

Abstract
In Alzheimer's disease (AD) brains, selected populations of neurons degenerate heavily, whereas others are frequently spared from degeneration. To address the cellular basis for this selective vulnerability of neurons in distinct brain regions, we compared gene expression between the severely affected inferior temporal lobes and the mostly unaffected fronto-parietal cortices by using an mRNA differential display. We identified seladin-1, a novel gene, which was downregulated in large pyramidal neurons in vulnerable regions in AD but not control brains. Seladin-1 is a human homolog of the DIMINUTO/DWARF1 gene described in plants and Caenorhabditis elegans. Its sequence shares similarities with flavin-adenin-dinucleotide (FAD)-dependent oxidoreductases. In human control brain, seladin-1was highly expressed in almost all neurons. In PC12 cell clones that were selected for resistance against AD-associated amyloid-β peptide (Aβ)-induced toxicity, both mRNA and protein levels of seladin-1 were approximately threefold higher as compared with the non-resistant wild-type cells. Functional expression of seladin-1 in human neuroglioma H4 cells resulted in the inhibition of caspase 3 activation after either Aβ-mediated toxicity or oxidative stress and protected the cells from apoptotic cell death. In apoptotic cells, however, endogenous seladin-1 was cleaved to a 40 kDa derivative in a caspase-dependent manner. These results establish that seladin-1 is an important factor for the protection of cells against Aβ toxicity and oxidative stress, and they suggest that seladin-1 may be involved in the regulation of cell survival and death. Decreased expression of seladin-1 in specific neurons may be a cause for selective vulnerability in AD.
Keywords: Alzheimer's disease, neurodegeneration, β-amyloid, seladin-1, human DIMINUTO/DWARF1 homolog, oxidative stress, differential display
Alzheimer's disease (AD) is characterized by a substantial loss of neurons and synapses in selective brain regions, by the generation of intracellular neurofibrillary tangles (NFT), and by extracellular and perivascular deposits of β-amyloid (Selkoe, 1999). These amyloid plaques are evenly distributed throughout the neocortex and the hippocampus (Arnold et al., 1991; Van Hoesen et al., 1991). Severe degeneration of neurons, however, occurs predominantly in such selectively vulnerable neuronal populations as the large pyramidal neurons of the inferior temporal cortex, the hippocampus, the amygdala, and the entorhinal cortex. Similar pyramidal neurons in the frontal cortex, the parietal cortex, and the occipital cortex are protected from degeneration, even in end-stage AD and when amyloid plaques are prevalent (Gomez-Isla et al., 1996, 1997). These observations indicate selective vulnerability of specific neurons, especially in brain regions involved in higher cognitive functions—including learning and memory—that are impaired early on in the course of AD. The pathological conditions underlying this selective vulnerability of neurons are incompletely understood, and differentially expressed genes that relate to this susceptibility are unknown. The identification of such genes will be important for understanding the pathophysiology of neurodegeneration in AD, and it may lead to novel targets for therapeutic strategies designed to halt—or prevent—neurodegeneration.
We used a differential mRNA display approach (von der Kammer et al., 1998, 1999) to identify genes that are differentially expressed in selective vulnerable cell populations. Therefore, we compared the gene expression in the inferior temporal cortices with confirmed neuronal degeneration with that in the largely unaffected frontal or sensorimotor cortices in brains from patients with a histopathologically confirmed diagnosis of AD and with postmortem time intervals of <4 hr.
MATERIALS AND METHODS
Brain tissues. The following AD brains were obtained from the Alzheimer's disease tissue resource center at the Massachusetts General Hospital: brain A93-80 (postmortem time interval 3 hr, male, 87 years; regions: sensorimotor cortex, inferior temporal cortex, and hippocampus), brain A95-34 (postmortem time interval 4 hr, female, 92 years; regions: frontal cortex, inferior temporal cortex, and hippocampus), brain A95-267 (postmortem time interval 3 hr, female, 65 years; regions: frontal cortex and inferior temporal cortex), and brain A45-271 (postmortem time interval 4 hr, female, 63 years; regions: frontal cortex and inferior temporal cortex). The following brains were obtained from the Kathleen Price Bryan brain bank from Duke University (Durham, NC): brain 803 (Braak IV, postmortem time interval 3:30 hr, male, 72 years; regions: frontal cortex and inferior temporal cortex, midfrontal cortex and superior temporal cortex), brain 765 (Braak V, postmortem time interval 1:30 hr, male, 62 years; regions: frontal cortex and inferior temporal cortex, midfrontal cortex and superior temporal cortex), brain 673 (normal control, postmortem time interval 1:10 hr, female, 80 years; regions: frontal cortex and inferior temporal cortex), brain A95-006 (normal control, postmortem time interval 2:35 hr, female, 92 years; regions: midfrontal cortex and superior temporal cortex), brain A98-046 (normal control, postmortem time interval 2 hr, male, 83 years; regions: midfrontal cortex and superior temporal cortex), brain A96-263 (normal control, postmortem time interval 2:22 hr, female, 78 years; regions: midfrontal cortex and superior temporal cortex).
Differential display and reverse Northern analysis. The differential display (DD) screen was done exactly as described (von der Kammer et al., 1998, 1999). Reverse Northern blotting was performed as described (Van Gelder et al., 1990; Poirier et al., 1997). Briefly, 2 μg of total RNA from each brain region were reverse-transcribed using the Superscript Plasmid System for cDNA Synthesis (Life Technologies, Karlsruhe, Germany). RNA amplification was performed with the whole cDNA reaction using the Ampliscribe T7 Transcription kit (Epicenter, Biozym, Hessisch Oldendorf, Germany). [32P]-labeled cDNA probes were made with the amplified RNA using Superscript Preamplification System (Life Technologies) and 0.5 mCi per reaction of [α32P]-dCTP (3000 Ci/mmol) (Amersham Pharmacia, Freiburg, Germany). The cloned cDNAs identified in the DD screen were spotted identically on two nylon membranes (Schleicher und Schüll, Dassel, Germany) and were hybridized with radiolabeled cDNA (107cpm/ml) from either frontal or temporal cortex of AD brains. Blots were analyzed by autoradiography, and signal intensities relative to β-actin were determined by densitometry.
Cloning and sequencing of seladin-1. The full-length cDNA for seladin-1 was isolated from a human brain library provided by the Resource Center Primary Database (RZPD) of the Human Genome Project. The cDNA sequence was confirmed by automated DNA sequence analysis using an ABI 377 sequencer.
Northern blot analysis. RNA from brain tissues or cells was extracted with Trizol (Life Technologies), separated on 0.8% formaldehyde-agarose gels, and blotted on Hybond-N+ nylon membranes (Amersham Pharmacia). Northern blots were hybridized with [32P]-labeled cDNA ofseladin-1 (nt 1–3505) and [32P]-labeled cDNA of human β-actin as control (Clontech, Heidelberg, Germany) in ExpressHyb solution (Clontech) at 68°C. Northern blot filters of distinct brain regions and peripheral tissues were obtained from Clontech.
In situ hybridization. In situ hybridization of human brain sections was performed as described (Hartmann et al., 1995; Susens et al., 1997). The following tissue specimens were used: 765; 803; A93-80; A95-34; A96-263; A95-006; and A98-046.
Fragments of 650 and 900 bp of the seladin-1 open reading frame (ORF) were amplified by PCR using the following primer pairs: 1s (nt 76–99: GCG CTT ACC GCG CGG CGC CGC ACC), 1as (nt 749–726: GAC CAG GGT ACG GCA TAG AAC AGG) and 3s (nt 803–826: AGA AGT ACG TCA AGC TGC GTT TCG), 3as (nt: 1749–1726: TTC TCT TTG AAA GTG TGG ATC TAG). PCR products were cloned into pGEM-Teasy (Promega, Heidelberg, Germany), excised with EcoRI, and cloned into pBluescript KS+. [35S]-UTP-labeled antisense and sense riboprobes were generated on NotI and ClaI linearized plasmids with T3 and T7-Polymerase by using the Ambion Maxiscript kit (Ambion, AMS Biotechnology, Wiesbaden, Germany). Hybridized sections were dipped in NTB-2 photographic emulsion (Kodak, Stuttgart, Germany), exposed for 4 weeks, and counterstained with Giemsa. All brain sections were hybridized with identical antisense or sense probes, respectively and processed in parallel. For quantification of neuronal grain density, neuronal grains were automatically counted in three fields from temporal and frontal cortices of three AD and three normal control brains, respectively, that contained on average identical amounts of neurons (∼50 per field). The fields were chosen by an investigator blinded to diagnosis and brain region. Statistically significant differences were calculated by applying Student's t test.
Cell culture and stable cell lines. H4 cells were cultured in DMEM medium (Life Technologies), supplemented with 10% calf serum, or in OptiMEM medium (Life Technologies), supplemented with 2 mm CaCl2. PC12 cells were cultured in DMEM medium, supplemented with 10% calf serum, 5% horse serum, 1% sodium pyruvate, and 1% penicillin–streptomycin (Life Technologies). Human umbilical vein endothelial cells (HUVECs) were cultured in RPMI 1640 medium (Life Technologies), supplemented with 15% calf serum, 3% endothelial cell growth supplement, and 50 μg/ml heparin. The seladin-1 open reading frame was amplified by PCR using the primers sel-Nhe (CCT AGC TAG CGG GCC AGG CGC GGA GCT GGC GGC) and sel-Kpn (GCG GTA CCG TGT GCC TGG CGG CCT TGC AGA TCT TGT C). The sequence was confirmed by DNA sequence analysis. The seladin-1 ORF was cloned at the Nhe/KpnI site of pEGFP-N1 (Clontech). H4 cells were stably transfected with the empty vector pEGFP-N1 or with the seladin-1-enhanced green fluorescence protein (EGFP) fusion construct using DOTAP transfection reagent (Roche, Mannheim, Germany). Stably expressing cells were selected under 500 μg/ml G418 (Roche) and cloned. Expression of EGFP or the fusion protein was confirmed in several selected clones by fluorescence microscopy.
Immunfluorescence. H4 human neuroglioma cells that stably express the seladin-1-EGFP fusion construct were grown on coverslips and fixed with 4% paraformaldehyde in PBS or treated for 45 min with 250 nm of the red fluorescent mitochondrial stain MitoTracker red CM-H2Xros (Molecular Probes, MoBiTec, Göttingen, Germany) or with 500 nmM of the red fluorescence Golgi marker Bodipy TR ceramide (Molecular Probes, MoBiTec) before fixation. The cells that were not prestained with the MitoTracker or Bodipy probe were fixed, permeabilized in 0.2% Triton X-100 in PBS, blocked overnight at 4°C in 5% low-fat milk, 0.1% Triton X-100 in PBS, and incubated for 2 hr at room temperature with a monoclonal antibody against mouse disulfide isomerase (anti-PDImAb; StressGen Biotechnologies Corp., Biomol, Hamburg, Germany), a marker for the endoplasmic reticulum. After washing, the cells were incubated for 1 hr with an anti-mouse IgG, CY3-labeled secondary antibody (Amersham Pharmacia). Subsequently, cells were visualized by confocal laser scanning microscopy.
Subcellular fractionation and enzyme assays. Subcellular organelle fractionation and enzyme assays were performed as described (Graham et al., 1994). Briefly, 10 confluent tissue culture plates of H4 cells were washed three times with ice-cold PBS and incubated for 15 min in 0.2 m sucrose. The cells were homogenized with 25 strokes of a Kontes homogenizer in medium A [8% (w/v) sucrose, 20 mm Tricine-NaOH, pH 7.4, 1 mm EDTA]. The homogenate was centrifuged at 1000 × g for 10 min, and the supernatant was recentrifuged at 17,000 × g for 15 min. The 17,000 × g pellet (P2) was resuspended with a Dounce homogenizer in 1 ml of medium A and subjected to an Iodixanol gradient (Optiprep, Nycomed Pharma, Oslo, Norway). To generate the Iodixanol gradients, 5 vol of Optiprep solution were diluted to a working solution of 50% by the addition of 1 vol of 8% (w/v) sucrose, 120 mm Tricine-NaOH, pH 7.4, 6 mm EDTA. The working solution was diluted to 10 and 30% gradient medium by addition of medium A. Three milliliters of P2 in medium A adjusted to 35% Iodixanol by addition of 50% working solution were layered under the gradient consisting of 5 ml of high-density medium (30%) followed by 5 ml of low-density medium (10%). The gradient was centrifuged at 52,000 ×gav for 1.5 hr using a Beckman SW60 rotor in a Beckman L7–55 ultracentrifuge. The gradients were fractionated into 26 fractions of 0.5 ml each by upward displacement using a gradient unloader (Nycomed Pharma, Oslo, Norway). All procedures were performed at 4°C. Each gradient fraction was directly assayed for mitochondrial succinate-dehydrogenase, and for endoplasmic reticulum NADPH-cytochrome c reductase activities exactly as described by Nycomed Pharma. Proteins of each fraction were separated by 10% SDS-PAGE and subjected to immunoblotting. Briefly, the Immobilon membranes (Millipore GmbH, Eschborn, Germany) were blocked in 5% low-fat milk for 4 hr at 4°C and incubated overnight at 4°C with a rabbit polyclonal peptide-specific seladin-1 antiserum (1:1000 diluted) or with a mouse monoclonal anti-Golgi 58K antibody (1:1000 diluted) (Sigma Aldrich, Munich, Germany). Peroxidase-coupled goat anti-rabbit (Vector, Alexis Biochemicals, Grünberg, Germany) or goat anti-mouse antibodies (Amersham Pharmacia) were used as secondary antibodies. Immunoblots were visualized by enhanced chemiluminescence (Amersham Pharmacia).
Flow cytometry. Ten and 16 hr after incubation of three seladin-1-EGFP clones (A6, B1, B5) and three EGFP-control clones (B5, B6, C1) in OptiMEM medium containing 200 μmH2O2, the cells remaining attached to the culture dish as well as the cells in the supernatant were harvested and stained with 7-amino-actinomycin D (ViaProbe, PharMingen, Becton Dickinson GmbH, Heidelberg, Germany) to distinguish viable from dead cells. Only membranes of dead and damaged cells are permeable to this DNA dye and stain positive. Live/dead counts were performed by using a FACSCalibur (Becton Dickinson) fluorescence-activated cell sorter. Cells (105 per clone) were counted in three independent experiments. Statistically significant differences were determined by using Student's t test.
Trypan blue and Hoechst dye staining. Three seladin-1-EGFP clones (A6, B1, B5) and three EGFP-control clones (B5, B6, C1) were grown to 50% confluence on coverslips in 24-well plates and treated either for 20 hr with 200 μmH2O2 or for 18 hr with 10 μm Aβ25–35 using OptiMEM, 2 mm CaCl2. Cells on one plate were stained with trypan blue. The trypan blue-positive and -negative cells were counted in five 20× fields of each well, and the percentage of trypan blue-positive cells for each clone was calculated. Cells on the second plate were fixed in 4% paraformaldehyde and stained with 1 μg/ml Hoechst dye 33342 (Molecular Probes, MoBiTec) after permeabilization of cell membranes with 0.1% Triton X-100. Hoechst-stained cells were visualized under epifluorescence illumination (350 nm excitation, 461 nm emission) with a 40× oil immersion objective. Two hundred cells were counted per well, and the percentage for apoptotic cells with condensed and fragmented nuclei in each well was determined. Statistically significant differences were determined by using Student's t test.
Caspase 3 activity. Caspase 3 activity was measured in cell lysates of three seladin-1-EGFP clones (A6, B1, B5) and three EGFP-control clones (B5, B6, C1) plated to identical densities by using the caspase 3 assay kit (PharMingen, Becton Dickinson GmbH). After exposure to 200 μmH2O2 for 2 and 4 hr or to 25 μm Aβ25–35 for 4 hr, cells were washed briefly in PBS and lysed in 100 μl 10 mmTris-HCl, pH 7.5, 10 mmNaH2PO4/NaHPO4, pH 7.5, 130 mm NaCl, 1% Triton-X-100, 10 nmNaPPi. Lysates (100 μg of protein) were incubated in 200 μl HEPES buffer for 1 hr at 37°C with 5 μg of the caspase 3 fluorogenic substrate Ac-DEVD-AMC or with 5 μg Ac-DEVD-AMC in the presence of 0.5 μg of the caspase 3 aldehyde inhibitor Ac-DEVD-CHO in a 96-multiwell plate. AMC liberated from Ac-DEVD by caspase cleavage was measured on a spectrofluorometer (Spectramax Gemini, Molecular Devices, Munich, Germany) at excitation wavelength of 380 nm and an emission wavelength spectrum from 420 to 460 nm. The means (±SEM) of caspase 3 activity of three independent experiments, each with three seladin-1-EGFP and three EGFP-control clones, are given in relative fluorescence units (RFUs). Statistically significant differences were calculated applying Student's t test.
Generation of seladin-1 antisera. For immunizations of rabbits, a peptide consisting of amino acid residues 203–218 of seladin-1 was synthesized and coupled to keyhole limpet hemocyanin at the C terminus: H2N-TPS ENS DLF YAV PWS C-CONH2 (Eurogentec, Seraing, Belgium). The rabbit immune serum was purified on HiTrap affinity columns (Amersham Pharmacia) to which the peptide was coupled according to the instructions of the manufacturer.
Protein analysis. For Western blotting, human brain tissues 673, 765, and 803 were used. Brain tissues were homogenized with mortar and pestle in liquid nitrogen and subsequently with a Dounce homogenizer in 20% glycerol, 2% SDS, 125 mm NaCl, 0.075m DTT, 1% Triton X-100. Protein concentrations were measured with a Bradford assay, and 25 μg of protein of each brain extract were separated by 10% SDS-PAGE. Proteins were transferred to Immobilon membranes (Millipore GmbH) that were subsequently blocked in 5% low-fat milk overnight at 4°C and incubated overnight at 4°C with a 1:200 dilution of the seladin-1-specific, affinity-purified antiserum, followed by 1 hr incubation with 1:30,000 of a goat anti-rabbit peroxidase-conjugated secondary antibody at room temperature (Vector, Alexis Biochemicals). To control for equal loading, blots were reprobed with a rabbit polyclonal anti-actin antibody (Sigma Aldrich). Immunoblots were visualized by enhanced chemiluminescence (Amersham Pharmacia). PC12 cells were extracted in 10 mm Tris/HCl, 10 mmNaH2PO4/NaHPO4, pH 7.5, 130 mm NaCl, 1% Triton X-100, 10 mmsodium pyrophosphate. Twenty micrograms of protein of the extracts were separated by 10% SDS-PAGE, transferred to Immobilon membrane (Millipore GmbH), and incubated with the seladin-1-specific antibody as described above. HUVEC cells were lysed in 50 mm Tris/HCl, pH 7.4, 250 mm NaCl, 0.5% NP40, 10% glycerol, 5 mm EDTA, 50 mm NaF, 0.5 mmNa3VO4, 10 mmglycerophosphate, 0.5 mm PMSF, 5 mg/ml leupeptin and aprotinin, and were subjected to 10% SDS-PAGE. Films were scanned and analyzed by using the NIH image software.
Apoptosis in HUVECs. Confluent HUVECs were deprived of growth factors for 12 hr. Cell lysates of control cells and surviving viable and apoptotic floaters were immunoblotted and analyzed with an antigen affinity-purified antibody against seladin-1. In addition, confluent HUVECs were deprived of growth factors for 16 hr in the absence or the presence of the cell-permeable caspase inhibitor ZVAD. Cell lysates including attached and floating cells were immunoblotted and analyzed with the seladin-1 antibody.
In vitro translation. The complete ORF of seladin-1 was amplified by PCR using the following primers: sel-BamHs (CGG GAT CCA TGG AGC CCG CCG TGT CGC TGG CC) and sel-Xhoas (CCG CTC GAG CTC AGT GCC TGG CGG CCT TGC AG), and cloned in theBamHI/XhoI site of pcDNA3.0 (Invitrogen, Groningen, Netherlands). The sequence of the seladin-1 ORF was confirmed by DNA sequencing. In vitro transcription and translation of seladin-1 was performed in the presence of [35S]-methionine (1000 Ci/mmol; Amersham Pharmacia) by using the TNT-coupled reticulocyte lysate system (Promega). Two microliters of the translation reaction (25 μl) were incubated at 37°C for 1 hr or 2.5 hr with 250 ng of recombinant caspases 3, 6, and 7 (provided by K. Orth and V. Dixit, University of Michigan, Ann Arbor, MI) in a total volume of 10 μl in 10 mm HEPES/KOH, pH 7.4, 2 mmEDTA, 5 mm DTT, 1% NP40, 5 μg/ml leupeptin and aprotinin, or with reaction buffer alone (control). The samples were separated on a 10% SDS-PAGE, and full-length seladin-1 as well as the resulting seladin-1 fragments were visualized by autoradiography.
RESULTS
Identification of seladin-1
By using 40 different primer combinations, we identified >30 differentially expressed cDNAs from inferior temporal cortex and frontal or sensorimotor cortex of AD patients. We cloned and sequenced these cDNAs and further analyzed them by reverse Northern blotting (Van Gelder et al., 1990; Poirier et al., 1997) to confirm differential gene expression within the distinct brain regions. We then prioritized the functional analyses according to confirmed differential expression and the predictions derived from sequence information. Among these clones was a novel cDNA with markedly reduced expression in inferior temporal lobes as compared with the sensorimotor cortex. We designated this new gene seladin-1 (selective AD indicator 1), and cloned its full-length cDNA from a human brain cDNA library. The cDNA sequence was deposited at GenBank under the accession number AF261758. It consists of 4248 nucleotides and encodes an open reading frame of 516 amino acid residues (Fig. 1). Because of a cytidine insertion at nucleotide position 1167, this sequence differs from the much shorter coding region of its homolog KIAA0018 (Nomura et al., 1994). Hydrophobicity analysis revealed a hydrophobic region in the N terminus consistent with a leader sequence as well as at least four possible transmembrane domains (Fig. 1). Seladin-1 sequence has homology to the “diminuto-like protein” or “dwarf-1,” a cell elongation factor in Arabidopsis thaliana (Takahashi et al., 1995). The respective sequence is highly conserved among plants,Caenorhabditis elegans, and mammals (Fig. 1). In addition, seladin-1 contains a domain characteristic for oxidoreductases (Fig.1), including a consensus sequence for non-covalent FAD binding (Mushegian and Koonin, 1995; Fraaije et al., 1998).
Fig. 1.

Amino acid sequence of seladin-1. The domains with homologies to oxidoreductases are marked in black, and regions with homologies to diminuto-like proteins inC. elegans, Arabidopsis thaliana,Pisum sativum, and Mycobacterium leprae/tuberculosis are underlined. Conserved regions within the family of FAD-dependent oxidases are printed initalics. The region for non-covalent FAD binding is marked with a dotted line. The first 21 amino acid residues represent a putative signal peptide. Two possible caspase recognition motifs are marked with a star.
Loss of seladin-1 expression in neurons of affected regions in AD brain
Northern blots of total RNA extracted from AD brains with short postmortem time intervals showed that the expression ofseladin-1 was substantially lower in the inferior temporal lobe as compared with the frontal cortex (Fig.2A). This difference was accompanied by low tissue concentrations of seladin-1 protein in extracts from the inferior temporal lobe (Fig. 2B). Thus, the differential expression of seladin-1 between temporal and frontal cortex within individual AD brains as initially observed by differential display and reverse Northern blotting was reflected by reduced protein levels and confirmed in additional patients. In contrast, in brains obtained from normal, non-demented control subjects, no differences in the seladin-1 mRNA or protein levels were detected among these brain regions (Fig.2A–D) indicating a disease-related decrease in seladin-1 expression. In brains of non-demented human control subjects,seladin-1 was strongly expressed with highest expression in cortical regions, substantia nigra, caudate nucleus, hippocampus, medulla oblongata, and spinal cord (Fig.2C,D). In peripheral human tissues,seladin-1 was almost ubiquitously expressed. High transcript levels were found in adrenal gland and moderate expression was detected in liver, lung, spleen, and prostate. Seladin-1 was rare in heart, uterus or intestine, and undetectable in blood cells (Fig.2D).
Fig. 2.
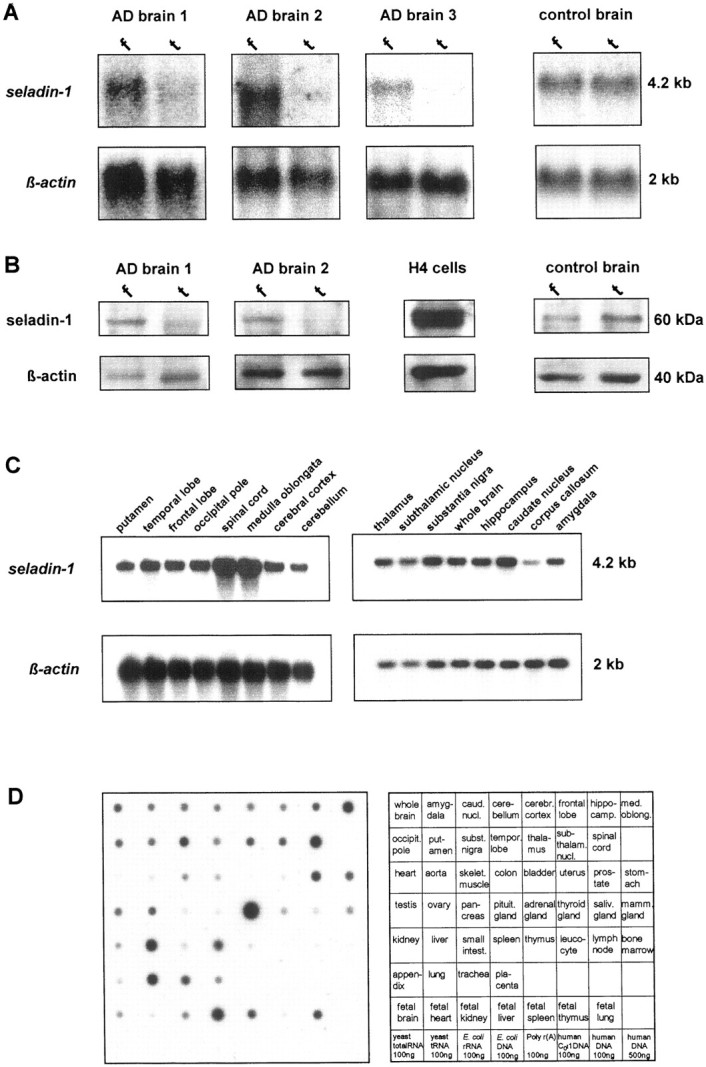
Levels of seladin-1 RNA and protein in Alzheimer's disease (AD) brain tissue, in normal control brain tissue, and in peripheral tissues. A, Total RNA of three different AD brains (10 μg per lane) was analyzed. The blots were hybridized with a [32P]-labeled cDNA probe of seladin-1 and a control probe of human β-actin (f, frontal cortex;t, temporal cortex). B, Protein (25 μg) of extracts from brain 1, brain 2, H4 cells, and control brain were subjected to 10% SDS-PAGE and subsequent immunoblot analysis for seladin-1 and β-actin (f, frontal cortex;t, temporal cortex). C, Human brain multiple tissue Northern blot II (Clontech 7755–1) and III (Clontech 7750–1) containing 2 μg of poly(A+) RNA per lane from 16 different human brain regions. Blots were hybridized with the same probes as described in A. D, Human RNA Master Blot (Clontech 7770–1) containing normalized loading of 89–514 ng of each poly(A+) RNA per dot from 50 different human tissues and six different control RNAs and DNAs. The blot was hybridized to a [32P]-labeled cDNA probe ofseladin-1.
In situ hybridization of human cortical brain sections demonstrated a neuronal expression pattern of seladin-1. In AD brains, seladin-1 mRNA was reduced in the remaining neurons of the degenerated temporal cortex in comparison to neurons in the frontal cortex from the same patients (Fig.3A–D,K). In contrast, in brains obtained from normal human control subjects, neuronal expression of seladin-1 was identical among frontal and temporal cortices (Fig.3E–H,K). Therefore, the reduced levels of seladin-1 mRNA in the temporal cortex of AD brains were caused not only by cell loss but also by low expression within the remaining neurons.
Fig. 3.

In situ hybridization of human AD (A–D) and normal brain (E–J). A, C,E, G, I, Dark-field illuminations; B, D, F,H, J, corresponding bright-field photomicrographs. Representative hybridization patterns of seladin-1 in midfrontal cortex of AD brains (A, B), and in superior temporal cortex of AD brains (C,D). Representative hybridization pattern of seladin-1 in midfrontal cortex of normal brains (E,F) and in superior temporal cortex of normal brains (G, H).Arrowheads indicate neurons packed with silver grains;arrows indicate the neurons with only few grains.I, J, Background hybridization of the sense control probe. K, Neuronal grain density in frontal versus temporal cortices of human AD and control brains. Means (±SEM) of neuronal grain counts from three fields containing on average equal amounts of neurons (∼50 neurons per field) of the respective brain regions of three AD and three normal control brains are given. Two asterisks indicate statistically significant difference at p < 0.001.
Localization of seladin-1 in membranes of the endoplasmic reticulum
To localize seladin-1 within cells, we stably transfected human H4 neuroglioma cells with seladin-1 fused at its C terminus to EGFP. Colocalization studies with markers and antibodies against several subcellular organelles indicated that seladin-1-EGFP was mainly localized in the endoplasmic reticulum (ER) (Fig.4A–C), and to lesser extent in Golgi complexes (data not shown), but not in mitochondria, despite the presence of a putative mitochondrial localization signal at the N terminus of seladin-1 (Fig. 4D–F). To study further the cellular localization of endogenous seladin-1, we separated subcellular organelles from H4 cells by Iodixanol gradient fractionation. Seladin-1 was clearly present in the Golgi and ER fractions but was not detectable in the mitochondrial fractions (Fig.5A,B). Thus, seladin-1 resides in a subcellular compartment in which processing of both the amyloid precursor protein and the presenilins takes place (Tomita et al., 1998; Zhang et al., 1998).
Fig. 4.

Subcellular localization of seladin-1 in H4 human neuroglioma cells. A, D, Subcellular distribution of the green fluorescent seladin-1-EGFP fusion protein.B, Staining of the endoplasmic reticulum with the anti-PDImAb and the red fluorescent CY3-labeled secondary antibody. C, Overlay fromA and B shows the colocalization of seladin-1 with the ER marker, indicated by yellowfluorescence. E, Staining of the mitochondria with thered fluorescence MitoTracker CM-H2Xros.F, Overlay of D andE.
Fig. 5.

Subcellular localization of endogenous seladin-1. Human H4 neuroglioma cells were homogenized, and the subcellular 17,000 × g pellet P2 was subjected to Iodixanol-gradient ultracentrifugation. The gradients were fractionated from the top into 26 fractions. A, Forty micrograms of protein of P2 and one-tenth of each gradient fraction were separated by SDS-PAGE and analyzed by immunoblotting for the presence of seladin-1 (top panel) and the Golgi marker p58 (bottom panel). B, Gradient fractions were assayed for activity of NADPH-cytochrome c reductase (dark dotted line) as a marker enzyme for the endoplasmic reticulum, and for activity of succinate dehydrogenase (faint rhomb-shaped line) as a marker enzyme for mitochondria.
Protection from programmed cell death by seladin-1 via inhibition of caspase 3 activation
The clonal cell lines that stably expressed the seladin-1-EGFP fusion construct consistently appeared to be more robust and faster growing than control cell lines expressing only EGFP. On the basis of the primary amino acid sequence, we reasoned that seladin-1 shares functions with oxidoreductases that may protect cells against oxidative stress. Several seladin-1-EGFP-expressing cell clones or EGFP-control clones were tested for resistance against the presence of H2O2 in the cell culture medium. All seladin-1-expressing clones tolerated H2O2-induced oxidative stress much better than nontransfected or EGFP-expressing clones. During the initial 10 hr of exposure to 200 μmH2O2, nearly 90% of the seladin-1-expressing cells and 75–80% of the control cells were viable. After 16 hr, however, 80% of the seladin-1-expressing cells but only 52% of the control cells survived (Fig.6A). Unchallenged control cells contained a maximum of 5% dead cells at equivalent time intervals. Improved survival of seladin-1-expressing cells during exposure to oxidative stress was confirmed by two independent approaches. First, live/dead counts were done by trypan blue exclusion of cells in culture. Second, nuclei of cells grown and fixed on coverslips were stained with Hoechst dye 33342, visualized by fluorescence microscopy, and counted. Results of both measurements were consistent with resistance of seladin-1-expressing cells against induction of cell death. To determine whether seladin-1 function is associated with markers for apoptotic cell death, we measured the activity of caspase 3 in three independent seladin-1-EGFP-expressing clones and in three EGFP-expressing control clones. Two hours after exposure to 200 μmH2O2, caspase 3 activity was not detectable in either seladin-1-EGFP clones or the EGFP-control clones. After 4 hr, however, the activity of caspase 3 strongly increased and was found to be twofold higher in the three EGFP-control clones as compared with the three seladin-1-EGFP clones (Fig.6B). This increase in caspase 3 activity was blocked in both cell lines by the addition of the caspase inhibitor Ac-DEVD-CHO (Fig. 6B). These results establish the ability of seladin-1 to reduce caspase 3 activity during apoptosis induced by oxidative stress.
Fig. 6.

Seladin-1-EGFP cells are resistant to oxidative stress and Aβ-toxicity. A, Ten and 16 hr after exposure of three seladin-1-EGFP clones (A6,B1, B5) and three EGFP-control clones (B5, B6, C1) to 200 μm H2O2, cells were harvested and stained with 7-AAD. Live versus dead cells were counted by using a FACSCalibur (Becton Dickinson) machine counting 105 cells per clone. Means (±SEM) of three experiments in triplicates are shown. B, Caspase 3 activity was measured in cell lysates of three seladin-1-EGFP clones (A6, B1, B5) and three EGFP-control clones (B5, B6, C1) after exposure to 200 μm H2O2 for 2 and 4 hr and after addition of the caspase inhibitor Ac-DEVD-CHO. Means (±SEM) of caspase 3 activity of three independent experiments, each with three seladin-1-EGFP and three EGFP-control clones, are shown in RFUs. C, Eighteen hours after exposure of three seladin-1-EGFP clones (A6, B1, B5) and three EGFP-control clones (B5, B6, C1) to 10 μm Aβ25–35, cells were stained by trypan blue. The trypan blue-positive and -negative cells were counted in five 20× fields of each clone. The means (±SEM) of the percentage of viable cells for the three seladin-1-EGFP clones and the three EGFP-control clones are given. D, Caspase 3 activity was measured in cell lysates of three seladin-1-EGFP clones (A6, B1, B5) and three EGFP-control clones (B5, B6, C1) after 4 hr exposure to 25 μm Aβ25–35. The means (±SEM) of caspase 3 activity of three independent experiments, each with three seladin-1-EGFP and three EGFP-control clones, are given in RFUs. One asterisk indicates statistically significant difference at p < 0.05; two asterisks indicate statistically significant difference atp < 0.001.
Seladin-1 mediates resistance against Aβ-induced apoptotic cell death
Aβ peptides can be toxic in several in vitro andin vivo systems, and Aβ aggregates can produce hydrogen peroxide through metal ion reduction (Behl et al., 1994; Huang et al., 1999). To determine whether seladin-1 can protect cells from Aβ-mediated toxicity, we exposed both the seladin-1-EGFP-expressing cell clones and the EGFP-control clones to 10 and 25 μm Aβ25–35. Again, all seladin-1-expressing clones tolerated Aβ-induced toxicity much better than the EGFP-expressing control clones. Eighteen hours after exposure to 10 μm Aβ25–35, 90% of the seladin-1-expressing cells but only 60% of the control cells were still viable (Fig. 6C). To test whether seladin-1 blocks Aβ-induced caspase 3 activation, we measured the activity of caspase 3 in three independent seladin-1-expressing clones and in three control clones. Four hours after exposure to 25 μm Aβ25–35, the activity of caspase 3 was more than two times higher in the EGFP-expressing control cells in comparison to the seladin-1-expressing cells (Fig. 6D). Again, in both cell lines the caspase 3 activity was blocked by addition of the caspase inhibitor Ac-DEVD-CHO (Fig. 6D). These data indicate that seladin-1 can protect cells against Aβ-induced apoptosis.
High expression of seladin-1 is associated with resistance against Aβ toxicity
To further investigate whether seladin-1 expression is related to cellular resistance against Aβ, we analyzed the expression of seladin-1 in PC12 cells that were selected for resistance against Aβ toxicity and H2O2-mediated oxidative stress (Behl et al., 1994; Sagara et al., 1996). We found that cellular levels of seladin-1 mRNA were severalfold higher in the Aβ-resistant cell lines as compared with the parental PC12 cells leading to approximately twofold higher levels of seladin-1 protein in protected cells (Fig.7A,B). This increase was accompanied by upregulation of catalase and glutathione peroxidase (Sagara et al., 1996), suggesting that seladin-1, catalase, and glutathione peroxidase function in a concerted manner in protecting cells from Aβ-induced toxicity.
Fig. 7.

Upregulation of seladin-1 in PC12 rat pheochromocytoma cells resistant to Aβ-induced oxidative stress.A, Protein (20 μg) of cell extracts of the PC12 resistant clone 8 (rCl8) and of the wild-type PC12 cells were separated by SDS-PAGE and analyzed for seladin-1 and actin by immunoblot analysis. B, Total RNA (20 μg) from rCl8 and PC12 cells was analyzed by Northern blotting. 28s RNA and 18s RNA is shown as a loading control.
Seladin-1 is a death substrate for caspases during apoptosis
To analyze the fate of endogenous seladin-1 in dying cells during apoptosis, we determined its expression in a well characterized cell system for the study of apoptosis: growth factor deprivation-induced apoptosis in HUVEC cells (Levkau et al., 1998, 1999). Twelve hours after growth factor withdrawal, these cells subdivide in two independent populations: apoptotic cells that detach from the cell culture dishes and float in the supernatant, and non-apoptotic, viable cells that remain firmly attached to the cell culture dishes. In lysates of HUVEC cells, the seladin-1 antiserum recognized a protein of 60 kDa, the predicted molecular mass of seladin-1. After preadsorption of the antibody with the cognate peptide before immunoblotting, the 60 kDa band was no longer detectable, indicating specificity of the antibody. Cellular levels of seladin-1 were similar in HUVEC cells cultured in the presence of growth factors and in growth factor-deprived but viable HUVEC cells (Fig.8A). Seladin-1 was cleaved to a distinct 40 kDa fragment (p40) in the population of HUVEC cells that underwent apoptosis, consistent with apoptosis-related endoproteolytic cleavage at one specific cleavage site (Fig.8A). Sequence analysis identified the presence of two putative caspase recognition motifs: LEVD at position 122–125 or VVQD at position 383–386 (Fig. 1). Both motifs are highly conserved in all seladin-1 homologs, strongly suggesting that seladin-1 may be a death substrate for caspase cleavage. Cleavage at either of these positions is consistent with the generation of the observed 40 kDa seladin-1 fragment p40 that could comprise the C terminus in case of cleavage after D125 or the N-terminal fragment after cleavage at D386, respectively. To demonstrate the formation of p40 by caspase activity during apoptosis, we treated HUVEC cells with the cell-permeable caspase inhibitor ZVAD during growth factor withdrawal. Addition of ZVAD inhibited the cleavage of seladin-1, and p40 was no longer detectable (Fig. 8B). These data indicate that seladin-1 is a caspase substrate. To directly demonstrate caspase cleavage, in vitro translated seladin-1 labeled with [35S]-methionine was incubated with the recombinant caspase 3, 6, and 7. Seladin-1 was readily cleaved by recombinant caspase 6 and, to lesser extent, caspase 3, but not caspase 7 (Fig. 8C). In vitro cleavage of seladin-1 by caspase 3 or 6 generated four different seladin-1 fragments of ∼50, 40, 30, and 20 kDa (Fig. 8C). The 40 kDa fragment corresponded in molecular mass to p40 generated in tissue culture during apoptosis in HUVEC cells (Fig. 8A).
Fig. 8.
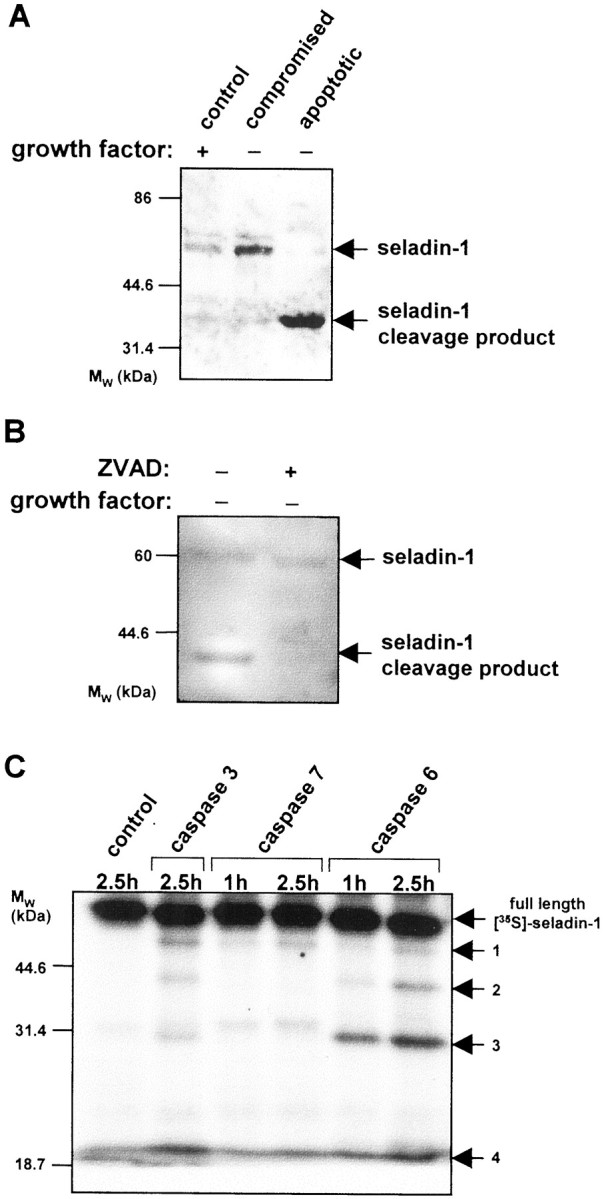
Seladin-1 is a substrate for caspases in tissue culture and in vitro. A, Confluent HUVECs were deprived of growth factors for 12 hr. Cell lysates of control cells, of surviving viable cells, and of apoptotic floaters were immunoblotted and analyzed with a specific, antigen affinity-purified, antiserum against seladin-1. B, Confluent HUVECs were deprived of growth factors for 16 hr in the absence (lane 1) or presence (lane 2) of ZVAD. Cell lysates including attached and floating cells were immunoblotted and analyzed with the seladin-1 antibody. C, In vitrotranslated and [35S]-methionine-labeled seladin-1 was incubated at 37°C for 1 hr and 2.5 hr with reaction buffer alone (control) or with recombinant caspase-3, caspase-6, or caspase-7. The samples were analyzed on 10% SDS-PAGE, and full-length seladin-1 as well as seladin-1 fragments 1–4 were visualized by autoradiography.
DISCUSSION
In this study, we identified the novel gene seladin-1that shares domain homologies with a gene family of FAD-dependent oxidoreductases. Seladin-1 was highly expressed in neuronal cells throughout mammalian brains, and its expression was low in neurons within selectively vulnerable regions of AD brains. Reduced levels of seladin-1 mRNA in affected areas of AD brains was related to reduced amounts of seladin-1 mRNA within remaining neurons and did not simply reflect neuronal cell loss. Reduced brain tissue levels of seladin-1 mRNA were paralleled by reduced levels of seladin-1 protein in affected regions. Reduced expression of seladin-1 in vulnerable brain regions in AD is in line with results of previous studies on antioxidant enzyme activities in AD brains that found lower activities of catalase and superoxide dismutase in temporal cortices from AD brains as compared with temporal cortices from non-demented normal controls (Marcus et al., 1998). Differential activity of antioxidant enzymes in nerve cell populations may be one important cause for the selective resistance of specific cells against degeneration on toxic factors such as Aβ, which is distributed throughout the brain in both vulnerable and protected regions. In cell culture, overexpression of seladin-1 protected cells from apoptosis induced not only by oxidative stress but also by Aβ, and high expression of endogenous seladin-1 was associated with resistance against Aβ-induced toxicity. Resistance of cultured cells to Aβ toxicity was previously found to be attributable to the transcriptional activation of antioxidant enzymes, including glutathione peroxidase or catalase (Behl et al., 1994; Sagara et al., 1996, 1998). Our data extend that proposal by adding seladin-1, which may function in concert with these enzymes in protecting cells from oxidative stress and Aβ toxicity.
Despite its activity on increasing resistance to apoptosis, seladin-1 itself is apparently cleaved to p40 by apoptosis-related endoproteolytic cleavage at one of two possible motifs at positions 122–125 or 383–386. Our antibody recognizes an epitope in between these two motifs and therefore detects cleavage at either position. Together, our results suggest that seladin-1 is an integral component of the cellular machinery that protects neurons from oxidative stress. Once oxidative stress becomes overwhelming, seladin-1 is cleaved and presumably inactivated because the putative domain for non-covalent FAD binding is located within the caspase cleavage motifs. Dual roles in apoptosis are known for several negative regulators of apoptosis. These antiapoptotic proteins include Bcl 2, ICAD (Cheng et al., 1997; Xue and Horvitz, 1997; Adams and Cory, 1998; Enari et al., 1998), and NfκB (Levkau et al., 1999), all of which are cleaved during apoptosis by caspases and turn into proapoptotic stimuli (Cheng et al., 1997; Xue and Horvitz, 1997; Adams and Cory, 1998; Enari et al., 1998; Levkau et al., 1999). It will be interesting to determine whether p40 also has pro-apoptotic functions.
Accumulating data underscore the importance of reactive oxygen species in the pathogenesis of neurodegenerative diseases, including AD (Beal, 1996; Multhaup et al., 1996; Browne et al., 1999). Our data raise the possibility that seladin-1 protects large pyramidal neurons from Aβ-induced toxicity via a mechanism that involves increased resistance against oxidative stress. Seladin-1 thus may link oxidoreductase activity to apoptosis and neurodegeneration, similar to the recently reported protein ERAB or ABAD, an endoplasmic reticulum-associated amyloid β-peptide binding protein that participates in fatty acid β-oxidation and is known to mediate apoptosis (He et al., 1998; Oppermann et al., 1999; Yan et al., 1999). In contrast to ERAB, which is mainly localized in mitochondria and at the cytosolic site of the ER, seladin-1 is predominantly localized within the ER, and to a lesser amount in Golgi complexes, suggesting that the ER/intermediate compartment (IC) is a possible site for interaction with Aβ, which is known to be produced in the ER/IC (Xia et al., 1998; Soriano et al., 1999; Skovronsky et al., 2000). Whether seladin-1 interacts with Aβ in the ER/IC and modifies Aβ toxicity within cells will be addressed in further studies.
Despite the observed potential of seladin-1 for neuroprotection, its precise physiological function has yet to be established. In plants, the seladin-1 homolog “Diminuto/Dwarf1” is required for the synthesis of brassinosteroids, which are important plant sterols essential for normal plant growth and development (McMorris, 1997;Klahre et al., 1998; Bishop et al., 1999). Thediminuto/dwarf1 mutant is defective in synthesizing the early precursor of brassinolide, campesterol (Klahre et al., 1998; Choe et al., 1999), leading to the accumulation of 24-methylencholesterol and resulting in severe growth defects in Arabidopsis thaliana (Klahre et al., 1998; Choe et al., 1999). Analogous to the function of diminuto/dwarf1, seladin-1 may well be required for FAD-dependent oxidation of a specific metabolic intermediate necessary for cell growth and differentiation.
The initial cause for the loss of seladin-1 expression in selectively vulnerable regions of AD brains remains to be investigated, as well as possible genetic heterogeneity at this locus. Taken together, our results provide evidence for an involvement of seladin-1 in neurodegeneration and offer a novel therapeutic strategy for delaying—or preventing—neurodegeneration in AD and other neurodegenerative diseases.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant Al 454/1-1 to I.G. We thank Dr. H. von der Kammer for his help in establishing the differential display, and Dr. R. Laas (Department of Neuropathology, University of Hamburg) for help in analyzing human brain samples. We acknowledge the Resource Center of the German Human Genome Project at the Max-Planck-Institute for Molecular Genetics, which provided us with clones and filters. We thank C. Hulette and C. Rosenberg (Duke University) and C. Fitch (Massachusetts General Hospital) for preparing AD and normal control brain tissue and sections. We thank Dr. J. Greeve and Dr. R. Reifegerste for advice in the course of the study and for help in preparing this manuscript.
Correspondence should be addressed to Dr. Isabell Greeve, Center for Molecular Neurobiology, University of Hamburg, Martinistraβe 52, 20246 Hamburg, Germany. E-mail:isabell.greeve@zmnh.uni-hamburg.de.
REFERENCES
- 1.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 2.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 4.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 5.Bishop GJ, Nomura T, Yokota T, Harrison K, Noguchi T, Fujioka S, Takatsuto S, Jones JD, Kamiya Y. The tomato DWARF enzyme catalyses C-6 oxidation in brassinosteroid biosynthesis. Proc Natl Acad Sci USA. 1999;96:1761–1766. doi: 10.1073/pnas.96.4.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington's disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- 8.Choe S, Dilkes BP, Gregory BD, Ross AS, Yuan H, Noguchi T, Fujioka S, Takatsuto S, Tanaka A, Yoshida S, Tax FE, Feldmann KA. The Arabidopsis dwarf1 mutant is defective in the conversion of 24-methylenecholesterol to campesterol in brassinosteroid biosynthesis. Plant Physiol. 1999;119:897–907. doi: 10.1104/pp.119.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 10.Fraaije MW, Van Berkel WJ, Benen JA, Visser J, Mattevi A. A novel oxidoreductase family sharing a conserved FAD-binding domain. Trends Biochem Sci. 1998;23:206–207. doi: 10.1016/s0968-0004(98)01210-9. [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 13.Graham J, Ford T, Rickwood D. The preparation of subcellular organelles from mouse liver in self-generated gradients of iodixanol. Anal Biochem. 1994;220:367–373. doi: 10.1006/abio.1994.1351. [DOI] [PubMed] [Google Scholar]
- 14.Hartmann D, Fehr S, Meyerhof W, Richter D. Distribution of somatostatin receptor subtype 1 mRNA in the developing cerebral hemispheres of the rat. Dev Neurosci. 1995;17:246–255. doi: 10.1159/000111293. [DOI] [PubMed] [Google Scholar]
- 15.He XY, Schulz H, Yang SY. A human brain l-3-hydroxyacyl-coenzyme A dehydrogenase is identical to an amyloid beta-peptide-binding protein involved in Alzheimer's disease. J Biol Chem. 1998;273:10741–10746. doi: 10.1074/jbc.273.17.10741. [DOI] [PubMed] [Google Scholar]
- 16.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 17.Klahre U, Noguchi T, Fujioka S, Takatsuto S, Yokota T, Nomura T, Yoshida S, Chua NH. The Arabidopsis DIMINUTO/DWARF1 gene encodes a protein involved in steroid synthesis. Plant Cell. 1998;10:1677–1690. doi: 10.1105/tpc.10.10.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM, Ross R. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol Cell. 1998;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- 19.Levkau B, Scatena M, Giachelli CM, Ross R, Raines EW. Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-kappa B loop. Nat Cell Biol. 1999;1:227–233. doi: 10.1038/12050. [DOI] [PubMed] [Google Scholar]
- 20.Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol. 1998;150:40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- 21.McMorris TC. Recent developments in the field of plant steroid hormones. Lipids. 1997;32:1303–1308. doi: 10.1007/s11745-006-0168-z. [DOI] [PubMed] [Google Scholar]
- 22.Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K. The amyloid precursor protein of Alzheimer's disease in the reduction of copper(II) to copper(I). Science. 1996;271:1406–1409. doi: 10.1126/science.271.5254.1406. [DOI] [PubMed] [Google Scholar]
- 23.Mushegian AR, Koonin EV. A putative FAD-binding domain in a distinct group of oxidases including a protein involved in plant development. Protein Sci. 1995;4:1243–1244. doi: 10.1002/pro.5560040623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nomura N, Miyajima N, Sazuka T, Tanaka A, Kawarabayasi Y, Sato S, Nagase T, Seki N, Ishikawa K, Tabata S. Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001-KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res. 1994;1:47–56. doi: 10.1093/dnares/1.1.47. [DOI] [PubMed] [Google Scholar]
- 25.Oppermann UC, Salim S, Tjernberg LO, Terenius L, Jornvall H. Binding of amyloid beta-peptide to mitochondrial hydroxyacyl-CoA dehydrogenase (ERAB): regulation of an SDR enzyme activity with implications for apoptosis in Alzheimer's disease. FEBS Lett. 1999;451:238–242. doi: 10.1016/s0014-5793(99)00586-4. [DOI] [PubMed] [Google Scholar]
- 26.Poirier GM, Pyati J, Wan JS, Erlander MG. Screening differentially expressed cDNA clones obtained by differential display using amplified RNA. Nucleic Acids Res. 1997;25:913–914. doi: 10.1093/nar/25.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sagara Y, Dargusch R, Klier FG, Schubert D, Behl C. Increased antioxidant enzyme activity in amyloid beta protein-resistant cells. J Neurosci. 1996;16:497–505. doi: 10.1523/JNEUROSCI.16-02-00497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagara Y, Dargusch R, Chambers D, Davis J, Schubert D, Maher P. Cellular mechanisms of resistance to chronic oxidative stress. Free Radic Biol Med. 1998;24:1375–1389. doi: 10.1016/s0891-5849(97)00457-7. [DOI] [PubMed] [Google Scholar]
- 29.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 30.Skovronsky DM, Pijak DS, Doms RW, Lee VM. A distinct ER/IC gamma-secretase competes with the proteasome for cleavage of APP. Biochemistry. 2000;39:810–817. doi: 10.1021/bi991728z. [DOI] [PubMed] [Google Scholar]
- 31.Soriano S, Chyung AS, Chen X, Stokin GB, Lee VM, Koo EH. Expression of beta-amyloid precursor protein-CD3 gamma chimeras to demonstrate the selective generation of amyloid beta(1–40) and amyloid beta(1–42) peptides within secretory and endocytic compartments. J Biol Chem. 1999;274:32295–32300. doi: 10.1074/jbc.274.45.32295. [DOI] [PubMed] [Google Scholar]
- 32.Susens U, Aguiluz JB, Evans RM, Borgmeyer U. The germ cell nuclear factor mGCNF is expressed in the developing nervous system. Dev Neurosci. 1997;19:410–420. doi: 10.1159/000111238. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi T, Gasch A, Nishizawa N, Chua NH. The DIMINUTO gene of Arabidopsis is involved in regulating cell elongation. Genes Dev. 1995;9:97–107. doi: 10.1101/gad.9.1.97. [DOI] [PubMed] [Google Scholar]
- 34.Tomita S, Kirino Y, Suzuki T. Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J Biol Chem. 1998;273:6277–6284. doi: 10.1074/jbc.273.11.6277. [DOI] [PubMed] [Google Scholar]
- 35.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Hoesen GW, Hyman BT, Damasio AR. Entorhinal cortex pathology in Alzheimer's disease. Hippocampus. 1991;1:1–8. doi: 10.1002/hipo.450010102. [DOI] [PubMed] [Google Scholar]
- 37.von der Kammer H, Mayhaus M, Albrecht C, Enderich J, Wegner M, Nitsch RM. Muscarinic acetylcholine receptors activate expression of the EGR gene family of transcription factors. J Biol Chem. 1998;273:14538–14544. doi: 10.1074/jbc.273.23.14538. [DOI] [PubMed] [Google Scholar]
- 38.von der Kammer H, Albrecht C, Mayhaus M, Hoffmann B, Stanke G, Nitsch RM. Identification of genes regulated by muscarinic acetylcholine receptors: application of an improved and statistically comprehensive mRNA differential display technique. Nucleic Acids Res. 1999;27:2211–2218. doi: 10.1093/nar/27.10.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia W, Zhang J, Ostaszewski BL, Kimberly WT, Seubert P, Koo EH, Shen J, Selkoe DJ. Presenilin 1 regulates the processing of beta-amyloid precursor protein C-terminal fragments and the generation of amyloid beta-protein in endoplasmic reticulum and Golgi. Biochemistry. 1998;37:16465–16471. doi: 10.1021/bi9816195. [DOI] [PubMed] [Google Scholar]
- 40.Xue D, Horvitz HR. Caenorhabditis elegans CED-9 protein is a bifunctional cell-death inhibitor. Nature. 1997;390:305–308. doi: 10.1038/36889. [DOI] [PubMed] [Google Scholar]
- 41.Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, Gibson L, Stern E, Collison K, Al-Mohanna F, Ogawa S, Roher A, Clarke SG, Stern DM. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Aβ-induced cytotoxicity. J Biol Chem. 1999;274:2145–2156. doi: 10.1074/jbc.274.4.2145. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH. Subcellular distribution and turnover of presenilins in transfected cells. J Biol Chem. 1998;273:12436–12442. doi: 10.1074/jbc.273.20.12436. [DOI] [PubMed] [Google Scholar]