amount of spontaneous apoptosis was detectable, as evidenced by normal, low levels of TUNEL-positive cells in the brains of newborn mutant mice. It is evident from the lack of obvious morphological abnormalities that a more detailed analysis is necessary, addressing for example possible deficits in synaptogenesis and/or electrophysiological alterations.
Neuronal survival and response to glutamate-induced excitotoxicity
One aim of this work was to assess the role of endogenous APP family members for neuronal survival and protection against excitotoxic stress that may contribute to the pathology of Alzheimer's disease. This study clearly demonstrates that cortical neurons, deficient for either APLP2 alone, or doubly deficient for APLP2/APP, or APLP2/APLP1 respectively, show unaltered survival rates under basal culture conditions, which confirms and extends previous studies in APP or APLP2 single knock-outs (Harper et al., 1998; Steinbach et al., 1998; White et al., 1998, 1999a). These data suggest that the presumed neurotrophic activities of endogenous APP/APLP proteins are not essential for survival, at least as long as one APP family member is still expressed.Perez et al. (1997) have reported a small (∼20%) difference in the rate of survival of hippocampal neurons lacking APP, contrasting with normal survival rates in this study. This discrepancy may either be attributable to differences in culture conditions or alternatively, endogenous APP may play a greater role in neurons derived from the hippocampus, a region especially vulnerable in AD.
Several studies suggested that APP (and possibly also APLPs) may exert neuroprotective functions. In vitro, exogenously added (hu)sAPP can protect cortical and hippocampal neurons against the toxic effects of glutamate and Aβ peptide, and may modulate K+ channels and suppress NMDA receptor-mediated ion currents (Mattson et al., 1993; Furukawa et al., 1996; Furukawa and Mattson, 1998). Ca2+influx via NMDA channels has in turn been implicated as a major mechanism underlying excitotoxic cell death. In vivo, moderate overexpression of huAPP in transgenic mice confers resistance to kainate-induced and chronic forms of excitotoxicity (Mucke et al., 1994; Masliah et al., 1997). The role of endogenous APP for neuroprotection seems less clear. We have previously shown that APP knock-out mice are more vulnerable to kainate-induced seizures, however no increase in the amount of tissue damage or reduced survival responses of primary neurons were found (Steinbach et al., 1998; White et al., 1998). To assess whether compensation by APLPs may have alleviated excitotoxicity, we investigated in this study the response of cortical neurons derived from double mutants against glutamate excitotoxicity. Our data, demonstrating no significant difference in the response of cortical neurons derived from APLP2−/−single mutants or APP−/−/APLP2−/−and APLP1−/−/APLP2−/−double mutants, as compared to wt neurons, do not support a prominent protective function of endogenous APP/APLPs against glutamate toxicity in vitro. We cannot exclude, however, that because of experimental variability (SEM of ∼10–15% of variable) more subtle effects may have been masked. Conversely, it is also possible that the third APP family member might still exert compensatory effects. It remains to be seen, whether neurons from APLP2−/−single mutants, or double mutants, will show altered responses to other forms of metabolic stress, including Aβ-mediated toxicity. In vivo, endogenous APP/APLPs secreted locally might reach higher concentrations and might play a role in ameliorating chronic forms of excitotoxic and oxidative stress that have been implicated as factors contributing to AD pathology (Furukawa and Mattson, 1998; White et al., 1998).
Redundancy within the gene family
Interestingly, lack of APLP2 in combination with a deficiency of either APP or APLP1 was perinatally lethal, whereas the third double mutant, APP−/−/APLP1−/−was viable and apparently normal (Table 2). These data corroborate a key physiological role for APLP2 and indicate redundancy between APLP2 and both other family members. The viability of APP−/−/APLP1−/−mice could not be explained by an upregulation of APLP2, because APLP2 expression was unaltered (Fig. 3). Likewise, viable heterozygous littermates of lethal double mutants showed no upregulation of APP/APLP expression. Even in the absence of APLP2 upregulation, viability of APP−/−/APLP1−/−mice might still be attributable to compensation by APLP2, a view that is further supported by the postnatal lethality of APP−/−/APLP1−/−/APLP2+/−mice. It will be interesting to see whether lack of the entire APP family will result in an even more pronounced phenotype.
The unexpected finding that APP−/−/APLP1−/−mice are viable has also crucial implications for the specific physiological functions exerted by APP and APLP2. So far, it has been widely assumed (von Koch et al., 1997; Steinbach et al., 1998; White et al., 1998) that both proteins may serve primarily overlapping functions because (1) APP and APLP2 are highly homologous, (2) they are similarly processed, (3) they are expressed in a virtually overlapping pattern, and (4) deficiency of both proteins results is a synthetic lethal phenotype suggesting functional redundancy. This view, however, is challenged by our data showing that double mutants obtained by crosses of APP−/− or APLP2−/−single mutants with APLP1−/−mice have clearly different phenotypes, being either viable (APP−/−/APLP1−/−) or lethal (APLP2−/−/APLP1−/−). We thus provide genetic evidence that APP and APLP2 must serve, besides overlapping functions, also at least some distinct, nonredundant functions in vivo.
Footnotes
This work was supported by grants from the Swiss National Science Foundation (NF31-42347.94) and the Volkswagen Foundation. We thank Nicole Fürst, Gerlinde Stark, Rosemarie Lang, Mariana Stagliar, and Walter Hofer for excellent technical assistance. We are grateful to Wilma Wasco for providing the muAPLP1 and huAPLP2 cDNAs and to Gopal Thinakaran for providing antibodies directed against APLP1 and APLP2. We thank Marcel Leist and Pierluigi Nicotera for advice on neuronal cell culture and cell viability assays. We thank Charles Weissmann and Heinrich Betz for critical advice and continuous support to U.M.
Correspondence should be addressed to Dr. Ulrike Müller, Max-Planck Institute for Brain Research, Deutschordenstrasse 46, D-60528 Frankfurt, Germany. E-mail: umueller@mpih-frankfurt.mpg.de.
REFERENCES
- 1.Banker G, Goslin K. Culturing nerve cells. MIT; Cambridge, MA: 1998. [Google Scholar]
- 2.Daigle I, Li C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA. 1993;90:12045–12049. doi: 10.1073/pnas.90.24.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fox WM. Reflex-ontogeny and behavioural development of the mouse. Anim Behav. 1965;13:234–241. doi: 10.1016/0003-3472(65)90041-2. [DOI] [PubMed] [Google Scholar]
- 4.Furukawa K, Mattson MP. Secreted amyloid precursor protein alpha selectively suppresses N-methyl-d-aspartate currents in hippocampal neurons: involvement of cyclic GMP. Neuroscience. 1998;83:429–438. doi: 10.1016/s0306-4522(97)00398-9. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa K, Barger SW, Blalock EM, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature. 1996;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- 6.Harper SJ, Bilsland JG, Shearman MS, Zheng H, Van der Ploeg L, Sirinathsinghji DJ. Mouse cortical neurones lacking APP show normal neurite outgrowth and survival responses in vitro. NeuroReport. 1998;9:3053–3058. doi: 10.1097/00001756-199809140-00025. [DOI] [PubMed] [Google Scholar]
- 7.Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornsten A, Hausmann RE, Li C. The C. elegans amyloid precursor-related gene has an essential function. Soc Neurosci Abstr. 1999;25:2054. [Google Scholar]
- 9.Ikin AF, Annaert WG, Takei K, De Camilli P, Jahn R, Greengard P, Buxbaum JD. Alzheimer amyloid protein precursor is localized in nerve terminal preparations in Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem. 1996;271:31783–31786. doi: 10.1074/jbc.271.50.31783. [DOI] [PubMed] [Google Scholar]
- 10.Li ZW, Stark G, Götz J, Rülicke T, Gschwind M, Huber G, Müller U, Weissmann C. Generation of mice with a 200-kb amyloid precursor protein gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. Proc Natl Acad Sci USA. 1996;93:6158–6162. doi: 10.1073/pnas.93.12.6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lorent K, Overbergh L, Moechars D, De Strooper B, Van Leuven F, Van den Berghe H. Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the alpha-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, alpha-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience. 1995;65:1009–1025. doi: 10.1016/0306-4522(94)00555-j. [DOI] [PubMed] [Google Scholar]
- 12.Magara F, Müller U, Li ZW, Lipp HP, Weissmann C, Stagljar M, Wolfer DP. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc Natl Acad Sci USA. 1999;96:4656–4661. doi: 10.1073/pnas.96.8.4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997;78:135–146. doi: 10.1016/s0306-4522(96)00553-2. [DOI] [PubMed] [Google Scholar]
- 14.Mattson MP. Secreted forms of beta-amyloid precursor protein modulate dendrite outgrowth and calcium responses to glutamate in cultured embryonic hippocampal neurons. J Neurobiol. 1994;25:439–450. doi: 10.1002/neu.480250409. [DOI] [PubMed] [Google Scholar]
- 15.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 16.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 17.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 18.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss PS, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 19.Müller U. Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis. Mech Dev. 1999;82:3–21. doi: 10.1016/s0925-4773(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 20.Müller U, Cristina N, Li ZW, Wolfer DP, Lipp HP, Rülicke T, Brandner S, Aguzzi A, Weissmann C. Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell. 1994;79:755–765. doi: 10.1016/0092-8674(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 21.Nicotera P, Leist M, Manzo L. Neuronal cell death: a demise with different shapes. Trends Pharmacol Sci. 1999;20:46–51. doi: 10.1016/s0165-6147(99)01304-8. [DOI] [PubMed] [Google Scholar]
- 22.Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer β/A4 amyloid precursor protein in clathrin-coated vesicles from PC12 cells. J Biol Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- 23.Ohshima T, Ward JM, Huh C-G, Longenecker G, Veerana, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paliga K, Peraus G, Kreger S, Durrwang U, Hesse L, Multhaup G, Masters CL, Beyreuther K, Weidemann A. Human amyloid precursor-like protein 1 - cDNA cloning, ectopic expression in cos-7 cells and identification of soluble forms in the cerebrospinal fluid. Eur J Biochem. 1997;250:354–363. doi: 10.1111/j.1432-1033.1997.0354a.x. [DOI] [PubMed] [Google Scholar]
- 25.Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The beta-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice DS, Curran T. Mutant mice with scrambled brains: understanding the signaling pathways that control cell positioning in the CNS. Genes Dev. 1999;3:2758–2773. doi: 10.1101/gad.13.21.2758. [DOI] [PubMed] [Google Scholar]
- 27.Ruffner H, Reis LF, Näf D, Weissmann C. Induction of type I interferon genes and interferon-inducible genes in embryonal stem cells devoid of interferon regulatory factor 1. Proc Natl Acad Sci USA. 1993;90:11503–11507. doi: 10.1073/pnas.90.24.11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandbrink R, Masters CL, Beyreuther K. Complete nucleotide and deduced amino acid sequence of rat amyloid protein precursor-like protein 2 (APLP2/APPH): two amino acids length difference to human and murine homologues. Biochimica et Biophysica Acta. 1994;1219:167–170. doi: 10.1016/0167-4781(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 29.Simons M, Ikonen E, Tienari PJ, Cid-Arregui A, Monning U, Beyreuther K, Dotti CG. Intracellular routing of human amyloid protein precursor: axonal delivery followed by transport to the dendrites. J Neurosci Res. 1995;41:121–128. doi: 10.1002/jnr.490410114. [DOI] [PubMed] [Google Scholar]
- 30.Slunt HH, Thinakaran G, Von KC, Lo AC, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP). J Biol Chem. 1994;269:2637–2644. [PubMed] [Google Scholar]
- 31.Sprecher CA, Grant FJ, Grimm G, O'Hara PJ, Norris F, Norris K, Foster DC. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry. 1993;32:4481–4486. doi: 10.1021/bi00068a002. [DOI] [PubMed] [Google Scholar]
- 32.Steinbach JP, Müller U, Leist M, Li Z-W, Nicotera P, Aguzzi A. Hypersensitivity to Seizures in β-amyloid precursor protein deficient mice. Cell Death Diff. 1998;5:858–866. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- 33.Tenneti L, Lipton SA. Involvement of activated caspase-3-like proteases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. 2000;74:134–142. doi: 10.1046/j.1471-4159.2000.0740134.x. [DOI] [PubMed] [Google Scholar]
- 34.Tienari PJ, De Strooper B, Ikonen E, Simons M, Weidemann A, Czech C, Hartmann T, Ida N, Multhaup G, Masters CL, Van Leuven F, Beyreuther K, Dotti C. The β-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J. 1996;15:5218–5229. [PMC free article] [PubMed] [Google Scholar]
- 35.Thinakaran G, Sisodia SS. Amyloid precursor-like protein 2 (APLP2) is modified by the addition of chondroitin sulfate glycosaminoglycan at a single site. J Biol Chem. 1994;269:22099–22104. [PubMed] [Google Scholar]
- 36.Thinakaran G, Kitt CA, Roskams AJ, Slunt HH, Masliah E, von Koch C, Ginsberg SD, Ronnett GV, Reed RR, Price DL, Sisodia SS. Distribution of an APP homolog, APLP2, in the mouse olfactory system: a potential role for APLP2 in axogenesis. J Neurosci. 1995a;15:6314–6326. doi: 10.1523/JNEUROSCI.15-10-06314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thinakaran G, Slunt HH, Sisodia SS. Novel regulation of chondroitin sulfate glycosaminoglycan modification of amyloid precursor protein and its homologue, APLP2. J Biol Chem. 1995b;270:16522–16525. doi: 10.1074/jbc.270.28.16522. [DOI] [PubMed] [Google Scholar]
- 38.Thinakaran G, Slunt HH, Spitzer L, Lee MK, Sisodia SS. Tissue distribution and developmental expression of the amyloid precursor protein homolog, APLP1. Soc Neurosci Abstr. 1995c;21:205. [Google Scholar]
- 39.Tremml P, Müller U, Lipp H-P, Wolfer DP. Neurobehvioral development, adult open-field activity and swimming navigation learning in mice underexpressing βAPP and in mice lacking APLP1. Soc Neurosci Abstr. 1997;23:1637. [Google Scholar]
- 40.Tremml P, Lipp HP, Müller U, Ricceri L, Wolfer DP. Neurobehavioral development, adult openfield exploration and swimming navigation learning in mice with a modified beta-amyloid precursor protein gene. Behav Brain Res. 1998;95:65–76. doi: 10.1016/s0166-4328(97)00211-8. [DOI] [PubMed] [Google Scholar]
- 41.von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- 42.Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA. 1992;89:10758–10762. doi: 10.1073/pnas.89.22.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet. 1993;5:95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- 44.White AR, Zheng H, Galatis D, Maher F, Hesse L, Multhaup G, Beyreuther K, Masters CL, Cappai R. Survival of culturedneurons from amyloid precursor protein knock-out mice against Alzheimer's amyloid-beta toxicity and oxidative stress. J Neurosci. 1998;18:6207–6217. doi: 10.1523/JNEUROSCI.18-16-06207.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H, Bush AI, Beyreuther K, Masters CL, Cappai R. The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci. 1999a;19:9170–9179. doi: 10.1523/JNEUROSCI.19-21-09170.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White AR, Reyes R, Mercer JFB, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knock-out mice. Brain Res. 1999b;842:439–444. doi: 10.1016/s0006-8993(99)01861-2. [DOI] [PubMed] [Google Scholar]
- 47.Xu X, Yang DS, Wyss-Coray T, Yan J, Gan L, Sun Y, Mucke L. Wild-type but not Alzheimer-mutant amyloid precursor protein confers resistance against p53-mediated apoptosis. Proc Natl Acad Sci USA. 1999;96:7547–7552. doi: 10.1073/pnas.96.13.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamazaki T, Selkoe DJ, Koo EH. Trafficking of cell surface beta-amyloid precursor protein: retrograde and transcytotic transport in cultured neurons. J Cell Biol. 1995;129:431–442. doi: 10.1083/jcb.129.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng H, Jiang MH, Trumbauer ME, Sirinathsinghji DJS, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisodia SS, Chen HY, Vanderploeg LHT. Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]