

Abstract
Nerve growth factor (NGF) supports target-dependent survival of sympathetic and other neurons during development; however, the NGF-regulated signaling pathways required for survival are not fully understood. Sympathetic neurons are able to abort acutely the cell death pathway initiated by NGF deprivation at early, as well as late, time points after readdition of NGF. We found that NGF-dependent phosphatidylinositol 3-kinase (PI-3-K) activity inhibited an early cell death event proximal to c-Jun phosphorylation. However, PI-3-K activity was not required for NGF to inhibit the translocation of Bax from the cytoplasm to the mitochondria, nor was it required for NGF to inhibit the subsequent release of mitochondrial cytochrome c,two events required for NGF deprivation-induced apoptosis. MEK/MAPK activity did not account for any of these NGF-dependent events. When subjected to long-term PI-3-K inhibition in the presence of NGF, the majority of sympathetic neurons did not die. Those that did die exhibited significant differences in the characteristics of death caused by PI-3-K inhibition as compared with NGF deprivation. Additionally, PI-3-K inhibition in the presence of NGF did not induce release of mitochondrial cytochrome c, indicating that these neurons were unable to complete the apoptotic program. In contrast to its modest effects on survival, inhibition of PI-3-K induced marked decreases in somal diameter and metabolic function, as measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphen-yltetrazolium bromide (MTT) reduction, suggesting that PI-3-K is required for the trophic effects of NGF. Therefore, although PI-3-K is important for the trophic effects of NGF, it is not required for survival. Other, or at least additional, signaling pathways contribute to NGF-mediated survival of sympathetic neurons.
Keywords: phosphatidylinositol 3-kinase, sympathetic neurons, NGF, Bax, cytochrome c, apoptosis
Target-dependent survival of sympathetic neurons is regulated during development by nerve growth factor (NGF) through activation of its receptor tyrosine kinase, TrkA (Levi-Montalcini, 1987; Korsching, 1993; Bothwell, 1995). A signaling molecule important for NGF-dependent survival of sympathetic neurons is phosphatidylinositol 3-kinase (PI-3-K). A mechanism by which PI-3-K promotes survival of neurons is via activation of the serine/threonine kinase Akt/protein kinase B (PKB) (Franke et al., 1997; Datta et al., 1999). Akt/PKB inhibits apoptosis through phosphorylation of cell death regulatory molecules such as BAD (Datta et al., 1997), caspase-9 (Cardone et al., 1998), and Forkhead transcription factors (Biggs et al., 1999; Brunet et al., 1999). Although PI-3-K-dependent survival of cerebellar granule neurons is well established (D'Mello et al., 1997;Dudek et al., 1997; Miller et al., 1997), the exact role of PI-3-K in sympathetic neuron survival is less clear. Constitutively active or overexpressed PI-3-K and Akt/PKB support survival of sympathetic neurons after NGF withdrawal, demonstrating that either is sufficient for survival (Philpott et al., 1997; Crowder and Freeman, 1998). Whether PI-3-K is necessary for the survival-promoting activity of NGF, however, is controversial. Although some have observed that inhibition of PI-3-K activity results in death of sympathetic neurons (Crowder and Freeman, 1998; Mazzoni et al., 1999; Vaillant et al., 1999), others have not (Philpott et al., 1997; Virdee et al., 1999).
NGF deprivation of sympathetic neurons initiates a sequence of molecular events that culminates in caspase activation and death (Deshmukh and Johnson, 1997). An early event is the phosphorylation and activation of c-Jun that may be responsible in part for the expression of death-promoting genes (Estus et al., 1994; Ham et al., 1995). Consistent with c-Jun activation, several genes are induced after NGF deprivation, such as c-jun, cyclin D1, and SM-20, that may be important for NGF deprivation-induced cell death (Estus et al., 1994; Freeman et al., 1994; Lipscomb et al., 1999). The expression of proapoptotic genes is thought to mediate translocation of Bax from the cytoplasm to the mitochondria, an event that promotes cytochrome c release from the mitochondria (Putcha et al., 1999). Cytoplasmic cytochromec is required, but not sufficient, for apoptosis of sympathetic neurons, which also must develop “competence-to-die,” the molecular nature of which is unclear (Deshmukh and Johnson, 1998;Neame et al., 1998).
NGF can abort the cell death pathway at multiple points after NGF deprivation. Sympathetic neurons can be deprived of NGF for 22 hr, a time when 50% of neurons have released cytochrome c from the mitochondria; readdition of NGF at this time halts any further cytochrome c release and death (Deckwerth and Johnson, 1993;Deshmukh and Johnson, 1998). Similarly, NGF can inhibit acutely any further Bax translocation after 18 hr of NGF deprivation, a time when 50% of rat neurons have already translocated Bax (Putcha et al., 1999). We examined which signaling pathways account for NGF-dependent inhibition of c-Jun phosphorylation, Bax translocation to mitochondria, and mitochondrial cytochrome c release. These experiments helped determine whether TrkA used multiple signaling pathways to promote survival of sympathetic neurons or relied solely on PI-3-K activity.
MATERIALS AND METHODS
Materials. LY294002, PD98059, SB203580, H89, and Wortmannin were obtained from Biomol (Plymouth Meeting, PA) and diluted in DMSO to the concentrations recommended. Boc-aspartyl fluoromethyl ketone (BAF) was obtained from Enzyme Systems Products (Livermore, CA) and diluted in DMSO as well. All other pharmacological agents were reagent grade and diluted into medium just before use. Anti-α-tubulin (Sigma, St. Louis, MO), anti-phosphoMAPK (New England Biolabs, Beverly, MA), anti-phosphoAKT (Ser472, New England Biolabs), anti-phosphoAKT (Thr308, Upstate Biotechnology, Inc., Lake Placid, NY), anti-LDH (Rockland Immunochemicals, Gilbertsville, PA), anti-COX IV (Molecular Probes, Eugene, OR), and anti-Bax (Upstate Biotechnology, Inc.) were used for Western blotting at the dilutions described below. Anti-phospho-c-Jun (New England Biolabs), anti-cytochrome c(PharMingen, San Diego, CA), and CM1 (IDUN, San Diego, CA) were used for immunocytochemistry.
Sympathetic neuronal cultures and treatments. The superior cervical ganglia from postnatal day 1 Sprague Dawley rats were dissected, dissociated, and seeded onto collagen-coated 35 mm culture dishes, 24-well culture plates, or two-well glass chamber slides (Nunc, Naperville, IL) as described previously (Martin et al., 1992). Cultures were maintained for 5 d in vitro (DIV) in medium (90% Minimum Essential medium, 10% fetal bovine serum, 2 mm glutamine, 20 μmuridine, 20 μm fluorodeoxyuridine, 100 U/ml penicillin, and 100 μg/ml streptomycin) containing NGF (50 ng/ml), after which time cultures were washed twice with medium without NGF (AM0). AM0 medium containing anti-NGF (goat polyclonal, 1:10,000 dilution) was then added. The neurons were maintained in this medium for the amount of time described in each experiment, washed twice with AM0, and next treated with AM0 containing NGF (300 ng/ml) in the presence or absence of the described pharmacological agent. For experiments examining the survival and metabolism of sympathetic neurons after PI-3-K blockade, LY294002 (50 μm) was added in the presence of NGF, and control cultures were treated with NGF and vehicle alone (0.1% DMSO). In these experiments the medium was replaced every 2 d. For c-Jun phosphorylation studies, neurons were maintained in anti-NGF for 4 hr, and the described pharmacological agent was added for an additional 0.5 hr before NGF readdition in the continued presence of the drug or vehicle alone (DMSO). For Bax translocation experiments, neurons were maintained in anti-NGF for 18 hr before drug pretreatment and NGF readdition. For cytochrome c translocation experiments, sympathetic neurons were maintained in anti-NGF for 22 hr before drug and NGF additions. For Bax, cytochrome c, and in some survival experiments, BAF (50 μm) was added to the cultures during the entire treatment regimen to prevent the neurons from lifting off the collagen substrate after caspase activation and apoptotic death.
Immunocytochemistry. All sympathetic neuronal cultures processed for immunofluorescence were grown on glass two-well chamber slides. After the described treatments, cultures were washed once with ice-cold PBS and fixed with fresh 4% paraformaldehyde in PBS for 30 min at 4°C. Cultures were next washed three times with Tris-buffered saline (TBS) and incubated in blocking solution [5% normal goat serum (NGS) in TBS that contained 0.3% Triton X-100] for 30−60 min at room temperature. The neurons were then incubated with primary antibodies in blocking solution overnight at 4°C. The phospho-c-Jun (P-Jun) antibody was used at a dilution of 1:400, and the cytochromec antibody was used at a dilution of 1:1000 (0.5 μg/ml). The cultures were next washed three times with TBS and incubated in blocking solution containing fluorescently labeled secondary antibodies for 2−4 hr at 4°C (1.5 μg/ml anti-rabbit Cy-3 and 2 μg/ml anti-mouse Alexa 488). The cultures were then washed twice with TBS, incubated for 20 min with Hoechst 33258 (1 μg/ml) in TBS, washed a final two times in TBS, and mounted for fluorescence microscopy. All counts of positively or negatively stained neurons were obtained in a blinded manner.
Survival assays. Toluidine blue staining was performed on sympathetic neuronal cultures grown in two-well chamber slides. After the indicated treatments, cultures were washed with ice-cold PBS and next fixed for 1−2 d with 4% paraformaldehyde. Cultures were then washed with deionized water, stained for 45−60 sec in toluidine blue-O (1 gm/l), and incubated for an additional 60 sec in deionized water. The cells were dehydrated by using successive 2 min washes in deionized water containing increasing concentrations of ethanol to reach 100%, after which the slides were washed in toluene, and coverslips mounted with Permount (Sigma). Neurons displaying smooth cell bodies that were Nissl-stained were considered alive and were counted. All survival counts were obtained in a blinded manner.
Subcellular fractionation. After the described treatments, sympathetic neurons (106) were harvested into 10 ml of isotonic fractionation buffer (250 mmsucrose, 0.5 mm EDTA, 20 mm HEPES, 500 μm Na3VO4, pH 7.2) supplemented with protease inhibitors (Inhibitor cocktail complete, Roche Molecular Biochemicals, Indianapolis, IN) and centrifuged at 900 × g for 5 min. All subsequent steps were conducted at 4°C. The pellet was resuspended into 200 μl of fractionation buffer, homogenized with a ball-bearing homogenizer, and centrifuged at 900 × g for 5 min to remove nuclei and intact cells. The postnuclear supernatant was transferred to a microfuge tube (Beckman, Palo Alto, CA) and centrifuged at 25,000 × g for 10 min to collect the heavy membrane (HM) fraction that is enriched in mitochondria, followed by centrifugation of the post-HM supernatant at 100,000 × g for 10−20 min to obtain the microsomal and cytosolic fractions. All pellets were resuspended in a volume of fractionation buffer equivalent to the cytosolic volume. These fractions were then resuspended to equivalent volumes with 2× sample buffer and evaluated by Western blotting.
Western blotting. Sympathetic neurons maintained in 35 mm culture dishes were washed twice with ice-cold TBS and incubated with lysis buffer (1% Nonidet P-40, 10% glycerol, 500 μmsodium orthovanadate, and protease inhibitors in TBS) for 20 min at 4°C with gentle agitation. The cell extracts were centrifuged at maximum speed in a microfuge to remove collagen and nuclei from the detergent extracts. The supernatants were next diluted into 2× sample buffer to arrive at 1×, and boiled for 5−10 min. SDS-PAGE was performed on these denatured extracts using 4−12% gradient gels (Novex, San Diego, CA). Resolved proteins were transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA) and washed with TBS containing 0.1% Tween-20 (TBST). All immunoblots were incubated with a blocking solution consisting of TBST containing 4% heat-inactivated horse serum (HS) for 1 hr at room temperature, and further incubated for 2 hr in TBST containing the primary antibody, 4% BSA, and 2% normal donkey serum. Immunoblots were washed three times with TBST containing 1% HS, incubated with the secondary antibody (1:10,000 dilution) in blocking solution for 1 hr, washed twice with TBST containing 1% HS, and finally washed once with TBST alone. Proteins were visualized by using the supersignal chemiluminescent detection system (Pierce, Rockford, IL). The α−tubulin antibody was used at a 1:50,000 dilution, whereas all other antibodies were used at a 1:1000 dilution. Immunoblots were reprobed by using the Western analysis protocol described above after stripping residual antibodies from the blots by incubation in 100 mm glycine, pH 2.75, for 1 hr at room temperature followed by two washes with TBST.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays. Sympathetic neurons maintained in 24-well culture plates (20,000 neurons per well) were treated as described in the text. After 2 d the plates were washed with PBS once and incubated for 10 min in L15 medium containing 10% fetal bovine serum and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (0.4 mg/ml, Sigma). The medium was then removed, and DMSO (250 μl) was added to each well, mixed, and transferred to a translucent microtiter plate. The absorbance at 550 nm was measured by using a microplate reader and subtracted from the background absorbance measured at 650 nm. The background level of metabolic activity of contaminating glial cells was subtracted from all conditions by performing the above analysis on sympathetic neuronal cultures maintained in anti-NGF for 7 d beginning on the first day of plating, which killed all neurons leaving only glial cells behind.
Lipid kinase assays. Sympathetic neurons (5 DIV) were deprived of NGF for 8 hr and then treated with medium alone or NGF (100 ng/ml) in the presence or absence of LY294002 (50 μm) or Wortmannin (1–10 μm) for 3 min. Both LY294002 and Wortmannin were added to the culture medium 30 min before NGF treatment. Because LY294002 is a reversible inhibitor, it had to be added to all subsequent buffers and washes, whereas Wortmannin, which is an irreversible inhibitor, was only added to the culture medium immediately before and during the NGF treatments. The sympathetic neurons were next washed twice with TBS, and detergent extracts were produced using the NP40-containing lysis buffer described above. The extracts were subjected to immunoprecipitation using anti-phosphotyrosine antibody conjugated to agarose (50 μl) (Calbiochem, San Diego, CA) overnight at 4°C. The immunoprecipitates were then subjected to lipid kinase analysis essentially as described previously (Auger et al., 1990). Briefly, the immunoprecipitates were washed at 4°C three times with PBS containing NP40 (1%), three times with PBS containing lithium chloride (0.5 m), and finally three times with TBS (20 mm Tris, 137 mm NaCl). The immunoprecipitates were then incubated with phosphatidylinositol (20 μg, Sigma), cold ATP (40 mm), and [γ32P]ATP (10 μCi) (ICN Radiochemicals, Irvine, CA) in TBS containing magnesium chloride (15 mm) for 15 min at room temperature under constant agitation. The reaction was stopped by adding HCl (6 m, 20 μl), and the lipids was extracted with addition of chloroform/methanol (1:1, 160 μl total). The lower organic phase, obtained after centrifugation, was subjected to TLC using oxalate-coated silica gel chromatography plates (Whatman Inc., Clifton, NJ) and developed in chloroform/methanol/water/ammonium hydroxide (60:47:11.3:2). The plate was then dried and subjected to phosphorimaging analysis.
RESULTS
PI-3-K inhibits a cell death event proximal to c-Jun phosphorylation
A critical early event in the cell death pathway initiated by NGF deprivation of sympathetic neurons is the Ser-63 phosphorylation and transcriptional activation of c-Jun (Estus et al., 1994; Ham et al., 1995). After NGF deprivation c-Jun was rapidly phosphorylated, with 30% of neurons displaying nuclear P-Jun staining after 4 hr, and almost 60% were P-Jun positive after 9.5 hr (Fig.1C, D). The number of P-Jun-positive neurons increased more slowly thereafter, reaching a maximum of 80% after 2 d (see Fig. 6B). To determine whether NGF could inhibit acutely c-Jun phosphorylation, sympathetic neurons were deprived of NGF for 4 hr to reach a half-maximal number of P-Jun-positive neurons. This medium was replaced with medium containing NGF or medium without NGF, and the cultures were incubated for an additional 5.5 hr (Fig. 1B). After treatment, the sympathetic neurons were fixed, and immunocytochemical detection of Ser-63-phosphorylated c-Jun was performed as before. Acute NGF readdition to cultured sympathetic neurons not only halted the number of P-Jun positive neurons (Fig.1C), it also reversed the number to basal levels . As expected, readdition of medium alone did not alter the number of neurons that became P-Jun positive (Fig. 1C). Therefore, NGF can reverse the phosphorylation of c-Jun after NGF deprivation.
Fig. 1.
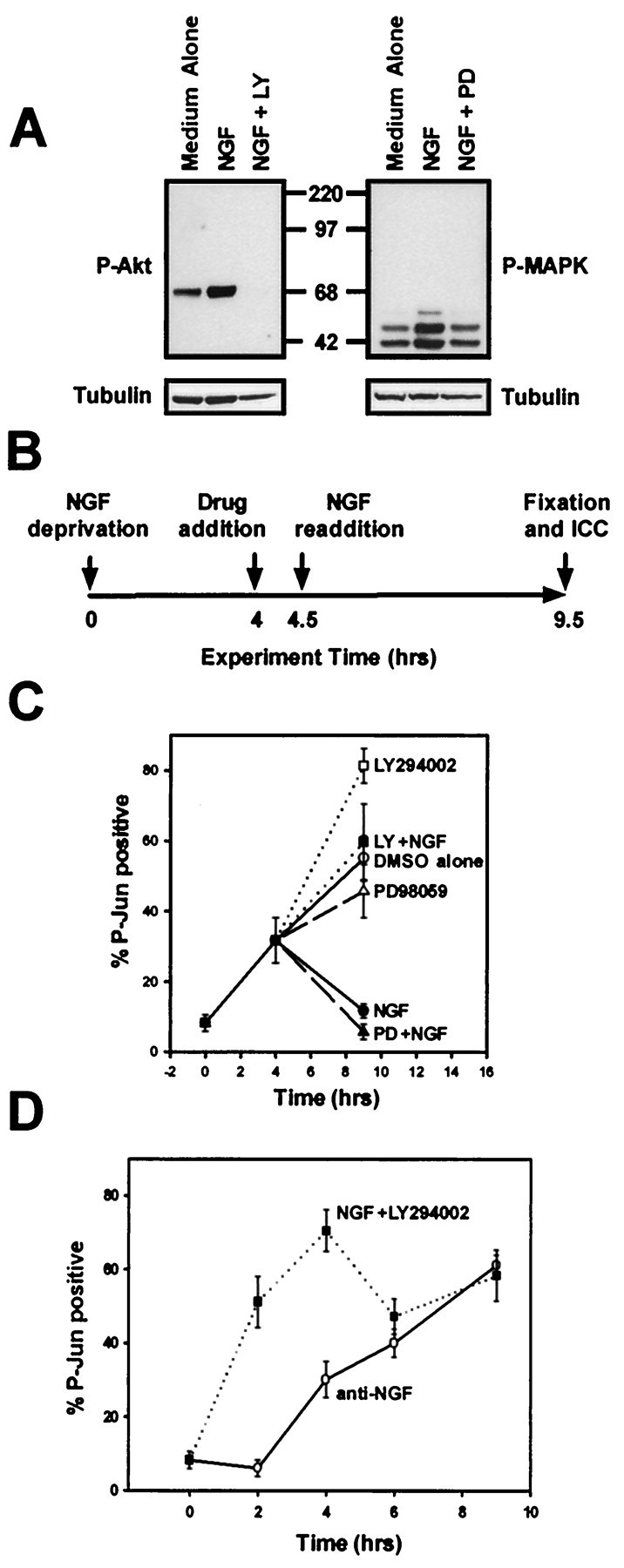
PI-3-K inhibits a cell death event proximal to c-Jun phosphorylation. A, Cultures of sympathetic neurons maintained in NGF (50 ng/ml) were treated with medium containing LY294002 (50 μm, left panel) or PD98059 (25 μm, right panel) to inhibit PI-3-K or MEK activities, respectively, or medium containing vehicle alone (DMSO, 0.1%) for 30 min. The cultures were then treated with medium alone, medium containing NGF (50 ng/ml), or medium containing NGF (50 ng/ml) in the continued presence of LY294002 or PD98059 for an additional 30 min. Detergent extracts were prepared, and equal amounts of the extracts were subjected to phospho-Akt (P-Akt) or phospho-MAPK (P-MAPK) immunoblotting. Reprobing the same blot with anti-α-tubulin after stripping demonstrates equal amounts of protein in the extracts. B, This schematic diagram depicts the experimental paradigm for the experiment described in C. Sympathetic neurons (5 DIV) were deprived of NGF for 4 hr, preincubated with medium containing LY294003 (50 μm), PD98059 (25 μm), or vehicle alone for 30 min, and next treated with medium containing NGF (300 ng/nl) in the continued presence of LY294002 or PD98059 for an additional 5 hr. C, Sympathetic neurons treated as in B were fixed after the treatments, and P-Jun was detected immunocytochemically. The number of neurons showing nuclear P-Jun staining is shown as a percentage of total cells. This experiment was performed in duplicate in three independent cultures.D, Sympathetic neurons were deprived of NGF (anti-NGF) or treated with LY294003 (50 μm) in the continued presence of NGF (NGF +LY29400). The cultures were fixed 2, 4, 6, or 9 hr after treatment and subjected to P-Jun immunocytochemistry. This experiment was performed in duplicate in three independent cultures. Error bars represent SEM.
Fig. 6.
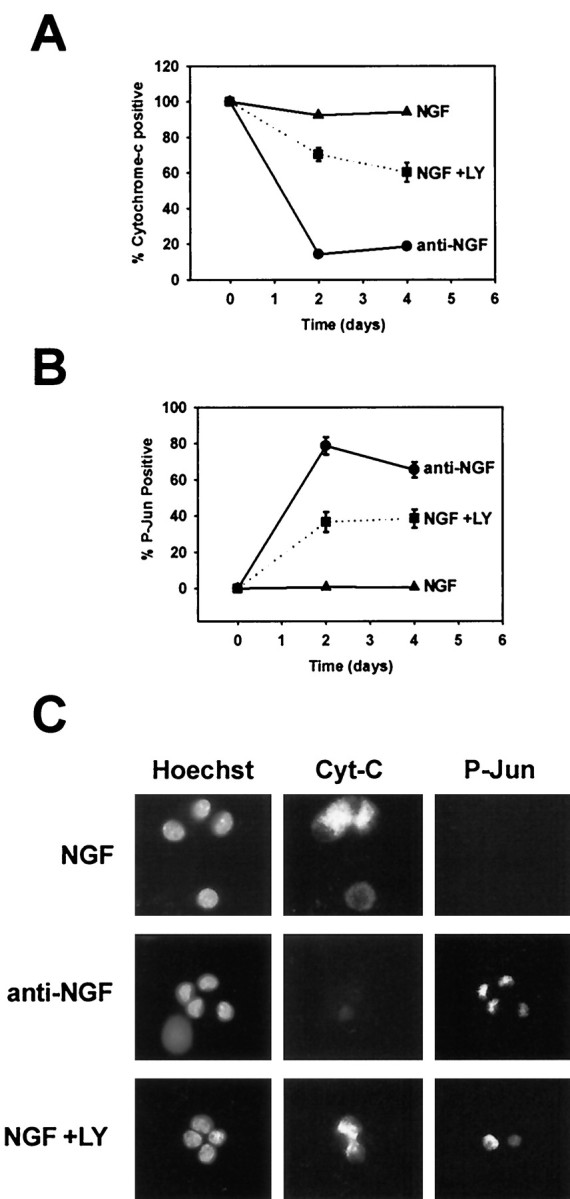
PI-3-K inhibition does not induce cytochromec release from most NGF-dependent sympathetic neurons. Sympathetic neurons were maintained in NGF (50 ng/ml), maintained in NGF with LY294002 (50 μm), or deprived of NGF for 2–4 d. The cultures were then fixed and subjected to simultaneous P-Jun and cytochrome c immunocytochemistry. Hoechst staining was also performed to identify nuclei. All conditions included BAF (50 μm) to prevent the loss of neurons. The number of cytochrome c-positive neurons (A) and the number of P-Jun-positive neurons (B) were scored in each condition and graphed as a percentage of total neurons. Note that initially more neurons are P-Jun positive a few hours after LY treatment (Fig. 1) but plateau to a lower level after 2−4 d of LY294002 treatment in the presence of BAF. C, Representative examples of immunofluorescent detection of cytochromec (Cyt-C, Alexa 488, middle column), P-Jun (Cy-3, right column), and nuclei (Hoechst, left column) in sympathetic neurons after 2 d of treatments as described above, which are listed to the left of each panel depicting each condition.
We next determined whether the ability of NGF to reverse c-Jun phosphorylation required PI-3-K or MEK activity. Sympathetic neurons were deprived of NGF for 4 hr. The neurons were then treated with LY294002 or PD98059 for 30 min to inhibit the activity of PI-3-K or MEK, respectively; controls were treated with vehicle (0.1−0.2% DMSO). NGF was next added in the continued presence of LY294002 or PD98059, and these cultures incubated for an additional 5 hr, after which time they were processed for P-Jun immunocytochemistry (Fig.1B). PD98059 did not inhibit NGF-mediated reversal of c-Jun phosphorylation (Fig. 1C). PD98059 inhibited NGF-dependent MAPK activation, as determined by phospho-MAPK Western analysis of extracts made from PD98059-treated sympathetic neurons (Fig. 1A), confirming that an effective dose of PD98059 was used. Treatment of sympathetic neurons with LY294002 completely inhibited NGF-dependent Akt phosphorylation, as determined by P-Akt (S473) Western analysis (Fig. 1A). In contrast to inhibition of MEK activity, inhibition of PI-3-K activity completely blocked NGF-mediated reversal of c-Jun phosphorylation (Fig.1C).
Because P-Jun staining was often intense when neurons were treated with LY294002 in either the presence or absence of NGF, we determined whether PI-3-K inhibition in the presence of NGF was sufficient to induce c-Jun phosphorylation in sympathetic neurons. Neurons were treated with LY294002 in the presence of NGF for 2−9 hr and fixed, and P-Jun immunocytochemistry was performed. To compare PI-3-K inhibition with NGF withdrawal, sympathetic neurons were deprived of NGF for the same amounts of time, and P-Jun immunocytochemistry was performed as well. PI-3-K inhibition was sufficient to induce rapid and sustained c-Jun phosphorylation in the presence of NGF that reached a maximum within 4 hr (Fig. 1D). NGF deprivation, in contrast, did not induce maximal c-Jun phosphorylation until at least 9 hr after NGF withdrawal (Fig. 1D). Therefore, PI-3-K activity inhibits an event in the cell death pathway proximal to c-Jun phosphorylation, whereas MEK/MAPK activity is not required for NGF-dependent inhibition of c-Jun phosphorylation initiated by NGF withdrawal. These results are consistent with reports demonstrating that PI-3-K inhibition induces c-Jun kinase activity in cerebellar granule neurons as well as c-Jun phosphorylation in DRG neurons (Vogelbaum et al., 1998; Shimoke et al., 1999).
Neither PI-3-K nor MEK activity is required for NGF-dependent inhibition of Bax translocation from cytoplasm to mitochondria
Bax, a proapoptotic Bcl-2 family member, is required for NGF deprivation-induced cell death of sympathetic neurons (Deckwerth et al., 1996). The translocation of Bax from the cytoplasm to the mitochondria is an event that occurs after NGF deprivation of sympathetic neurons and is required for the release of cytochromec from the mitochondria and subsequent activation of caspases (Putcha et al., 1999, 2000). Because NGF can acutely inhibit the translocation of Bax to the mitochondria, we determined whether PI-3-K or MEK activity is required for the ability of NGF to regulate this event. Sympathetic neurons were deprived of NGF for 18 hr, after which time they were treated with LY294002, PD98059, or vehicle alone (0.1−0.2% DMSO) for 30 min. These cultures were then treated with NGF for an additional 12 hr in the continued presence of LY294002 or PD98059 (Fig.2A). Cultures were then lysed, and subcellular fractionation was performed to separate cytoplasmic proteins from lysosomal and mitochondrial proteins. Cytoplasmic and HM fractions were subjected to SDS-PAGE, and Western analysis was performed with antibodies directed against Bax and cytochrome c. NGF deprivation resulted in the loss of Bax from the cytoplasmic fraction and the coincident accumulation of Bax in the HM fraction (containing mitochondria), consistent with its translocation from the cytoplasm to the mitochondria (Fig.2B). If NGF was replaced after 18 hr of deprivation, Bax redistribution was halted as compared with replacement of medium alone (Fig. 2B). Inhibition of PI-3-K or MEK activity did not alter the ability of NGF to inhibit redistribution of Bax (Fig.2B), although these pharmacological agents did block downstream Akt and MAPK phosphorylation, respectively (Fig.1A). Therefore, neither the PI-3-K nor MEK/MAPK pathways are required for NGF-dependent inhibition of Bax translocation from the cytoplasm to mitochondria.
Fig. 2.

Neither PI-3-K nor MEK/MAPK is required for NGF-dependent inhibition of Bax translocation from cytoplasm to mitochondria. A, This schematic diagram depicts the treatment paradigm used in B. Sympathetic neurons were deprived of NGF for 18 hr before a 30 min incubation with LY294002 (50 μm), PD98059 (25 μm), or vehicle alone (DMSO, 0.1%). Neurons were next washed and treated with NGF (300 ng/ml) in the continued presence of LY294002, PD98059, or DMSO for an additional 12 hr. B, Sympathetic neurons treated as inA were homogenized in an isotonic buffer and subcellular fractions produced by differential centrifugation. Equal volumes of the cytosolic (Cyto) fractions and heavy membrane (HM) fractions containing mitochondria were subjected to Western analysis by using antibodies directed against Bax, cytochrome c, lactate dehydrogenase (LDH, a cytosolic protein), and cytochrome oxidase IV (COX IV, a mitochondrial protein). LDH and COX IV immunoblots demonstrate not only that the cytoplasmic and heavy membrane fractions do not contain significant amounts of contamination, but also that equal amounts of protein from each of these fractions were analyzed. This experiment was performed on two independent cultures with similar results.
Neither PI-3-K nor MEK activity is required for NGF-dependent inhibition of cytochrome c release in sympathetic neurons
NGF can act late in the cell death pathway to inhibit the release of cytochrome c from the mitochondria, thus inhibiting activation of caspases (Deshmukh and Johnson, 1998; Putcha et al., 1999). Because the signaling pathway that NGF uses to inhibit release of mitochondrial cytochrome c is unknown, we determined whether NGF-dependent inhibition of cytochrome c release required PI-3-K or MEK activity. Sympathetic neurons were deprived of NGF for 22 hr, after which time LY294002, PD98059, or vehicle alone (DMSO) was added for an additional 30 min. The neurons were then subjected to NGF readdition, or to readdition of medium alone, in the continued presence of LY294002 or PD98059 for an additional 12 hr (Fig.3A). The sympathetic neurons were then fixed, and cytochrome c immunocytochemistry was performed. As observed previously, acute NGF readdition after 22 hr of NGF deprivation inhibited any further release of cytochromec from the mitochondria (Fig. 3B). When sympathetic neurons were treated with levels of LY294002 that completely inhibited Akt phosphorylation (data not shown), PI-3-K inhibition did not alter NGF-dependent inhibition of cytochromec release (Fig. 3B). NGF readdition after 22 hr of deprivation induced Akt phosphorylation (data not shown), indicating that signaling downstream of TrkA is still intact after 22 hr of NGF deprivation. NGF-mediated inhibition of cytochrome c release was also not blocked by concentrations of PD98059 that completely blocked MEK activity (Fig. 3B). Therefore, neither PI-3-K activity nor the MEK/MAPK cascade was required for NGF-dependent inhibition of cytochrome c release from the mitochondria. In fact, inhibition of both PI-3-K and MEK activities simultaneously by treatment with both LY294002 and PD98059 failed to alter NGF-dependent inhibition of cytochrome c release (data not shown). These findings are consistent with the data from subcellular fractionations demonstrating that inhibition of PI-3-K activity or MEK activity did not affect NGF-dependent inhibition of cytochrome crelease (Fig. 2B).
Fig. 3.
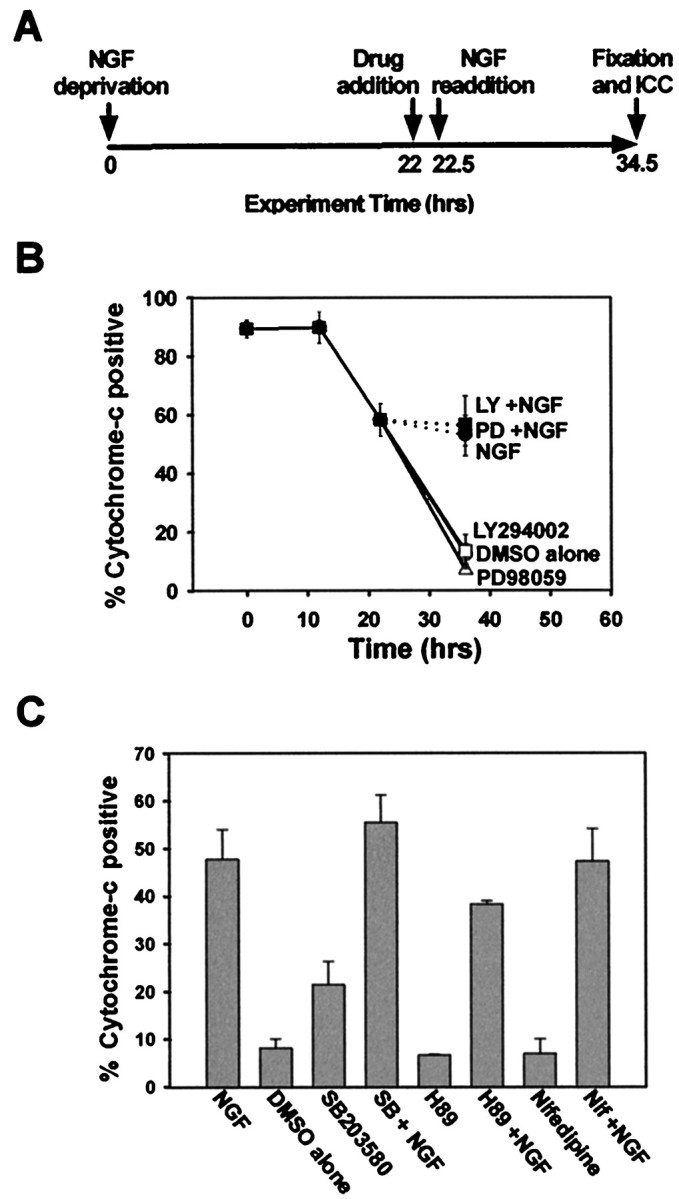
Neither PI-3-K nor MEK/MAPK mediates NGF-dependent inhibition of mitochondrial cytochrome c release.A, This schematic diagram represents the experimental paradigm used in B and C. Sympathetic neurons were deprived of NGF for 22 hr and next treated with vehicle alone (DMSO, 0.1%), LY294002 (50 μm), or PD98059 (25 μm) for 30 min. The cultures were then treated with medium alone or medium containing NGF (300 ng/ml) in the continued presence of LY294002 or PD98059 for an additional 12 hr before fixation. B, Cultures treated as in Awere immunostained for cytochrome c, and the number of neurons displaying punctate cytochrome c are shown as the percentage of total cells. This experiment was performed in duplicate in three independent cultures. Error bars represent SEM.Open symbols indicate cultures that were treated with medium alone in the presence of the listed pharmacological agent;closed symbols represent cultures treated with NGF.C, Sympathetic neuronal cultures were treated as inA, but the pharmacological agents added were the p38MAPK inhibitor SB203580 (30 μm), the PKA inhibitor H89 (5 μm), the L-type calcium channel antagonist nifedipine (200 nm), or vehicle alone, before NGF readdition. The number of neurons displaying punctate cytochrome c is displayed as a vertical bar chart depicting only the percentage of cytochrome c-positive cells remaining at the end of the treatments. This experiment was performed in duplicate in three independent cultures, with error bars representing SEM.
Because neuroprotectants such as cAMP and potassium depolarization are able to inhibit cytochrome c release after BAX translocation to the mitochondria (Putcha et al., 1999), we determined whether NGF-dependent inhibition of cytochrome c release might use similar signaling pathways. Sympathetic neurons were deprived of NGF for 22 hr and treated with nifedipine (200 nm) to block L-type channel-dependent calcium influx or with H-89 (5 μm) to block protein kinase A (PKA) activity, both of which are downstream effectors of potassium depolarization and cAMP, respectively. The neurons were next treated with NGF in the continued presence of nifedipine or H-89 for an additional 12 hr, and the number of mitochondrial cytochrome c-positive neurons was determined. Neither calcium influx nor PKA activity altered the ability of NGF to inhibit cytochrome c release from mitochondria (Fig. 3C). Nifedipine and H-89, however, were able to inhibit >50% of KCl-dependent and cAMP-dependent inhibition of cytochrome c release, respectively, thus demonstrating the effectiveness of these compounds (data not shown).
We also determined whether p38MAPK, another NGF-regulated kinase, might mediate the ability of NGF to block cytochrome c release. Treatment of sympathetic neurons with a specific inhibitor of p38MAPK activation, SB203580, with concentrations that completely inhibit p38MAPK activity did not alter NGF-dependent inhibition of cytochromec release (Fig. 3C). Therefore, p38MAPK did not mediate NGF inhibition of mitochondrial cytochrome crelease. In summary, although acute application of NGF can inhibit the release of mitochondrial cytochrome c, activation of PI-3-K, MEK/MAPK, or p38MAPK was not required. Furthermore, neither PKA nor calcium influx through L-type channels was responsible for NGF-dependent inhibition of cytochrome c release.
PI-3-K activity contributes to NGF-dependent metabolism of sympathetic neurons
Although the above experiments demonstrated that PI-3-K was not required for the later anti-apoptotic actions of NGF, sympathetic neurons treated with LY294002 atrophied, even in the presence of NGF. Therefore, the metabolic effects of PI-3-K were examined. To determine the minimum concentration of LY294002 that is effective in inhibiting PI-3-K activity over prolonged periods of time, a dose–response analysis of LY294002 was performed. As a first step, sympathetic neurons were stimulated with NGF (100 ng/ml) or medium alone, in the presence of either LY294002 (50 μm) or Wortmannin (1–10 μm), and the activity of PI-3-K was measured directly using lipid kinase assays. NGF treatment lead to a ∼2.5-fold induction of PI-3-K activity (Fig.4A), similar to what has been reported previously for sympathetic neurons (Virdee et al., 1999). LY294002 completely inhibited NGF-dependent PI-3-K activity (Fig. 4A), but because LY294002 is a reversible inhibitor, its inhibitory effect could be washed out of the immune complexes if LY294002 was not included in subsequent immunoprecipitation and kinase buffers (data not shown). An irreversible PI-3-K inhibitor, Wortmannin, when added only to the culture medium immediately before and during the NGF treatment, also blocked PI-3-K activity at concentrations known to be maximal in intact cells (10 μm), with a lower concentration (1 μm) leading to a ∼60% inhibition (Fig.4A). In conjunction with the PI-3-K assays, Western analysis was performed on the supernatants from the lipid kinase assays performed above with antibodies specific for phosphorylated threonine 308 (T308) or phosphorylated serine 473 (S473) of Akt. Importantly, the phosphorylation of both residues, and thus the kinase activity, correlated with the level of catalytic activity of PI-3-K (Fig.4A). Therefore, Akt phosphorylation and activity were completely dependent on PI-3-K activity in 5 DIV sympathetic neurons, and thus monitoring the phosphorylation state of Akt was a reliable measure of the activity of PI-3-K in these neurons. To perform a dose–response analysis of LY294002, sympathetic neurons were treated with increasing concentrations of LY294002 in the presence of NGF for 2 d, after which detergent extracts were prepared from the cultures that were then subjected to P-Akt (S473) Western analysis (Fig. 4B). LY294002 (25 μmand 50 μm) completely inhibited Akt phosphorylation in the presence of NGF. Similar results were obtained with P-Akt (T308) immunoblotting as well (data not shown). Therefore, as in previous experiments, a concentration of 50 μm LY294002 was used for all subsequent treatments to inhibit PI-3-K activity completely in sympathetic neurons maintained in NGF.
Fig. 4.

PI-3-K regulates NGF-dependent metabolism of sympathetic neurons. A, Sympathetic neurons were treated for 3 min with medium alone, medium containing NGF (100 ng/ml), or pretreated with LY294002 (50 μm) or Wortmannin (1–10 μm) for 30 min and then treated with NGF in the continued presence of the respective PI-3-K inhibitor. Detergent extracts were produced, and lipid kinase assays (top panel) were performed on phosphotyrosine immunoprecipitates. P-Akt (S473 or T308) immunoblotting was performed on the supernatants from the lipid kinase assays (middle panels), and α-tubulin immunoblotting (bottom panel) was also performed to confirm equal protein loading. This experiment was performed twice with identical results, and the bar graph represents the means. B, Cultures of sympathetic neurons were treated with NGF (50 ng/ml) in the presence of increasing concentrations of LY294002 for 2 d. After this time, detergent extracts produced from these cultures were subjected to P-Akt (S473) immunoblotting (top panel). Equal protein loading was confirmed by reprobing the same immunoblot with antibodies directed against α-tubulin (bottom panel). C, Sympathetic neurons were treated with NGF (50 ng/ml), vehicle (DMSO, 0.1%), anti-NGF in the presence of BAF (50 μm), or NGF (50 ng/ml) with LY294002 (50 μm) and BAF (50 μm) for 2 d. The neurons were fixed and subjected to toluidine blue staining as described in Materials and Methods. Somal diameters of 100−120 neurons were determined from each condition from three independent cultures. Error bars represent SEM. The somal diameter of neurons in NGF medium was smaller than that normally observed because of the fixation, staining, and dehydration of the neurons. D, Sympathetic neurons were treated with NGF containing vehicle alone (DMSO, 0.1%), NGF with LY294002 (50 μm), NGF with LY294002 and BAF (50 μm), or anti-NGF with BAF for 2 d. Cultures were then subjected to MTT analysis as described in Materials and Methods. The data are presented as the percentage of MTT reduction compared with NGF-maintained neurons, and each condition was performed in quadruplicate in four independent cultures. Error bars represent SEM.
To determine whether PI-3-K activity regulates NGF-dependent growth of the cell body, the somal diameter of sympathetic neurons was measured after PI-3-K inhibition or NGF deprivation. BAF was included in all conditions to inhibit neuronal death. Neurons maintained in NGF for 7 d had an average somal diameter of 15 μm, whereas neurons maintained in NGF for 5 d and then deprived of NGF for 2 d had an average somal diameter of 8 μm (Fig. 4C). PI-3-K inhibition of 5 DIV, NGF-maintained sympathetic neurons caused a marked atrophy of the cell bodies to 10 μm (Fig. 4C). PI-3-K inhibition induced less somal atrophy than NGF deprivation, suggesting that although PI-3-K is required, it did not completely account for NGF-dependent growth of the neuronal cell body.
To examine whether PI-3-K also accounts for the metabolic effects of NGF, we performed MTT reduction assays, which measure the level of mitochondrial oxidative phosphorylation, on NGF-maintained sympathetic neurons subjected to PI-3-K inhibition. Two days of NGF deprivation in the presence of BAF resulted in a 70% loss of metabolic activity (Fig.4D). PI-3-K inhibition for the same amount of time in the presence of NGF decreased the metabolic activity of sympathetic neurons by 35% (Fig. 4D). Similar to LY294002 treatment alone, PI-3-K inhibition of NGF-maintained sympathetic neurons in the presence of BAF, which completely prevents LY-dependent death after 2 d (Fig.5A), displayed a 45% loss of metabolic activity, demonstrating that LY294002-dependent inhibition of metabolic function was not the cause of neuronal death (Fig.4D). Therefore, PI-3-K contributes to the growth-promoting and anabolic effects of NGF on sympathetic neurons.
Fig. 5.
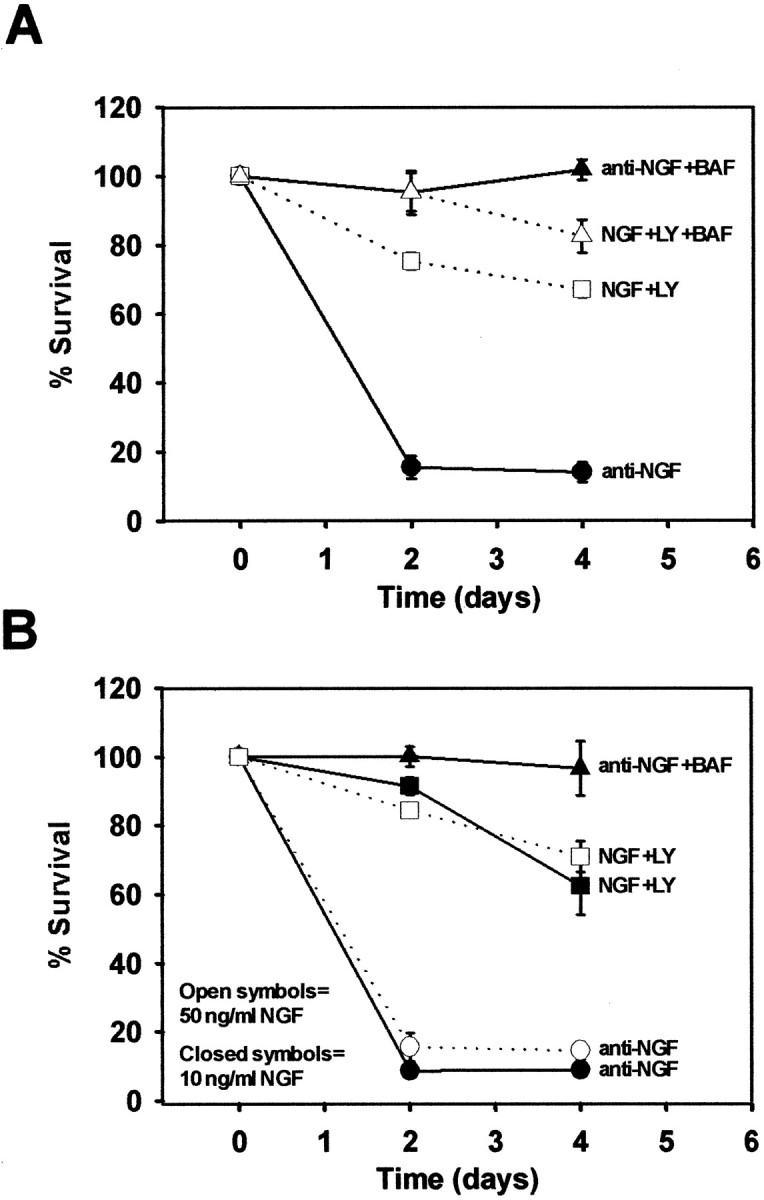
PI-3-K is not solely required for the survival-promoting activity of NGF. A, Sympathetic neurons were deprived of NGF in the absence or presence of BAF (50 μm) or were treated with LY294002 (50 μm) in the presence of NGF (50 ng/ml) with and without BAF. The neurons were fixed after 2−4 d. The neurons were stained with toluidine blue, and the number of surviving neurons was counted and graphed as a percentage (compared with age-matched NGF-maintained neurons). Each condition was performed in duplicate in four independent cultures.B, Sympathetic neurons maintained for 5 d in either 50 ng/ml NGF (open symbols) or 10 ng/ml NGF (closed symbols) were deprived of NGF or maintained in NGF in the presence of LY294002 (50 μm). In addition, sympathetic neurons maintained in 10 ng/ml NGF were also deprived of NGF in the presence of BAF (50 μm) to confirm that they could be rescued from apoptosis by a caspase inhibitor. Surviving neurons were counted from duplicate wells of each condition from two independent cultures. Error bars represent SEM.
PI-3-K is not required for the survival-promoting effects of NGF on sympathetic neurons
Because PI-3-K activity is not required for NGF-mediated inhibition of Bax translocation or cytochrome c release in sympathetic neurons, we hypothesized that PI-3-K inhibition in the presence of NGF would not activate caspases or induce apoptosis. To test this hypothesis, NGF-maintained sympathetic neurons were incubated with LY294002 for 2−4 d. The number of neurons that survived these treatments was ascertained by counting the number of Nissl-stained cells in each condition. In most experiments, detergent extracts were produced from parallel cultures treated with NGF or NGF in the presence of LY294002 for 2 d. These extracts were subjected to P-Akt Western analysis to confirm that LY294002 blocked PI-3-K activity (data not shown). We consistently observed that 70% of sympathetic neurons treated with LY294002 in the presence of NGF survived after 4 d, whereas <20% of neurons survived only 2 d of NGF deprivation (Fig. 5A). To determine whether the LY294002-induced death required activation of caspases, parallel NGF-maintained cultures were treated with LY294002 and BAF. Caspase inhibition blocked LY294002-dependent death after 2 d of treatment, but only partially blocked LY294002-dependent death after 4 d, in contrast to BAF treatment of NGF-deprived neurons, which rescued all neurons from death after 4 d. Taken together, these results indicate that PI-3-K did not completely account for NGF-dependent survival of sympathetic neurons in culture. All neurons treated with LY294002 in the presence of NGF atrophied (Fig. 5B). Therefore, although LY294002 was able to inhibit PI-3-K in all sympathetic neurons, resulting in a loss of somal diameter and metabolic activity, only 30% died after PI-3-K inhibition.
One explanation for the inability of PI-3-K inhibition to induce apoptosis is that maintaining sympathetic neurons in high concentrations of NGF (50 ng/ml) results in activation of survival pathways that would not normally be used in vivo. To test this hypothesis, sympathetic neurons were maintained in 50 ng/ml NGF as before, or in a lower concentration of NGF (10 ng/ml). Cultures maintained in 10 ng/ml NGF were phase bright and projected complex neuritic arborizations, as did the neurons maintained in 50 ng/ml NGF. However, the somal diameter of sympathetic neurons maintained in 10 ng/ml NGF was smaller than neurons maintained in 50 ng/ml NGF (data not shown). In addition, cell counts revealed that 85% of neurons survived in 10 ng/ml NGF as compared with neurons maintained in 50 ng/ml NGF (data not shown). When cultures maintained in 50 or 10 ng/ml NGF for 5 d were deprived of NGF, those maintained in 50 ng/ml NGF as well as those maintained in 10 ng/ml NGF underwent rapid apoptotic death (Fig. 5B). Sympathetic neurons maintained in 10 ng/ml NGF were completely rescued from NGF deprivation-induced apoptosis by caspase inhibition (Fig. 5B), similar to cultures maintained in 50 ng/ml NGF. To determine whether their susceptibility to PI-3-K inhibition differed, sympathetic neurons were treated with LY294002, as before, in the continued presence of either 50 or 10 ng/ml NGF for 2−4 d. In cultures maintained in 50 ng/ml NGF, only 25−30% of neurons died after 4 d of PI-3-K inhibition (Fig. 5B). A similar number of neurons died after 4 d of PI-3-K inhibition when maintained in only 10 ng/ml NGF (Fig. 5B). Therefore, susceptibility to PI-3-K inhibition does not depend on the concentration of NGF used to maintain the survival of sympathetic neurons, suggesting that even at a lower concentration of NGF, sympathetic neurons rely on additional NGF-regulated survival signals.
PI-3-K inhibition in the presence of NGF does not induce cytochromec release in sympathetic neurons
Because PI-3-K inhibition did not kill the majority of sympathetic neurons and because PI-3-K did not account for NGF-dependent inhibition of Bax translocation or cytochrome c release, we determined whether LY294002-treated sympathetic neurons were arrested in the cell death pathway after c-Jun phosphorylation, but before cytochromec release. Sympathetic neurons were deprived of NGF, maintained in NGF (50 ng/ml), or maintained in NGF with LY294002 for 2−4 d, all in the presence of BAF. After 2−4 d the neurons were fixed, and simultaneous immunocytochemical detection of cytochromec and P-Jun was assessed. NGF deprivation induced release of cytochrome c from 85% of sympathetic neurons after 2 d (Fig. 6A). PI-3-K inhibition in the presence of NGF, on the other hand, induced cytochrome c release in only 35% of neurons after 4 d (Fig. 6A). Although as many as 80% of sympathetic neurons were P-Jun positive shortly after LY294002 treatment (Fig.1D), many neurons lost c-Jun phosphorylation after several days, leaving only 35−40% of sympathetic neurons c-Jun positive after 4 d (Fig. 6B). In contrast, sympathetic neurons deprived of NGF maintained c-Jun phosphorylation in the presence of BAF (Fig. 6B), suggesting that PI-3-K inhibition, in contrast to NGF deprivation, did not induce sustained c-Jun phosphorylation in most neurons. Taken together, these data indicate that most sympathetic neurons do not complete the cell death pathway after PI-3-K inhibition but arrest, or perhaps abort, the cell death process before cytochrome c release. This conclusion is consistent with the observation that PI-3-K inhibition did not account for the ability of NGF to block Bax translocation or cytochromec release.
A similar percentage of neurons released cytochrome c after PI-3-K inhibition that maintained c-Jun phosphorylation for several days (Fig. 6B). In fact, many of the neurons that maintained c-Jun phosphorylation also released mitochondrial cytochromec, suggesting that these neurons completed the cell death pathway after PI-3-K inhibition (Fig. 6C). Interestingly, the size of this population of neurons is similar to the number of neurons that die after long-term PI-3-K inhibition (Fig.5A), suggesting that this population of sympathetic neurons may undergo an apoptotic cell death similar to NGF deprivation. LY294002-induced death, however, differed significantly from NGF deprivation-induced death in that 50% of the LY294002-susceptible neurons were not rescued by caspase inhibition (Fig. 5A). To determine whether protein synthesis inhibition protected sympathetic neurons from LY294002-induced death, NGF-maintained neurons were treated with LY294002 in the presence or absence of 1 μg/ml cycloheximide (CHX). In contrast to NGF deprivation-induced death, which is protected by protein synthesis inhibitors (Martin et al., 1988), CHX treatment exacerbated LY-induced death (Fig.7). In fact, only 40% of neurons survived concurrent treatment with LY294002 and CHX after 4 d (Fig. 7). Therefore, although PI-3-K inhibition-induced death displayed characteristics similar to the cell death pathway initiated by NGF deprivation, this death clearly differed from NGF deprivation-induced death in that it was not fully protein synthesis or caspase dependent.
Fig. 7.

Inhibition of protein synthesis exacerbates LY294002-induced death. Sympathetic neurons were maintained in NGF with vehicle alone (0.1% DMSO) or maintained in NGF with LY294002 (50 μm) in the presence or absence of CHX (1 μg/ml). In addition, neurons were deprived of NGF, and all conditions were maintained for 4 d. The cultures were then fixed and stained with toluidine blue, and the number of surviving neurons was counted. Each condition was performed in duplicate in two independent cultures, and error bars represent SEM.
DISCUSSION
TrkA activation regulates multiple signaling molecules such as PI-3-K, phospholipase C-γ (PLC-γ), and the MEK/MAPK cascade (Kaplan and Stephens, 1994; Greene and Kaplan, 1995; Segal and Greenberg, 1996). Sympathetic neurons are able to abort acutely the cell death pathway initiated by NGF deprivation at early and late time points on readdition of NGF. We found that NGF-dependent PI-3-K activity inhibited an early cell death event proximal to c-Jun phosphorylation. PI-3-K activity, however, was not required for NGF to inhibit the translocation of Bax from the cytoplasm to the mitochondria or the ability of NGF to inhibit the release of mitochondrial cytochrome c. Furthermore, the MEK/MAPK pathway did not account for any of these NGF-dependent events, suggesting that the MAPK pathway may be more important in regulating neuronal phenotype rather than survival, as suggested earlier (Virdee and Tolkovsky, 1995;Creedon et al., 1996; Virdee and Tolkovsky, 1996). Consistent with the observations that PI-3-K is not required for all of the survival-promoting effects of NGF, PI-3-K inhibition did not kill the majority of sympathetic neurons in the presence of NGF. In contrast to its effects on survival, PI-3-K did contribute to the NGF-dependent trophic status of sympathetic neurons as measured by the loss of somal diameter and metabolic function in the presence of PI-3-K inhibitors. The mechanisms by which NGF or PI-3-K promotes growth and metabolism are not understood. Consistently, 30% of sympathetic neurons died after PI-3-K inhibition in the presence of NGF, and this death displays some, but not all, of the properties of NGF withdrawal-mediated apoptosis.
PI-3-K is not required for the survival-promoting effects of NGF in sympathetic neurons
Three observations reported here support the conclusion that PI-3-K alone is not responsible for NGF-dependent survival of sympathetic neurons and that additional signaling pathways cooperate to maintain survival. First, PI-3-K did not mediate NGF-dependent inhibition of late cell death events such as Bax translocation and cytochrome c release (Fig. 8). Second, PI-3-K inhibition did not kill the majority of sympathetic neurons in the presence of NGF. Third, LY294002-induced death did not mimic apoptosis induced by NGF deprivation. Specifically, the time course of LY294002-stimulated death was considerably slower (Fig. 5), as reported previously (Crowder and Freeman, 1998). In addition, LY294002-induced death was not fully dependent on caspases (Fig. 5) and in fact was exacerbated by inhibition of protein synthesis (Fig. 7). This is in contrast to apoptosis of sympathetic neurons induced by NGF deprivation, which is completely protein synthesis and caspase dependent (Martin et al., 1988; Deshmukh et al., 1996). The inability of BAF and CHX to protect against LY294002-mediated death could be attributable in part to inhibition of other PI-3-K functions such as cytoskeletal regulation (Virdee et al., 1999) or metabolic activity (Fig. 4), although these functions should be lost after NGF deprivation as well. LY294002-stimulated death, however, had many of the features observed in NGF deprivation-induced apoptosis such as phosphorylation of c-Jun (Fig. 1), expression of proapoptotic genes such as cyclin D1 and c-jun (Estus et al., 1994; Freeman et al., 1994;Crowder and Freeman, 1998), and release of mitochondrial cytochromec (Fig. 6). Additionally, in the few neurons that did die, LY294002 treatment led to caspase activation as measured by CM-1 staining (data not shown), nuclear condensation, and terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) positivity (Crowder and Freeman, 1998; Vaillant et al., 1999).
Fig. 8.

Multiple signaling pathways regulate the cell death pathway in sympathetic neurons. This schematic diagram depicts three points at which NGF can inhibit further progression of the cell death pathway. PI-3-K activity is required for NGF-dependent inhibition of c-Jun phosphorylation but is not required for inhibition of Bax translocation to the mitochondria or release of cytochromec from the mitochondria. MEK/MAPK does not account for any of these NGF-dependent events, and the signaling pathways required for these two events remain unknown.
Although overexpression of PI-3-K is sufficient to prevent the death of sympathetic neurons deprived of NGF (Philpott et al., 1997; Crowder and Freeman, 1998), whether PI-3-K is necessary for NGF-dependent survival has been controversial (Philpott et al., 1997; Crowder and Freeman, 1998; Mazzoni et al., 1999; Vaillant et al., 1999; Virdee et al., 1999). Several methodological differences, such as variations in the culture conditions, complicate direct comparisons between these studies. Using culture conditions similar to those reported previously (Crowder and Freeman, 1998; Mazzoni et al., 1999; Vaillant et al., 1999), we found that PI-3-K inhibition with LY294002 promoted modest death of sympathetic neurons over 4 d. In all experiments reported here we confirmed that NGF deprivation in these cultures led to the rapid death of >80% of neurons after 2 d and that this death was completely prevented by caspase inhibition (Fig. 5). In addition, we frequently confirmed that each lot of LY294002 used was able to inhibit completely downstream Akt activation by P-Akt immunoblotting. The amount of death that we observed after LY294002 treatment was not greatly different from that reported by Crowder and Freeman (1998). However, we are more impressed with the differences than with the similarities between LY294002 treatment and NGF deprivation, which suggests that PI-3-K inhibition does not mimic NGF deprivation. Furthermore, because much more is now known about the terminal events involved in NGF deprivation-induced apoptosis, we were able to examine the function of PI-3-K in these events in greater detail, drawing us to the conclusion that PI-3-K alone is not required for NGF-dependent survival. Because PI-3-K inhibition decreased metabolic activity even when death of sympathetic neurons was prevented by caspase inhibitors (Fig. 6), relying solely on metabolic assays such as MTT reduction would lead to the erroneous conclusion that many neurons have died, when in fact they are atrophic but surviving. Furthermore, in experiments reported here, we used a concentration of 50 μm LY294002, which completely inhibited downstream Akt phosphorylation but was a lower concentration than that reported in several other studies. Although higher concentrations of LY294002 (100 μm) are reported to be more effective in causing death of sympathetic neurons (Crowder and Freeman, 1998), inhibition of PI-3-K is maximal by 25−50 μm LY294002, suggesting that higher concentrations of LY294002 may inhibit additional kinases that contribute to survival.
Cerebellar granule neurons require PI-3-K activity for IGF-I-mediated and insulin-mediated survival (D'Mello et al., 1997; Dudek et al., 1997; Miller et al., 1997). When treated with PI-3-K inhibitors in the presence of IGF-I or insulin, granule cells undergo apoptosis with a time course similar to survival-factor deprivation and exhibit similar morphological characteristics as well (D'Mello et al., 1997; Dudek et al., 1997; Miller et al., 1997). Furthermore, PI-3-K inhibition and survival-factor deprivation kill the same percentage of neurons, demonstrating that PI-3-K-independent subpopulations of neurons do not exist. The sensitivity of granule cells to PI-3-K inhibition under the conditions used in these experiments is in stark contrast to sympathetic neurons, which die more slowly and at a lower percentage as compared with NGF deprivation (Figs. 5, 6) (Crowder and Freeman, 1998). Taken together, the results reported here, in addition to other studies on NGF-dependent survival of sympathetic neurons, contrasted with insulin- and IGF-I-dependent survival of granule cells, support the conclusion that PI-3-K is not solely responsible for NGF-dependent survival of sympathetic neurons.
What additional signaling pathways cooperate to promote sympathetic neuron survival?
Inhibition of PI-3-K activity in NGF-maintained sympathetic neurons resulted in an arrest in the cell death pathway before mitochondrial cytochrome c release. The fact that PI-3-K was not required for NGF-inhibition of Bax translocation or cytochromec release suggests that an additional NGF-regulated pathway inhibits cell death at this point. This function could not be accounted for by L-type channel, p38MAPK, MEK, and PKA activity because inhibition of these molecules did not alter NGF-dependent inhibition of cytochrome c release. Although the signaling pathways used by TrkA to block these later cell death events are currently unknown, two possibilities include PLC-γ and Src family kinases (Atkinson et al., 1996; al-Ramadi et al., 1998; Hiwasa et al., 1998). In addition, although this study has focused on three NGF-regulated events in the cell death pathway, additional NGF-dependent signaling pathways exist, such as cAMP response element-binding protein-dependent transcriptional pathways (Bonni et al., 1999; Riccio et al., 1999) and the competence-to-die pathway (Deshmukh and Johnson, 1998). How NGF inhibits competence, as well as other cell death events, awaits further investigation.
Footnotes
This work was supported by National Institutes of Health Grants AG12947 and NS38651-01 (E.M.J.) and a National Institutes of Health Training Grant 5T32-NS07129 (B.A.T.-P.). We thank Patricia A. Osborne for expert technical assistance and critical reading of this manuscript. We also thank Dr. Mohanish Deshmukh, Krista Moulder, Charles Harris, Louis Chang, and Dr. Cynthia Tsui-Pierchala for their thoughtful scientific discussions and critical reading of this manuscript, and Mary Bloomgren for secretarial assistance.
Correspondence should be addressed to E. M. Johnson Jr, Department of Molecular Biology and Pharmacology, Washington University School of Medicine, 4566 Scott Avenue, Box 8103, St. Louis, MO 63110. E-mail:ejohnson@pcg.wustl.edu.
REFERENCES
- 1.al-Ramadi BK, Zhang H, Bothwell AL. Cell-cycle arrest and apoptosis hypersusceptibility as a consequence of Lck deficiency in nontransformed T lymphocytes. Proc Natl Acad Sci USA. 1998;95:12498–12503. doi: 10.1073/pnas.95.21.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkinson EA, Ostergaards H, Kanes K, Pinkoski MJ, Caputo A, Olszowy MW, Bleackley RC. A physical interaction between the cell death protein Fas and the tyrosine kinase p59fynT. J Biol Chem. 1996;271:5968–5971. doi: 10.1074/jbc.271.11.5968. [DOI] [PubMed] [Google Scholar]
- 3.Auger KR, Serunian LA, Cantley LC. Separation of novel polyphosphoinositides. In: Irvine RF, editor. Methods in inositide research. Raven; New York: 1990. pp. 159–166. [Google Scholar]
- 4.Biggs I, WH, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 6.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 7.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 8.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 9.Creedon DJ, Johnson EM, Lawrence JC. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- 10.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 12.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 13.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 15.Deshmukh M, Johnson EM., Jr Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- 16.Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 17.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudek H, Datta SA, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:628–830. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 20.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 22.Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 23.Greene LA, Kaplan DR. Early events in neurotrophin signaling via Trk and p75. Curr Opin Neurobiol. 1995;5:579–587. doi: 10.1016/0959-4388(95)80062-x. [DOI] [PubMed] [Google Scholar]
- 24.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 25.Hiwasa T, Nakamura Y, Sakiyama S. Isolation of a variant which has reduced Src activity from SH-SY5Y neuroblastoma cells. Anticancer Res. 1998;18:3311–3314. [PubMed] [Google Scholar]
- 26.Kaplan DR, Stephens RM. Neurotrophin signal transduction by the trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 27.Korsching S. The neurotrophic concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 29.Lipscomb EA, Sarmiere PD, Crowder RJ, Freeman RS. Expression of the SM-20 gene promotes death in nerve growth factor-dependent sympathetic neurons. J Neurochem. 1999;73:429–432. doi: 10.1046/j.1471-4159.1999.0730429.x. [DOI] [PubMed] [Google Scholar]
- 30.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal cell death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Biochemical characterization of programmed cell death in NGF-deprived sympathetic neurons. J Neurobiol. 1992;23:1205–1220. doi: 10.1002/neu.480230911. [DOI] [PubMed] [Google Scholar]
- 32.Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci. 1999;19:9716–9727. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor 1-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 34.Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Putcha GV, Deshmukh M, Johnson EM., Jr Bax translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Putcha GV, Deshmukh M, Johnson EM., Jr Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J Cell Biol. 2000;149:1011–1017. doi: 10.1083/jcb.149.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 39.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 40.Shimoke K, Yamagishi S, Yamada M, Ikeuchi T, Hatanaka H. Inhibition of phosphatidylinositol 3-kinase activity elevates c-Jun N-terminal kinase activity in apoptosis of cultured cerebellar granule neurons. Dev Brain Res. 1999;112:245–253. doi: 10.1016/s0165-3806(98)00172-2. [DOI] [PubMed] [Google Scholar]
- 41.Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Virdee K, Tolkovsky AM. Activation of p44 and p42 MAP kinases is not essential for the survival of rat sympathetic neurons. Eur J Neurosci. 1995;7:2159–2169. doi: 10.1111/j.1460-9568.1995.tb00637.x. [DOI] [PubMed] [Google Scholar]
- 43.Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- 44.Virdee K, Xue L, Hemmings BA, Goemans C, Heumann R, Tolkovsky AM. Nerve growth factor-induced PKB/Akt activity is sustained by phosphoinositide 3-kinase dependent and independent signals in sympathetic neurons. Brain Res. 1999;837:127–142. doi: 10.1016/s0006-8993(99)01643-1. [DOI] [PubMed] [Google Scholar]
- 45.Vogelbaum MA, Tong JX, Rich KM. Developmental regulation of apoptosis in dorsal root ganglion neurons. J Neurosci. 1998;18:8928–8935. doi: 10.1523/JNEUROSCI.18-21-08928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]