

Abstract
GluR3 autoantibodies have been implicated in the development of Rasmussen's encephalitis, a rare neurodegenerative disease of humans characterized by epilepsy and degeneration of a single cerebral hemisphere. GluR3 autoantibodies are found in some Rasmussen's encephalitis patients, and GluR3 antibodies raised in rabbits destroy cultured cortical cells in a complement-dependent manner. In this study, the cellular targets of anti-GluR3 antisera-mediated cytotoxicity were examined in mixed primary neuronal–glial cultures of rat cortex. Unexpectedly, astrocytes were the principal target of the cytotoxic effects as assessed by immunohistochemistry and lactate dehydrogenase activity; neurons were destroyed to a lesser extent. Astrocyte vulnerability was rescued by transfection with complement regulatory proteins, and neuronal resistance was defeated by impairing complement regulatory protein function. Astrocyte death may occur in Rasmussen's encephalitis, and destruction of this cell type may play a critical role in the progression of this disorder. The present findings suggest complement regulatory protein expression may in part determine the nature and severity of Rasmussen's encephalitis and other complement-dependent nervous system diseases and thus underscore the need for a systematic investigation of the expression of all known complement regulatory proteins in healthy and diseased nervous system tissues.
Keywords: GluR3 autoantibody, Rasmussen's encephalitis, complement, complement regulatory protein, autoimmunity, epilepsy, glutamate receptor
Rasmussen's encephalitis (RE) is an intractable epilepsy generally beginning during childhood or adolescence and characterized by progressive degeneration of a single cerebral hemisphere. Progressively worsening unilateral seizures and unihemispheric neurological problems, such as language deficits, hemiparesis, and hemisensory loss, develop in parallel with the hemispheric atrophy. Microscopic examination of resected brain tissue reveals focal inflammatory changes localized primarily to the cortex of the affected hemisphere, including microglial nodules, perivascular lymphocytic cuffing, meningeal infiltrates, neuronal loss, and gliosis (Rasmussen et al., 1958). Because the seizures are unresponsive to standard antiseizure pharmacotherapy, hemispherectomy is the standard treatment, typically leaving the child free of seizures but with a severe neurological deficit (Rasmussen et al., 1958; Oguni et al., 1991; Robitaille, 1991).
Although the etiology and pathogenesis of RE and the mechanism limiting the process to one hemisphere have yet to be completely elucidated, a series of recent studies has established autoantibodies and complement (C′) as possible agents in disease progression. Immunization of rabbits with the glutamate receptor subunit GluR3 serendipitously led to an animal model recapitulating many of the principal features of RE and in turn to the discovery that many RE patients harbor antibodies directed at GluR3 (Rogers et al., 1994). Importantly, seizure severity and frequency are reduced in some RE patients treated with plasmapheresis or IgG-selective immunoadsorption, in parallel with reduction of GluR3 antibody titers (Rogers et al., 1994; Andrews et al., 1996; Antozzi et al., 1998). Additionally, some antibodies collected from immunized rabbits or humans with RE elicit excitatory currents in cultured neurons (Twyman et al., 1995). We have also found that rabbit anti-GluR3 can destroy primary mixed neuronal–glial cultures in a C′-dependent manner (He et al., 1998). Additional support for IgG and C′ in RE pathogenesis emerged from immunohistochemical studies revealing IgG and C′ deposition on neurons in resected tissue from a subset of RE but not complex partial epilepsy patients (He et al., 1998; Whitney et al., 1999). These observations, along with the fact that most GluR3 in mixed neuronal–glial cultures are found on neurons (He et al., 1998), led to the hypothesis that anti-GluR3 binds selectively to neurons in culture, leading to antibody-dependent C′ activation in the vicinity of the neurons and selective neuronal destruction. The experiments described here reveal that anti-GluR3 unexpectedly does not selectively destroy neurons. Not only does anti-GluR3 destroy both neurons and astrocytes, neuronal death is more restricted and occurs more slowly than does astrocyte destruction. Neuronal resistance is reduced by impairing complement regulatory protein (CRP) function, and astrocyte resistance is improved by overexpression of CRPs.
MATERIALS AND METHODS
Unless otherwise indicated, all reagents were from Sigma (St. Louis, MO).
Production of antisera and purification of IgG. White New Zealand male rabbits were immunized as described previously (Rogers et al., 1994; He et al., 1998). Briefly, rabbits weighing 2.5 kg were injected subcutaneously with 100 μg of glutathioneS-transferase (GST)-GluR3, GST,trpE-GluR3, or trpE in complete Freund's adjuvant. The GST-GluR3 fusion protein contains GluR3 residues 246–455, whereas trpE-GluR3 encompasses residues 246–458 (Keinanen et al., 1990). Booster injections, given in incomplete Freund's adjuvant, were given 2 weeks after the initial immunization and then at 4 week intervals. Serum was obtained by allowing blood to clot at 4°C followed by centrifugation. Sephadex G-50 chromatography was used to reduce the concentration of glutamate and other small molecular weight substances in serum. Serum diluted 1:1 in HBSS–HEPES (Life Technologies, Rockville, MD) was passed sequentially over two Sephadex G-50 columns (Amersham Pharmacia Biotech, Piscataway, NJ). Serum filtrates were filter-sterilized, aliquotted, and stored at −80°C. BecausetrpE-GluR3 and GST-GluR3 antisera are functionally indistinguishable in the experiments reported here, they will be collectively referred to as “anti-GluR3” or “GluR3 antisera.” TrpE and GST antisera will be referred to as “control antisera” for similar reasons.
Purified IgG was prepared with protein G agarose (Pierce, Rockford, IL) affinity chromatography according to the instructions of the manufacturer. The eluted immunoglobulin fraction was concentrated using Centricon centrifugal concentrators with a nominal molecular weight cutoff of 50 kDa (Millipore, Bedford, MA) and then dialyzed in four exchanges of PBS. Filter-sterilized purified IgG was stored until use at 4°C.
Tissue culture. Mixed neuronal–glial cultures were prepared as described previously (He et al., 1998). Briefly, day 18 embryos were removed by cesarean section, the cerebral cortices were dissected, and the meninges were removed with forceps. Cortices were rinsed with Ca2+- and Mg2+-free HBSS (containing 1 mm HEPES; Life Technologies), minced, and then incubated with 0.25% trypsin in HBSS for 20 min at 37°C. Brain tissue was then rinsed twice in HBSS followed by two washes in plating media (minimal essential medium [MEM] supplemented with 3 mg/ml glucose, 5% fetal bovine serum, and 5% horse serum; all from Life Technologies) containing 10 μg/ml DNase (Life Technologies). The tissue was then dispersed by trituration through a fire-polished Pasteur pipette. After centrifugation and resuspension, cells were plated at 860,000 cells/ml in 90 μl/well in 96-well plates or glass 16-well chamber slides in plating media. Cultures were maintained at 37°C in 5% CO2–95% air in a humidified incubator until used for experiments on 14–16 days in vitro(DIV). The media was not exchanged during this time period. The numbers of neurons and glia as assessed by quantification of GFAP- and neuronal-specific nuclear protein (NeuN)-immunostained cells (see below) were found to vary by <10% from experiment to experiment; astrocytes represented ∼43% of the cells, with neurons making up the rest.
Transfection. Cultures were transfected using the Fugene 6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN) according to a method developed by VanDongen and colleagues (T. VanDongen [Duke University, Durham, NC], personal communication). Briefly, Fugene 6 was diluted 1:14 into Neurobasal medium (Life Technologies) and incubated at room temperature (RT) for 10 min. In a separate tube containing Neurobasal, each plasmid to be transfected was added at 2 μg/100 μl. Diluted Fugene was mixed 1:1 with the DNA mix, incubated at RT for 5 min, and then added to DIV 10 cultures. After 30 min at 37°C, the transfection mix was replaced with the original growth medium. Transfected cells were used on DIV 14. This method produced transfection efficiencies of ∼1% with minimal toxicity.
Expression vectors containing green fluorescent protein (GFP), CD8, or CD59 were kindly provided, respectively, by Drs. Donald Lo (Duke University), Brian Seed (Massachusetts General Hospital, Boston, MA), and B. P. Morgan (University of Cardiff, Cardiff, UK). A Crry expression vector was constructed using a PCR amplification product obtained from reverse-transcribed rat primary glial culture poly(A+) RNA. The primers used for the PCR amplification were as follows (5′ to 3′): (upstream primer) GAT CGA GCG GCC GCT ATG GAG GCT TCT TCG CCT CTG GAC CCC and (downstream primer) AAT ATC CTC GAG TGG CGG CTA TTA GAC TTC TTG AGT GAG. The upstream primer contains a NotI site ahead of the start codon, and the downstream primer contains an XhoI site downstream of the stop codon. The PCR product was gel-purified, digested withNotI and XhoI, and inserted at these sites into pcDNA3 (Invitrogen, Carlsbad, CA). The construct was confirmed by restriction and sequence analysis. The Crry cDNA isolated contains three divergent nucleotides compared with the original published sequence (Sakurada et al., 1994; Quigg et al., 1995) (the first, at base 1145 encodes a silent mutation; the second, at base 1272, encodes a methionine in place of a valine; and the third, at base 1545, encodes leucine in place of phenylalanine). In preliminary immunocytochemistry experiments in which GFP was cotransfected with CD59 or CD8, ∼80% of GFP-positive astrocytes were also positive to varying degrees for the second transfected gene. GFP fluorescence was therefore used in cell counting (see below) as a surrogate indicator of transfection with the second gene.
HEK-293 cells maintained in DMEM (Life Technologies) containing 10% FBS were transfected using the calcium phosphate method as described previously (He et al., 1998). HEK cultures were transfected with either pCMV-GluR3 or pCMV (generous gifts of Ray Dingledine, Emory University, Atlanta, GA).
Toxicity assays. On the day of the experiment, cultures were exposed to various compounds in MEM containing 100 μm APV (Tocris Cookson, Ballwin, MO). Control wells were incubated in MEM–APV alone. In some experiments, before addition of toxins, cultures were incubated for 30 min with phosphatidylinositol-specific phospholipase C (PI-PLC) (ICN Biochemicals, Costa Mesa, CA) diluted to 0.3–0.6 U/ml in MEM–APV followed by one wash in MEM. PI-PLC was heat-inactivated by boiling for 1 hr. In other experiments, cultures were exposed to vehicle or anti-GluR3 in the presence of 100 μg/ml anti-CD59 (monoclonal antibody clone 6D1, IgG1 isotype, generously provided by B. P. Morgan) or a nonspecific IgG1 isotype control (American Type Culture Collection clone MOPC-31C). This anti-CD59 antibody neutralizes cell-surface CD59 without itself activating C′ (Hughes et al., 1992;Rushmere et al., 1997). The soluble C′ inhibitor sCR1 (Avant Immunotherapeutics, Needham, MA) was added in some experiments at a final concentration of 200 μg/ml. Thirty minutes to 24 hr after the beginning the experiment, cell death was assessed by measuring lactate dehydrogenase (LDH) activity released from cells, by a spectrophotometric method (Vassault, 1983). In some experiments, propidium iodide was added to a final concentration of 5 μg/ml 30 min before the end of the experiment to label dying cells.
Immunocytochemistry. All steps were performed at room temperature on a shaker plate unless otherwise indicated. In some experiments, unfixed cells were exposed to antisera for 30 min on ice to minimize nonspecific antibody internalization, followed by three washes in PBS. In other experiments, cultures were exposed to primary antibody after fixation. Cells were fixed for 30 min in 4% paraformaldehyde–0.075% glutaraldehyde in 0.1 mphosphate buffer, pH 7.4. After three washes in PBS, cells were permeabilized in blocking buffer (PBS containing 2% BSA, 0.1% Triton X-100, and 2% donkey serum). Cells were then incubated with primary antibody [13 μg/ml anti-GFAP or 8 μg/ml anti-microtubule-associated protein 2 (MAP-2) (Sigma); 2.5 μg/ml anti-NeuN (Chemicon, Temecula, CA); anti-growth-associated protein 43 (GAP-43), 1:200 ascites (J. H. P. Skene, Duke University); and 1.25 μg/ml anti-Crry or 1.6 μg/ml anti-CD59 clone 1B4 (B. P. Morgan)] diluted in blocking buffer for 1 hr at RT or overnight at 4°C. Cultures were washed in blocking buffer and incubated for 1 hr in secondary antibody [1 μg/ml biotinylated anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA); 2 μg/ml anti-mouse IgG1-FITC (ICN Biochemicals); and 1.2 μg/ml biotinylated anti-mouse IgG2b (Zymed, South San Francisco, CA)] diluted in blocking buffer. To visualize biotinylated secondary antibodies, cultures were washed three times in PBS containing 2% BSA and 0.1% Triton X-100 (PBS+) and incubated for 1 hr in Vectastain ABC reagent (1:100; Vector Laboratories, Burlingame, CA) diluted in PBS containing 0.1% Triton X-100. In some experiments, the immunocytochemical signal was developed in 175 mm sodium acetate containing 0.001% H202, 0.25 mg/ml 3–3′ diaminobenzidine tetrahydrochloride, and 6.25 mg/ml NiSO4. In other experiments, cells were treated with biotinyl tyramide reagent (1:200; NEN Life Science Products, Boston, MA) in PBS for 15 min, followed by three washes and a 60 min incubation with streptavidin-linked rhodamine (4 μg/ml; Molecular Probes, Eugene, OR) in PBS+. After three washes in 175 mm sodium acetate, cultures were dried, mounted in Vectashield (Vector Laboratories), and examined in a fluorescent microscope. Control experiments confirming the specificity of staining involved omitting the primary antibody or substituting the primary antibody with one of a different species or isotype; in all cases, no specific cellular labeling was observed.
Cell counting. In a subset of experiments, cells transfected with GFP, labeled with propidium iodide, or stained with various antibodies were counted in a blinded manner. For cells transfected with GFP, all GFP-positive astrocytes and neurons present in each well were counted. For the other labeling techniques, three random fields of antibody-stained cells or propidium iodide-stained nuclei were counted in each well and averaged. Astroglia, labeled with anti-GFAP, and neuronal somata, stained with anti-MAP-2, were counted if the cell body was intact. Neuronal nuclei, labeled with anti-NeuN, were counted if the nucleus was not shrunken. A nucleus was defined as shrunken if it was <10 μm in diameter and was considered indicative of neuronal damage because the majority of neuronal nuclei were of this size after treatment with the neurotoxin kainic acid (KA). In contrast, most nuclei in untreated or vehicle-treated cultures were 12–16 μm in diameter.
Data analysis. Differences among experimental conditions were ascertained by t test and ANOVA with post hoc Tukey t tests and are summarized in Tables1 and 2. Statistical significance was established at p < 0.05.
Table 1.
Summary of cell count statistical comparisons performed
| Endpoint | Condition 1 | Condition 2 | pvalue | Condition 3 | p value | Condition 4 | p value |
|---|---|---|---|---|---|---|---|
| Experiment#1 | Vehicle | Anti-GluR3 | Control sera | l-AA (*) | |||
| % Astrocyte death after 24 hr | 0 | 85 | <0.001 | 4 | NS | 66 | <0.001 |
| Vehicle | Anti-GluR3 | Control sera | l-AA (*) | ||||
| % Neuron death after 24 hr | 0 | 47 | <0.001 | 0 | NS | 38 | <0.05 |
| Astrocyte | Neuron | ||||||
| % Cell death after GluR3 antisera for 24 hr | 85 | 47 | <0.001 | ||||
| Experiment #2 | Without sCR1 | With sCR1 (*) | |||||
| % Astrocyte death after GluR3 antisera for 24 hr | 85 | 22 | <0.01 | ||||
| Without sCR1 | With sCR1 (*) | ||||||
| % Neuron death after GluR3 antisera for 24 hr | 47 | 10 | <0.03 | ||||
| Experiment #3 | Vehicle | Anti-GluR3 | |||||
| % Propidium iodide+ astrocytes after 4 hr | 12 | 47 | <0.01 | ||||
| Vehicle | Anti-GluR3 | ||||||
| % Propidium iodide+ neurons after 4 hr | 6 | 7 | NS | ||||
| Astrocytes | Neurons | ||||||
| % Propidium iodide+ cells after anti-GluR3 for 4 hr | 47 | 7 | <0.001 | ||||
| Experiment #4 | Astrocytes | Neurons | |||||
| % Cells labeled by anti-GluR3 | <1 | 66 | <0.001 | ||||
| Control sera | Anti-GluR3 | ||||||
| % Neurons labeled by antisera | 7 | 66 | <0.001 | ||||
| Experiment #5 | Astrocytes | Neurons | |||||
| % Cells labeled by anti-CD59 | 4 | 97 | <0.0001 | ||||
| Experiment #7 | Vehicle treat | PI-PLC treat | CD59 treat | MOPC treat | |||
| % Propidium iodide+ neurons after anti-GluR3 for 4 hr | 7 | 72 | <0.001 | 37 | <0.01 | 11 | NS |
| Vehicle treat | PI-PLC treat | CD59 treat | MOPC treat | ||||
| % Propidium iodide+ astrocytes after anti-GluR3 for 4 hr | 47 | 80 | <0.05 | 49 | NS | 32 | NS |
| Experiment #10 | |||||||
| CD8 transfection | CD59 transfection | Crry transfection | |||||
| % Astrocyte death after anti-GluR3 for 4 hr | 40 | 8 | <0.05 | 8 | <0.05 | ||
| CD8 transfection | CD59 transfection | Crry transfection | |||||
| % Astrocyte death afterl-AA for 4 hr | 82 | 86 | NS | 82 | NS | ||
| CD59 transfection | Crry transfection | ||||||
| % Astrocyte death after anti-GluR3 for 4 hr | 8 | 8 | NS |
Cell count data was collected for seven of the sets of experiments reported in this paper. Within each experiment, one or more comparisons were made between the various experimental conditions tested in each experiment. Each line represents one set of comparisons between the cell count data for each of the experimental conditions (Conditions 1–4, labeled individually for each entry), and thep values, derived from Student's t tests or Tukey t tests performed post hoc to one- or two-way ANOVA, reflect the results of statistical comparisons among the conditions. For all but the last three lines, all cells in three random fields per well were counted, commonly amounting to ∼200 cells per field in the control conditions (and fewer in the toxin-treated wells). For the experiments summarized in the last three lines, all GFP-transfected cells were counted in each well. Typically, at least three wells were tested per condition per experiment. Each experiment was performed at least three times, except where indicated with an asterisk, when the experiment was performed twice with equivalent results. The data represent pooled results from replicate experiments.
Table 2.
Summary of LDH assay statistical comparisons performed
| Endpoint | Condition 1 | Condition 2 | p value |
|---|---|---|---|
| Experiment#6 | Vehicle pretreat | PI-PLC pretreat | |
| LDH release (% vehicle) after anti-GluR3 antisera for 24 hr | 205 | 549 | <0.001 |
| Experiment #8 | Vehicle cotreat | CD59 cotreat | |
| LDH release (% vehicle) after anti-GluR3 antisera for 24 hr | 165 | 218 | <0.01 |
| Vehicle cotreat | CD59 cotreat | ||
| LDH release (% vehicle) after KA for 24 hr | 250 | 250 | NS |
| Experiment #9 | Vehicle | Anti-GluR3 IgG | |
| LDH release (% vehicle) in presence of C6d serum + C6 | 100 | 132 | <0.05 |
Each line presents the results of comparisons between Conditions 1 and 2; p values are from Tukey t tests performed post hoc to two-way ANOVA run for each experiment. Each number represents enzymatic activity units present in culture supernatants, expressed as percent LDH values from vehicle-treated control wells, and is derived from at least three experiments in which three wells tested per condition were tested.
RESULTS
Cellular specificity of GluR3 antiserum cytotoxicity
To test the prediction that exposure of mixed neuronal–glial cultures to GluR3 antisera should selectively destroy neurons, GFAP-immunoreactive glia and Neu-N-immunopositive neurons were counted in cultures exposed for 24 hr to various antiserum preparations. Unexpectedly, most astrocytes (85%) and significantly fewer neurons (47%) were destroyed by each of six GluR3 antisera raised against either the trpE-GluR3 or GST-GluR3 fusion proteins (Fig.1; Table 1, Experiment #1). One anti-GluR3 antiserum was not toxic at the concentrations tested here (0.5–1.0 mg/ml) and was excluded from this cell death analysis. In contrast, neither vehicle alone, equal concentrations of four control antisera (Fig. 1; Table 1, Experiment #1), nor commercial C′ (data not shown) produced significant destruction of cells when compared with untreated cultures. Importantly, the C′ inhibitor sCR1 reduced anti-GluR3-mediated death of astrocytes and neurons to 22 and <10%, respectively (Table 1, Experiment #2). sCR1 also prevented cell death as assessed by LDH assays (data not shown), confirming our previous report (He et al., 1998). Interestingly, the selective astrotoxinl-amino adipate (l-AA) (Khurgel et al., 1996) likewise destroyed not only most astrocytes (66%) but also a significant number of neurons (38%) (Table 1, Experiment #1). Our recent demonstration (He et al., 1998) that cytotoxicity is eliminated by selective removal of GluR3 antibodies from GST-GluR3 antisera, together with the present observations of equivalent cytotoxicity with antisera raised against two distinct GluR3 fusion proteins, strongly implicate antibodies against GluR3 as the critical initiating factor.
Fig. 1.

Anti-GluR3 destroys astrocytes and neurons after 24 hr exposure. Mixed cultures were incubated with vehicle, various serum samples, or l-AA for 24 hr, fixed, and then immunostained for GFAP (white bars) or NeuN (black bars) to label astrocytes or neuronal nuclei, respectively. Data presented are averaged cell counts from three random fields per well from several experiments. Whereas anti-GluR3 (n = 6 antisera) destroy the majority of astrocytes and approximately half the neurons, control antisera (n = 5) destroy <10% of cells. The astrotoxinl-AA also destroys substantial numbers of both glia and neurons. *p < 0.05 compared with control groups; **p < 0.001 compared with vehicle or control serum-treated groups.
To begin to investigate the unexpected preferential destruction of astrocytes, cells were examined after 4 hr exposure to anti-GluR3 or other serum samples. In preliminary work, cell death as assessed by analysis of LDH release was first detected at 4 hr, increasing in magnitude during the subsequent 20 hr. Cultures were incubated with propidium iodide for 30 min before fixation to label dead or dying cells. In these experiments, astrocytes were labeled with anti-GFAP and neurons were stained for MAP-2 to facilitate qualitative evaluation of modest morphological changes. Although neurons and astrocytes both appeared normal in cultures exposed to vehicle alone, scattered confluent patches of astrocytes in cultures exposed to anti-GluR3 at 1 mg/ml were swollen or fragmented and propidium iodide-positive (Fig.2), indicating that the cells were either dead or dying. The remaining astrocytes in surrounding areas were propidium iodide-negative and of normal stellate morphology. Theleft panel in Figure 2 highlights the morphological changes seen in damaged (red arrows) compared with normal (green arrow) astrocytes. In contrast to the astrocytes, the great majority of neurons were healthy-appearing and propidium iodide-negative at this time point (Fig. 2, right panel, green arrows). Blinded cell counts (Table 1, Experiment #3) revealed that, whereas exposure to vehicle alone resulted in propidium iodide uptake in 12% of astrocytes, 47% of astrocytes had taken up propidium iodide after treatment with antisera. In contrast, <7% of neurons were stained by propidium iodide, irrespective of treatment.
Fig. 2.
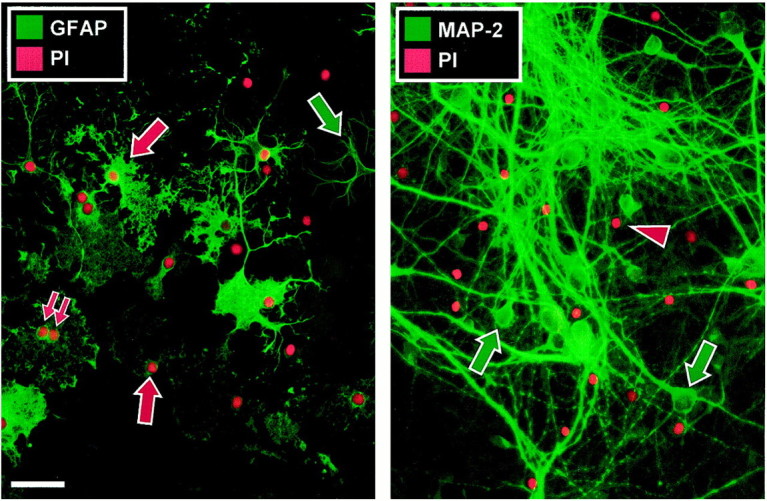
Anti-GluR3 selectively destroys astrocytes after 4 hr exposure. Mixed cultures exposed to vehicle or various antisera for 4 hr were incubated with propidium iodide (PI) for 30 min, fixed, and immunostained for GFAP or MAP-2. In cultures incubated with anti-GluR3 (1 mg/ml), approximately half the astrocytes (left) display a deranged morphology characterized by swelling, fragmentation, and lysis (red arrows). Binucleated astrocytes are a common feature (double red arrows). Other neighboring astrocytes appear morphologically normal (green arrow). Only deranged astrocytes are propidium iodide-positive, indicating damage to the plasma membrane occurring presumably as the cell begins to die (red nuclei). In contrast, most neurons appear morphologically normal and propidium iodide-negative at this time point (right, green arrows). The red arrowhead marks a propidium iodide-positive nucleus, possibly a dying astrocyte unlabeled by the MAP-2 antibody. Both astrocytes and neurons were comparatively unperturbed in vehicle-treated cultures (data not shown). The images presented are representative of seven experiments. Scale bar, 50 μm (pertains to this and all subsequent images).
One possible explanation for selective destruction of astrocytes at early time points by anti-GluR3 is that large numbers of antibodies might bind to these cells and fix C′. To explore antibody binding to glia, unfixed neuronal–glial cultures were incubated with various antisera or purified IgG for 30 min and then counterstained for GFAP or MAP-2. The percent of GFAP- or MAP-2-positive cells labeled by the various antisera was then determined by blinded cell counting. Whereas no more than 3% of astrocytes were labeled by any antiserum or antibody tested, on average 66% of neurons were labeled on their somata and processes by the cytotoxic GluR3 antisera (Fig.3; Table 1, Experiment #4). This staining pattern was observed with six different cytotoxic antisera and purified IgG analyzed in multiple experiments and is consistent with the predominantly neuronal expression of GluR3 in mixed neuronal–glial cultures (Eshar et al., 1993; He et al., 1998). Only 7% of neurons were labeled in cultures exposed to three control antisera (Table 1, Experiment #4), two commercial rabbit serum sources, or the single anti-GluR3 serum that was not cytotoxic at the concentrations used here. This last antiserum also failed to stain GluR3-transfected HEK cells, whereas the other GluR3 antisera stained GluR3 (but not mock-transfected) HEK cells (data not shown). Thus, only cytotoxic antisera contain anti-GluR3 antibodies capable of binding the antigen in a living cell, and when exposed to mixed neuronal–glial cultures, these antibodies preferentially bind neurons.
Fig. 3.
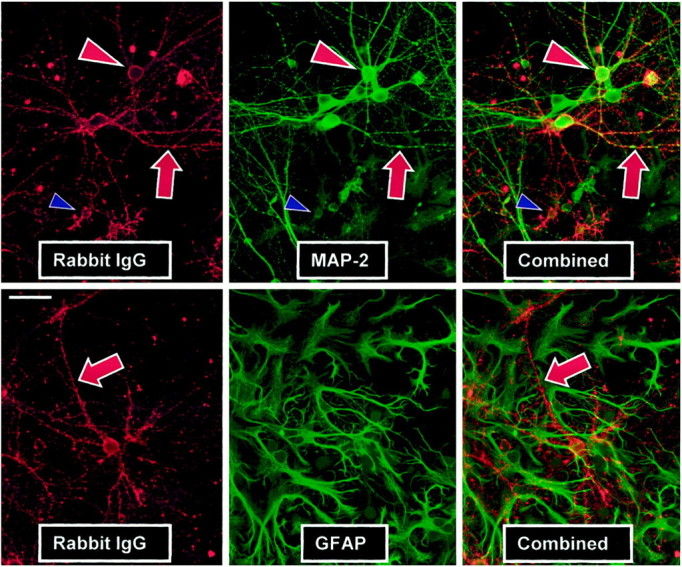
GluR3 antibodies bind exclusively to living neurons. Unfixed mixed cultures were incubated with various antisera, washed, fixed, and double-immunostained for rabbit IgG (left panels) and MAP-2 (top middle panel) or GFAP (bottom middle panel) to reveal neurons and astrocytes, respectively.Yellow structures in the overlaid or combined images (right panels) reveal concordant staining. All cytotoxic anti-GluR3 label neurons; results from a representative antiserum are presented. Cytotoxic antisera label most MAP-2-positive neurons but few GFAP-positive astrocytes (red arrowheads andarrows, respectively). Two of six GluR3 antisera that label neurons also label a small number of cells negative for GFAP and MAP-2 of possible oligodendroglial or microglial morphology (blue arrowheads). Six distinct noncytotoxic control rabbit sera all fail to substantially label cells, even at higher concentrations, with staining being limited to punctate labeling of particulate, noncellular debris similar to that seen in the background of the left panels (data not shown).
Cellular specificity of C′ regulatory protein expression
That neurons abundantly bind the IgG in GluR3 antisera predicts they would experience the greatest C′ attack. Yet neurons are not the cells most rapidly killed by GluR3 antisera. Instead, astrocytes, which apparently bind little high-affinity IgG in the GluR3 antisera, are rapidly destroyed by anti-GluR3. We hypothesized that neurons may resist the toxic effects of anti-GluR3 better than astrocytes because they express higher levels of CRPs than do astrocytes. CRPs are a family of proteins that protect self tissues from attack by inhibiting C′ activation and deposition (for review, see Ashgar, 1998). The fact that dying astrocytes appeared to be swollen suggested that CRP deficiencies had allowed membrane insertion of the cytotoxic membrane attack complex (MAC). The MAC is a multimeric 100 Å pore composed of the C′ factors C5b, C6, C7, C8, and multiple copies of C9, which allows ions and water free passage across the membrane of an attacked cell, causing cell swelling and lysis (Austyn and Wood, 1993). The CRP CD59 prevents MAC formation by preventing incorporation of C9 into the developing MAC (Morgan and Meri, 1994). To explore whether neurons express higher levels of CD59 than do astrocytes, cultures were stained with an anti-CD59 antibody (Fig. 4; Table1, Experiment #5). CD59 was found on virtually all (97%) GAP-43-positive axonal processes. In contrast, only 4% of GFAP-positive astrocytes expressed CD59. Importantly, substantial immunoreactivity was also found in these experiments on subsets of MAP-2-positive neuronal dendrites and neuronal somata (data not shown). Thus, whereas astrocytes express little CD59, neurons are coated from axon to dendrite with this protective protein.
Fig. 4.
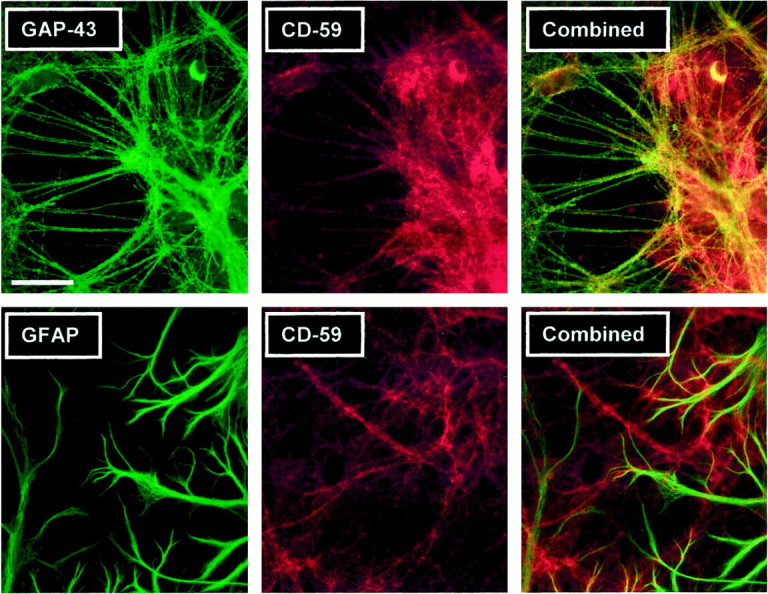
CD59 is expressed predominantly on neuronal structures. Mixed cultures were double-immunostained for CD59 (anti-CD59 antibody clone 1B4) and glial or neuronal markers. Most GAP-43-positive axonal structures are also CD59-positive (top panels), as seen when the left and middle panels are combined (right panel). In contrast, few GFAP-positive cells are CD59-positive (bottom panels). Staining with MAP-2 returned results similar to the GAP-43 staining (data not shown). These images are representative of four different experiments.
CRP inhibition, MAC formation, and cell death
The fact that CD59 is anchored to the cell membrane through a glycosylphosphatidyl inositol (GPI) linker can be exploited to test whether neuronal anti-GluR3 resistance is attributable to high CD59 expression levels. Cultures were treated with PI-PLC to remove GPI-anchored proteins such as CD59. As measured by LDH assay, PI-PLC-pretreated cultures were far more sensitive to low concentrations of anti-GluR3 than were vehicle pretreated cultures (Fig. 5, black vs white bars; Table 2, Experiment #6). The effects of PI-PLC were specific in that there was no significant effect on the toxicity produced by the neurotoxin KA or the astrotoxin l-AA. Moreover, basal cell viability, assessed in vehicle-challenged cultures, was unaffected by PI-PLC pretreatment. Heat-inactivated PI-PLC had no effect on the toxicity produced by any agent.
Fig. 5.

PI-PLC sensitizes cultures to C′-dependent toxins. Whereas exposure of vehicle-pretreated mixed cultures (white bars) to anti-GluR3 (0.4 mg/ml) produces only modest cytotoxicity, PI-PLC-pretreated cultures (0.6 U/ml for 30 min;black bars) are destroyed by anti-GluR3 (*p < 0.05). Pretreatment with PI-PLC does not dramatically enhance the toxicity produced by the non-C′-dependent toxins KA (80 μm) or l-AA (1 mm), and PI-PLC treatment is not toxic on its own. Heat-inactivated PI-PLC has no effect on the cytotoxicity produced by any compound (data not shown). Data are presented as percent unpretreated, vehicle-challenged control and are pooled from three experiments.
To determine whether PI-PLC pretreatment sensitizes otherwise resistant neurons to anti-GluR3, a set of immunocytochemistry experiments was performed in which vehicle- and PI-PLC-pretreated cultures were challenged for 4 hr with vehicle or anti-GluR3. Dying cells were labeled by incubation with propidium iodide during the last 30 min. The percent of GFAP- and MAP-2-positive cells also positive for propidium iodide was then determined by blinded cell counting. Whereas <7% of neurons in cultures pretreated with vehicle and challenged with anti-GluR3 were propidium iodide-positive, 72% of neurons in PI-PLC-pretreated cultures challenged with anti-GluR3 were propidium iodide-positive, bereft of neurites, and swollen (Table 1, Experiment #7). Images representative of three experiments are shown in Figure6 (top row). Astrocytes were also sensitized by PI-PLC pretreatment, with the percentage of astrocytes labeled by propidium iodide after 4 hr incubation with anti-GluR3 rising from ∼50% in the vehicle-pretreated cultures to >80% in the PI-PLC-pretreated cultures (Table 1, Experiment #7). No significant changes were observed in the numbers of propidium iodide-labeled astrocytes or neurons, or in their morphology, in vehicle-challenged wells, irrespective of pretreatment.
Fig. 6.
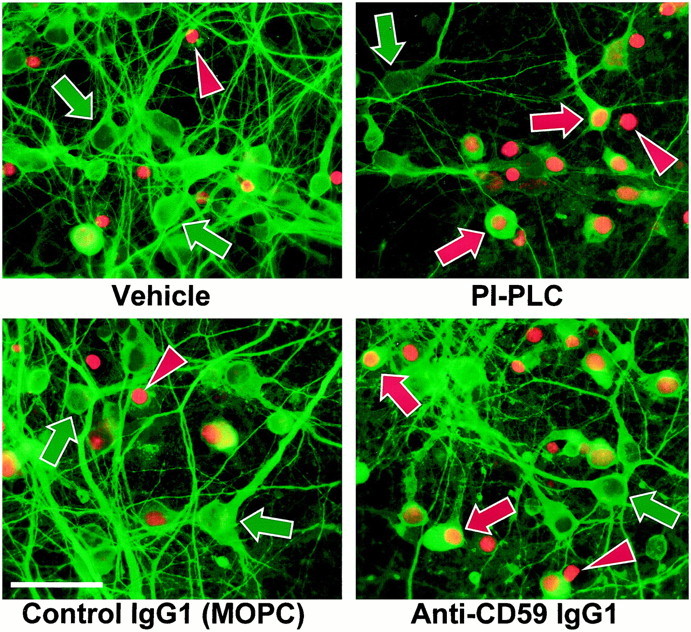
CRP debilitation sensitizes neurons to GluR3 antisera. In several experiments, mixed cultures (top panels) were pretreated with either vehicle or 0.3 U/ml PI-PLC and then challenged with either vehicle (data not shown) or GluR3 antisera (0.8 mg/ml), or were (bottom panels) exposed to either vehicle (data not shown) or GluR3 antisera (0.8 mg/ml) in the presence of 100 μg/ml of either neutralizing anti-CD59 (clone 6D1) or a nonspecific control IgG1 (MOPC). Note anti-CD59 and anti-MOPC are non-C′-fixing IgG1 (Hughes et al., 1992; Rushmere et al., 1997). Propidium iodide was added at 3.5 hr, and 30 min later the cultures were fixed and stained for GFAP (data not shown) or MAP-2. Whereas the majority of neurons in anti-GluR3-challenged cultures pretreated with vehicle or coincubated with anti-MOPC are normal-appearing and propidium iodide-negative (green arrows in left panels), many neurons in the PI-PLC-pretreated or anti-CD59 cotreated cultures appear swollen, deficient in neurites, and propidium iodide-positive after anti-GluR3 treatment (red arrows in right panels). A minority of neurons remains unaffected and healthy-appearing (green arrows). MAP-2-negative, propidium iodide-positive nuclei may represent lysed astrocytes (red arrowheads). In vehicle-challenged cultures, the numbers of propidium iodide-positive neurons and glia were not significantly affected by any pretreatment or cotreatment regimen (data not shown).
CD59 is just one of multiple proteins removed by PI-PLC. To directly test the role of CD59, a neutralizing anti-CD59 monoclonal antibody was used. Cultures were treated with GluR3 antisera in the presence of 100 μg/ml of this neutralizing antibody (clone 6D1) or a nonspecific control antibody (clone MOPC-31C). Note that neither of these antibodies can itself fix C′, being of the IgG1 isotype (Hughes et al., 1992; Rushmere et al., 1997). Whereas only 7% of neurons were propidium iodide-positive in cultures challenged with GluR3 antisera alone, 37% were positive in cultures challenged with GluR3 antisera in the presence of anti-CD59 (Table 1, Experiment #7). In contrast, only 11% of neurons were stained by propidium iodide in cultures challenged with GluR3 antisera in the presence of the MOPC control antibody. Images representative of four experiments are shown in the bottom row of Figure 6. Astrocyte death after challenge with anti-GluR3 was not appreciably affected by cotreatment with anti-CD59 (or anti-MOPC) (Table 1, Experiment #7), presumably reflecting the fact that these cells express little CD59. Furthermore, no significant changes in the numbers of propidium iodide-positive neurons or astrocytes were noted in cultures challenged with vehicle in either cotreatment group. As an independent indicator of cell death, LDH release was measured in cultures subjected to these cotreatment regimens. LDH release in anti-CD59, GluR3 antiserum cotreated cultures was 132% of that from cultures treated with anti-GluR3 alone (218 vs 165% of baseline control, respectively); cotreatment with anti-MOPC had no significant effect on cell death. Notably, KA toxicity was not affected by either antibody (LDH values ∼230% of baseline in each KA group) (Table 2, Experiment #8).
Together, these results indicate that GPI-anchored proteins such as CD59 play an important role in neuronal C′ defense. The greater neuronal death in GluR3-challenged cultures after PI-PLC treatment than anti-CD59 treatment (79 vs 37%) may reflect the fact that PI-PLC removes all GPI-anchored proteins, including other CRPs that may provide additional C′ defense. Similarly, enhanced astrocyte death after PI-PLC treatment may reflect removal of additional GPI-anchored CRPs from the membrane of this cell type.
Although the results of the CD59 inhibition experiments implicate MAC-mediated swelling and lysis as the mechanism of C′-mediated death, we could not directly assess MAC deposition on astrocytes (or on neurons with debilitated CD59) because the majority of commercially available anti-C′ antibodies are neither specific for rabbit C′ nor sufficiently robust to enable useful immunocytochemical analyses. Moreover, C′ cytotoxicity can in principle also result from the biological actions of small peptides liberated during the proteolytic processing of C′ factors C3 and C5. These peptides, C3a and C5a, potently activate cells bearing specific receptors such as T-cells, macrophages, neurons, astrocytes, and microglia (Armstrong et al., 1990; Yao et al., 1990; Austyn and Wood, 1993; Nolte et al., 1996;Nataf et al., 1999). Therefore, to determine by a functional endpoint whether anti-GluR3-mediated cell death results specifically from MAC deposition as opposed to C5a-mediated activation of microglia for example, cultures were exposed to IgG purified fromtrpE-GluR3 or trpE antisera in the presence of low concentrations of rabbit serum deficient in the C′ factor C6. Although C6-deficient serum (C6d) can be activated like normal serum, activation halts after processing C5 because incorporation of the downstream factors C7, C8, and C9 into the MAC is impossible without previous C6 incorporation. Thus, this serum produces normal amounts of C3a and C5a but no MACs. Cytotoxicity was observed only when C6d was coincubated with C6 and an anti-GluR3 antibody (Fig.7; Table 2, Experiment #9). The highly sensitive LDH assay was used in these experiments as opposed to direct cell counts because the effect was relatively modest compared with that presented, for example, in Figure 1. In the absence of C6, C6d was not toxic at concentrations up to 1.5 mg/ml, sevenfold more C6d than used in this experiment (data not shown). These findings indicate that GluR3 antibody-mediated cell death requires not only activation of the C′ cascade but also formation of the complete MAC channel.
Fig. 7.

GluR3 cytotoxicity requires MAC formation. In five experiments, mixed cultures were incubated with vehicle (white bars), purified trpE-GluR3 IgG; 200 μg/ml;hatched bars) or trpE IgG (200 μg/ml;black bars) alone (left set of bars) or in the presence of C6-deficient rabbit serum (C6d, 213 μg/ml;middle set of bars) or C6d reconstituted with human C6 (25 μg/ml; right set of bars). Elevated cytotoxicity is observed only when cultures are incubated with anti-GluR3 and reconstituted serum. *p < 0.05–0.001 compared with all other groups; #no difference compared with other unmarked groups.
CRP overexpression, cytoprotection, and the bystander hypothesis
The findings presented so far suggest that neuronal resistance results from high-level CRP expression and that astrocytes are lysed by MACs inserted into their membranes. However, although the C6d experiments indicate that MAC formation is essential for cell death, they do not prove MAC formation must occur specifically on astrocytes for astrocytes to die. For instance, MAC deposition on neurons may trigger the release of a compound that then kills astrocytes. One way to distinguish these possibilities is to examine the ability of transiently overexpressed CD59 to protect astrocytes against anti-GluR3-mediated destruction, because the role of CD59 is to prevent MAC formation. To address this question, cultures were cotransfected with GFP and CD59 and then challenged with anti-GluR3; the number of surviving GFP-positive astrocytes were compared with cultures cotransfected with GFP and CD8 as a control. Whereas anti-GluR3 destroyed ∼40% of CD8-cotransfected astrocytes (Fig.8, hatched vs white bar in left group of bars), anti-GluR3 destroyed <8% of CD59-cotransfected astrocytes (Fig. 8, middle set of bars; Table 1, Experiment #10). CD59 specifically protects against C′ attack becausel-AA destroyed >80% of astrocytes in both transfection conditions (black bars) (Table 1, Experiment #10). When combined with the C6d work, these observations indicate that MAC formation on astrocytes is required for astrocyte destruction.
Fig. 8.

Expression of either CD59 or Crry protects glia. Cultures transfected with GFP and CD8, CD59, or Crry were incubated for 4 hr with vehicle (white bars), anti-GluR3 (hatched bars; 0.8 mg/ml), or l-AA (black bars; 1 mm). Cultures were then fixed, and the total number of GFP-positive astrocytes were counted. Pooled data from seven experiments are presented and expressed as percent vehicle control. Compared with transfection with CD8, transfection with CD59 or Crry protects astrocytes against GluR3-mediated toxicity. l-AA destroys the majority of astrocytes in all transfection conditions. @ or *p< 0.05; # or ns, no difference.
If cell death is initiated by anti-GluR3 binding to GluR3 protein on neurons, with consequent C′ activation on this same cell type, how can MAC-dependent astrocyte death occur? One possibility is that C′ originally activated on neurons envelops and ultimately kills neighboring astrocytes. According to this “bystander” process, as the C′ cascade activates on neurons, a transiently water-soluble, incomplete MAC (C5b-7) deposits on both neurons and bystander astrocytes. To the extent a cell expresses CD59, construction of the complete MAC will be inhibited. The relative lack of CD59 on astrocytes suggests they would permit more construction of the full MAC than neurons and therefore suffer more C′-mediated lysis. This bystander mechanism has been established in C′-mediated destruction of other closely juxtaposed cell types (Park et al., 1997). An alternative possibility is that antibodies binding to neurons have no effect on astrocyte death at all; instead, small numbers of antibodies may bind to astrocytes and fix C′ directly on them. To distinguish these possibilities, the protective effect of overexpressing a CRP that acts at earlier stages of the C′ cascade was examined. Crry was chosen because (1) it is a multifunctional CRP that potently inhibits cascade activation and (2) immunocytochemistry experiments revealed Crry is selectively expressed on neuronal processes and not astrocytes, an expression pattern exactly matching that of endogenous CD59 (Fig. 4). If astrocytes are destroyed by the insertion of C5b-7, then Crry overexpression should fail to protect because this CRP acts upstream of C5b-7 formation. If the bystander process is not involved, however, and C′ activation is initiated directly on astrocytes, then astrocytes expressing Crry should be protected because the protein will halt the cascade as soon as it is activated. Only 8% of Crry-transfected astrocytes were destroyed by anti-GluR3, a level of protection equivalent to that afforded by CD59 transfection (Fig. 8,right set of bars; Table 1, Experiment #10). Importantly, l-AA destroyed >80% of astrocytes in this group as well, confirming the C′-specificity of Crry protection (Table 1, Experiment #10). That transfection of either CD59 or Crry protects astrocytes against the toxic effects of GluR3 antiserum implies that both early and late C′ cascade components are deposited on astrocytes and that bystander processes do not appear to contribute significantly to astroglial death.
DISCUSSION
Overview
This study investigates the cytotoxic actions of anti-GluR3 in primary cultures containing a mixture of astrocytes and neurons. The principal findings are fivefold. (1) Whereas the majority of GluR3 antibodies binds rapidly to neurons and not astrocytes, anti-GluR3 antisera preferentially and unexpectedly destroy astrocytes. Prominent neuronal damage is only seen at later time points, and even after 24 hr, the proportion of astrocytes destroyed exceeds that of neurons. (2) GluR3 antiserum-mediated cell death requires complement (C′) activation because cytotoxicity can be inhibited by sCR1. (3) The C6-deficient serum experiments reveal that anti-GluR3 cytotoxicity specifically requires construction of the MAC. (4) Neuronal resistance to C′ attack is enabled by high levels of CRPs because resistant neurons can be sensitized by interfering with CD59 function. (5) Conversely, astrocytes can be protected from C′ attack by transfection with either of two CRPs, CD59, or Crry. Together, these observations suggest a model explaining how GluR3 antisera damage both neurons and astrocytes (Fig. 9). Succinctly, anti-GluR3 binding to both neurons and astrocytes activates C′ on both cell types; that most antibodies bind to neurons suggests that extensive C′ deposition occurs on this cell type. However, high CRP expression levels by neurons protect them from C′-mediated destruction, whereas low astrocytic CRP expression levels leave this cell type incapable of preventing MAC construction. Consequently, astrocytes and not neurons suffer lytic death at early time points.
Fig. 9.

Model of GluR3 antibody-mediatedin vitro cytotoxicity. The experiments presented here support a multistep process: (1) anti-GluR3 antibodies bind to GluR3 antigen, which is expressed on neurons and, to a lesser extent, astrocytes; (2) although C′ activation occurs on both cell types, (3) neurons are initially protected from C′-mediated lysis by high expression levels of CRPs such as CD59; (3′) astrocytes, in contrast, lack CRPs and the protection they afford; thus, (4) MACs form on their surfaces, leading ultimately to their lytic demise; (5) at later time points, neurons suffer damage either by virtue of previous damage to astrocytes or finally succumbing to long-term low-level C′ attack.
There are several plausible explanations for the neuronal death that occurs at later time points. One is that neurons die principally as a consequence of previous destruction of astrocytes by anti-GluR3, as suggested by the similar pattern of neuronal and glial damage caused in the present work by the l-AA, a selective astrotoxin (Khurgel et al., 1996). Potential mechanisms of neuronal death include loss of astrocyte-derived BDNF or other growth factors upon which neurons are known to depend for survival in primary culture (Banker, 1980; Barde, 1989; Eddleston and Mucke, 1993) or from toxic accumulations of, for example, extracellular glutamate normally buffered by astrocytes (Choi, 1992; Rothstein et al., 1996; Tanaka et al., 1997). Alternatively, neurons may die only after exhausting their ability to resist C′ attack. For instance, CD59 complexes stoichiometrically with membrane-deposited C8 and C9 in preventing MAC formation, and because the CD59 stock of a cell is finite, it may eventually be depleted (Rollins et al., 1991; Morgan and Meri, 1994).
The bystander hypothesis, CRP expression levels, and neurological disease
The experiments with sCR1 and C6-deficient serum indicate that astrocyte destruction requires not only activation of the C′ cascade but also construction of the MAC channel. However, these experiments do not prove that cytotoxic MAC deposition occurs directly on astrocytes; conceivably, neuronal MAC deposition triggers release of a compound that then kills astrocytes. The CD59 transfection experiments were undertaken to investigate this possibility and revealed that astrocytes are protected by CD59 expression, implying that MAC deposition does occur directly on astrocytes. These findings beg the additional interesting question of whether astrocyte MAC deposition could result from C′ activation occurring in the vicinity of GluR3 on neurons. The bystander hypothesis predicts that MAC deposition on astrocytes occurs when incomplete MACs spread from neurons to nearby defenseless astrocytes. This possibility was tested by transfection with Crry, which will not protect astrocytes if the bystander hypothesis is correct because Crry acts at a stage earlier than formation of the free-floating nascent MAC. That expression of Crry on astrocytes enhances astrocyte resistance implies that early C′ components are deposited directly on astrocytes.
Together, these transfection experiments indicate that the C′ cascade is initiated and completed directly on astrocytes and that the bystander hypothesis is incorrect. Because of the paucity of astrocyte CRPs and the catalytic manner in which C′ cascade activation proceeds, even a small number of antibodies binding to astrocytes may trigger sufficient C′ activation to kill these cells. The antibodies may be so few in number or of such low affinity that they remain immunocytochemically undetectable. Based on previous immunodepletion experiments in which removal of GluR3 but not GST antibodies from GST-GluR3 antiserum completely abrogated toxicity, the astrocyte-binding antibodies are likely directed against GluR3 (He et al., 1998). Astrocytes express multiple types of glutamate receptorsin vitro, albeit at lower levels than neurons, including AMPA receptors containing GluR3 (for review, see Steinhäuser and Gallo, 1996).
Astrocytes express GluR3 in vivo and may likewise suffer antibody-mediated C′-dependent destruction in RE given the low CRP expression levels described by immunocytochemistry and in situ hybridization studies in the literature (Meri et al., 1991;Johnstone et al., 1993; Funabashi et al., 1994; Vedeler et al., 1994;Zajicek et al., 1995; Koski et al., 1996; Davoust et al., 1999; Spiller et al., 1999). However, the CRP family is a large one, and a comprehensive investigation of the cellular (i.e., neuronal vs glial) localization of all known family members in diseased and normal nervous system tissues from humans and the rodents used for neurological disease models remains to be completed. The pivotal role cellular CRP expression plays in vitro in protecting against C′-mediated attack underscores the need for such a systematic description. Indeed, deficient CRP expression or expression of a defective isoform of a CRPin vivo may constitute an important risk factor for RE, myasthenia gravis, multiple sclerosis, Alzheimer's disease, and other neurological disorders in which autoantibodies and C′ have been implicated (McRae et al., 1996; Whitney and McNamara, 1999). Although the ideas of CRP function and dysfunction are a central concept in the organ transplantation field (Baldwin et al., 1995; Cozzi and White, 1995; Ryan, 1995), comparatively little effort has been expended investigating their relationship to neurological disease. Determining whether CRP function is impaired in such diseases may prove helpful in understanding their pathogeneses and therefore in developing novel therapies, perhaps involving gene therapy to enhance surface expression of the deficient CRP.
Astrocyte death and RE progression
Whether or not RE patients suffer from defective CRP expression, this disease is classically defined by broad neuronal death and reactive astrogliosis. However, the drastic hemispheric degeneration common in advanced RE cases in which large parts of a hemisphere are destroyed is not likely attributable to the exclusive loss of neurons, because glia are the predominant cell type and probably are a major contributor to tissue volume in the CNS. Additionally, the magnitude of astrocyte loss may be obscured by the proliferation of normal glia responding to tissue damage. Damage to astrocytes in the context of neurodegeneration and reactive astrogliosis has been observed in other pathological conditions. For instance, degenerating astroglia have been detected in the brains of patients with frontotemporal dementia, a disease characterized by widespread layer-specific neuron loss and reactive astrogliosis (Martin et al., 1998). In this study, degenerating astrocytes were found scattered among proliferating reactive astroglia with the overall number of intact astrocytes being similar to control specimens.
How might such astrocyte death contribute to the neuronal loss, seizures, gliosis, and CNS inflammation typical of RE? Perhaps most simply, as alluded to above, neurons may die for lack of astrocyte-derived trophic support or from exposure to toxic compounds no longer sequestered by astrocytes. Also, seizures may result from deficient glutamate buffering (Rothstein et al., 1996; Tanaka et al., 1997). Additionally, cell death will activate neighboring astrocytes and microglia, both of which rapidly respond to even minute changes in the CNS environment by assuming an activated posture that includes proliferation, upregulation of numerous cell-surface receptors, and other genes, and for microglia the additional capacity to phagocytose cellular debris (Eddleston and Mucke, 1993; Kreutzberg, 1996; Streit, 1996). Fcγ, CR1, and CR3, which bind to IgG, C3b, and C4b, respectively, are among the upregulated microglial receptors, and interactions between these receptors and their membrane-bound ligands may trigger phagocytosis (Austyn and Wood, 1993). Brain sections from RE patients often contain neurons surrounded by microglia thought to be engaged in neuronophagia (Robitaille, 1991). We speculate that, after proliferating and clearing away cellular debris, the glia remain as the microglial nodules and gliotic scars often noted in RE specimens (Robitaille, 1991). Finally, high local concentrations of C5a may also underlie the formation of perivascular lymphocytic cuffs, because T-cells bear C5a receptors that mediate chemotaxis up C5a gradientsin vitro (Nataf et al., 1999).
A recent study of the role reactive astrocytes play in wound repair in the CNS provides evidence in support of some of these suggestions (Bush et al., 1999). Transgenic mice were generated whose astrocytes expressed the viral thymidine kinase gene driven by a GFAP promoter. Reactive astrocytes were selectively destroyed after an experimental stab wound with systemically administered ganciclovir because thymidine kinase levels rose in parallel with GFAP transcription around the wound site. Reactive astrocyte ablation was associated with exacerbated inflammation at the wound site, defective repair of the blood–brain barrier (BBB) as judged by prolonged permeability to circulating proteins, and destruction of neighboring neurons in a glutamate receptor activation-dependent manner. Extrapolating these findings to RE, astrocyte destruction may enable inflammatory cells, pathogenic IgG, and C′ easy access to the neural environment through a persistently damaged BBB. The phenomena that we postulate stem from these agents (inflammation, seizures, and cell death) would represent not only an exacerbation of the disease but also a means of disease perpetuation, because activated inflammatory cells, seizures, and C′ activation products can all disrupt the BBB (Cornford and Oldendorf, 1986; Hollerhage et al., 1989; Kadurugamuwa et al., 1989; Steinman, 1996). Consistent with the notion that peripheral pathogenic factors enter the CNS via a disrupted BBB are the immunocytochemical findings of preferential perivascular IgG and C′ deposition in brain samples from a subset of RE patients (Whitney et al., 1999) and responsiveness of some RE patients to plasma exchange or removal of circulating IgG (Rogers et al., 1994; Andrews et al., 1996; Antozzi et al., 1998). Examined in the context of these studies, C′-mediated astrocyte damage may be a pivotal event in RE pathogenesis that ensures the development of a chronic disease marked by seizures, neuronal and glial death, inflammation, and gliosis.
Footnotes
This work was supported by National Institutes of Health Grant NS 036808. K.D.W. was supported by a predoctoral fellowship from the Howard Hughes Medical Institute. We thank Wei Hua Qian for her excellent technical support in this project.
Correspondence should be addressed to Dr. James O. McNamara, Epilepsy Research Laboratory, Campus Box 3676, Duke University Medical Center, Durham, NC 27710. E-mail: jmc@neuro.duke.edu.
REFERENCES
- 1.Andrews PI, Dichter MA, Berkovic SF, Newton MR, McNamara JO. Plasmapheresis in Rasmussen's encephalitis. Neurology. 1996;46:242–246. doi: 10.1212/wnl.46.1.242. [DOI] [PubMed] [Google Scholar]
- 2.Antozzi C, Granata T, Aurisano N, Zardini G, Confalonieri P, Airaghi G, Mantegazza R, Spreafico R. Long-term selective IgG immunoadsorption improves Rasmussen's encephalitis. Neurology. 1998;51:302–305. doi: 10.1212/wnl.51.1.302. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RC, Harvath L, Dubois-Dalcq ME. Type 1 astrocytes and oligodendrocyte-type 2 astrocyte glial progenitors migrate toward distinct molecules. J Neurosci Res. 1990;27:400–407. doi: 10.1002/jnr.490270319. [DOI] [PubMed] [Google Scholar]
- 4.Ashgar SS, Pasch MC. Complement as a promiscuous signal transduction device. Lab Invest. 1998;78:1203–1225. [PubMed] [Google Scholar]
- 5.Austyn JM, Wood KJ. Principles of cellular and molecular immunology. Oxford UP; Oxford: 1993. [Google Scholar]
- 6.Baldwin WM, III, Pruitt SK, Brauer RB, Daha MR, Sanfilippo F. Complement in organ transplantation. Transplantation. 1995;59:797–808. [PubMed] [Google Scholar]
- 7.Banker GA. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science. 1980;209:809–810. doi: 10.1126/science.7403847. [DOI] [PubMed] [Google Scholar]
- 8.Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- 9.Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 11.Cornford EM, Oldendorf WH. Epilepsy and the blood–brain barrier. In: Delgado-Escuelta AV, Ward AA Jr, Woodbury DM, Porter RJ, editors. Advances in neurology. Raven; New York: 1986. pp. 787–812. [PubMed] [Google Scholar]
- 12.Cozzi E, White DJG. The generation of transgenic pigs as potential organ donors for humans. Nat Med. 1995;1:964–966. doi: 10.1038/nm0995-964. [DOI] [PubMed] [Google Scholar]
- 13.Davoust N, Nataf S, Holers VM, Barnum SR. Expression of the murine complement regulatory protein Crry by glial cells and neurons. Glia. 1999;27:162–170. doi: 10.1002/(sici)1098-1136(199908)27:2<162::aid-glia6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 14.Eddleston M, Mucke L. Molecular profile of reactive astrocytes: implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eshar N, Petralia RS, Winters CA, Niedzielinski AS, Wenthold RJ. The segregation and expression of glutamate receptor subunits in cultured hippocampal neurons. Neuroscience. 1993;57:943–964. doi: 10.1016/0306-4522(93)90040-m. [DOI] [PubMed] [Google Scholar]
- 16.Funabashi K, Okada N, Matsuo S, Yamamoto T, Morgan BP, Okada H. Tissue distribution of complement regulatory proteins in rats. Immunology. 1994;81:444–451. [PMC free article] [PubMed] [Google Scholar]
- 17.He X-P, Patel M, Whitney KD, Janumpalli S, Tenner A, McNamara JO. Glutamate receptor GluR3 antibodies and death of cortical cells. Neuron. 1998;20:153–163. doi: 10.1016/s0896-6273(00)80443-2. [DOI] [PubMed] [Google Scholar]
- 18.Hollerhage HG, Walter GF, Stolke D. Complement-derived polypeptide C3adesArg as a mediator of inflammation at the blood–brain barrier in a new experimental cat model. Acta Neuropathol. 1989;77:307–313. doi: 10.1007/BF00687583. [DOI] [PubMed] [Google Scholar]
- 19.Hughes TR, Piddlesden SJ, Williams JD, Harrison RA, Morgan BP. Isolation and characterization of a membrane protein from rat erythrocytes which inhibits lysis by the membrane attack complex of rat complement. Biochem J. 1992;284:169–176. doi: 10.1042/bj2840169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnstone RW, Loveland BE, McKenzie IFC. Identification and quantification of complement regulator CD46 on normal human tissues. Immunology. 1993;79:341–347. [PMC free article] [PubMed] [Google Scholar]
- 21.Kadurugamuwa JL, Hengstler B, Bray MA, Zak O. Inhibition of complement-factor-5a-induced inflammatory reactions by prostaglandin E2 in experimental meningitis. J Infect Dis. 1989;160:715–719. doi: 10.1093/infdis/160.4.715. [DOI] [PubMed] [Google Scholar]
- 22.Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn T, Sakmann B, Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- 23.Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16:351–358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 24.Koski CL, Estep AE, Sawant-Mane S, Shin ML, Highbarger L, Hansch GM. Complement regulatory molecules on human myelin and glial cells: differential expression affects the deposition of activated complement proteins. J Neurochem. 1996;66:303–312. doi: 10.1046/j.1471-4159.1996.66010303.x. [DOI] [PubMed] [Google Scholar]
- 25.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 26.Martin JA, Su JH, Cotman CW. Astrogliosis, astrodegeneration, and neuron loss in frontotemporal dementia. Soc Neurosci Abstr. 1998;24:1472. [Google Scholar]
- 27.McRae A, Ling EA, Wigander A, Dahlstrom A. Microglial cerebrospinal fluid antibodies. Significance for Alzheimer's disease. Mol Chem Neuropathol. 1996;28:89–95. doi: 10.1007/BF02815209. [DOI] [PubMed] [Google Scholar]
- 28.Meri S, Waldmann H, Lachmann PJ. Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Lab Invest. 1991;65:532–537. [PubMed] [Google Scholar]
- 29.Morgan BP, Meri S. Membrane proteins that protect against complement lysis. Springer Semin Immunopathol. 1994;15:369–396. doi: 10.1007/BF01837366. [DOI] [PubMed] [Google Scholar]
- 30.Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol. 1999;162:4018–4023. [PubMed] [Google Scholar]
- 31.Nolte C, Moller T, Walter T, Kettenmann H. Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience. 1996;73:1091–1107. doi: 10.1016/0306-4522(96)00106-6. [DOI] [PubMed] [Google Scholar]
- 32.Oguni H, Andermann F, Rasmussen TB. The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MNI series of 48 cases. In: Andermann F, editor. Chronic encephalitis and epilepsy. Butterworth-Heinemann; Boston: 1991. pp. 7–35. [PubMed] [Google Scholar]
- 33.Park CC, Shin ML, Simard JM. The complement membrane attack complex and the bystander effect in cerebral vasospasm. J Neurosurg. 1997;87:294–300. doi: 10.3171/jns.1997.87.2.0294. [DOI] [PubMed] [Google Scholar]
- 34.Quigg RJ, Lo CF, Alexander JJ, Sneed AE, III, Moxley G. Molecular characterization of rat Crry: widespread distribution of two alternative forms of Crry mRNA. Immunogenetics. 1995;42:362–367. doi: 10.1007/BF00179397. [DOI] [PubMed] [Google Scholar]
- 35.Rasmussen T, Olszweski J, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology. 1958;8:435–455. doi: 10.1212/wnl.8.6.435. [DOI] [PubMed] [Google Scholar]
- 36.Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Andermann F, editor. Chronic encephalitis and epilepsy. Butterworth-Heinemann; Boston: 1991. pp. 79–110. [Google Scholar]
- 37.Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, Hughes TE, Heinemann SF, McNamara JO. Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science. 1994;265:648–651. doi: 10.1126/science.8036512. [DOI] [PubMed] [Google Scholar]
- 38.Rollins SA, Zhao J, Ninomiya H, Sims PJ. Inhibition of homologous complement by CD59 is mediated by a species-specific recognition conferred through binding to C8 within C5b-8 or C9 within C5b-9. J Immunol. 1991;146:2345–2351. [PubMed] [Google Scholar]
- 39.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kunci RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 40.Rushmere NK, Tomlinson S, Morgan BP. Expression of rat CD59: functional analysis confirms lack of species selectivity and reveals that glycosylation is not required for function. Immunology. 1997;90:640–646. doi: 10.1046/j.1365-2567.1997.00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryan US. Complement inhibitory therapeutics and xenotransplantation. Nat Med. 1995;1:967–968. doi: 10.1038/nm0995-967. [DOI] [PubMed] [Google Scholar]
- 42.Sakurada C, Seno H, Dohi N, Takizawa H, Nonaka M, Okada N, Okada H. Molecular cloning of the rat complement regulatory protein, 5I2 antigen. Biochem Biophys Res Commun. 1994;193:819–826. doi: 10.1006/bbrc.1994.1117. [DOI] [PubMed] [Google Scholar]
- 43.Spiller OB, Hanna SM, Morgan BP. Tissue distribution of the rat homolog of decay-accelerating factor. Immunology. 1999;97:374–384. doi: 10.1046/j.1365-2567.1999.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinhäuser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- 45.Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 46.Streit WJ. The role of microglia in brain injury. Neurotoxicology. 1996;17:671–678. [PubMed] [Google Scholar]
- 47.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 48.Twyman RE, Gahring LC, Spiess J, Rogers SW. Glutamate receptor antibodies activate a subset of receptors and reveal an agonist binding site. Neuron. 1995;14:755–762. doi: 10.1016/0896-6273(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 49.Vassault A. Lactate dehydrogenase. In: Bergmeyer HU, editor. Methods of enzymatic analysis. Verlag Chemie; Weinheim, Germany: 1983. pp. 118–126. [Google Scholar]
- 50.Vedeler C, Ulvestad E, Bjorge L, Conti G, Williams K, Mork S, Matre R. The expression of CD59 in normal human nervous tissue. Immunology. 1994;82:542–547. [PMC free article] [PubMed] [Google Scholar]
- 51.Whitney KD, McNamara JO. Humoral autoimmunity and modulation of synaptic transmission. Annu Rev Neurosci. 1999;22:175–195. doi: 10.1146/annurev.neuro.22.1.175. [DOI] [PubMed] [Google Scholar]
- 52.Whitney KD, Andrews PI, McNamara JO. Immunoglobulin G and complement immunoreactivity in the cerebral cortex of patients with Rasmussen's encephalitis. Neurology. 1999;53:699–708. doi: 10.1212/wnl.53.4.699. [DOI] [PubMed] [Google Scholar]
- 53.Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res. 1990;27:36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]
- 54.Zajicek J, Wing M, Skepper J, Compston A. Human oligodendrocytes are not sensitive to complement. Lab Invest. 1995;73:128–138. [PubMed] [Google Scholar]