

Abstract
To define activity-dependent release of endogenous brain-derived neurotrophic factor (BDNF), we developed an in vitromodel using primary sensory neurons and a modified ELISA, termed ELISAin situ. Dissociate cultures of nodose-petrosal ganglion cells from newborn rats were grown in wells precoated with anti-BDNF antibody to capture released BDNF, which was subsequently detected using conventional ELISA. Conventional ELISA alone was unable to detect any increase in BDNF concentration above control values following chronic depolarization with 40 mm KCl for 72 hr. However, ELISA in situ demonstrated a highly significant increase in BDNF release, from 65 pg/ml in control to 228 pg/ml in KCl-treated cultures. The efficacy of the in situ assay appears to be related primarily to rapid capture of released BDNF that prevents BDNF binding to the cultured cells. We therefore used this approach to compare BDNF release from cultures exposed for 30 min to either continuous depolarization with elevated KCl or patterned electrical field stimulation (50 biphasic rectangular pulses of 25 msec, at 20 Hz, every 5 sec). Short-term KCl depolarization was completely ineffective at evoking any detectable release of BDNF, whereas patterned electrical stimulation increased extracellular BDNF levels by 20-fold. In addition, the magnitude of BDNF release was dependent on stimulus pattern, with high-frequency bursts being most effective. These data indicate that the optimal stimulus profile for BDNF release resembles that of other neuroactive peptides. Moreover, our findings demonstrate that BDNF release can encode temporal features of presynaptic neuronal activity.
Keywords: BDNF release, chronic depolarization, electrical field stimulation, ELISA, ELISA in situ, frequency, patterned stimulation, P-CREB, primary sensory neurons
There is increasing evidence that brain-derived neurotrophic factor (BDNF) plays a trans-synaptic role in regulating transmission between primary sensory neurons and second-order sensory relay cells. BDNF is expressed by subsets of sensory ganglion cells (Schecterson and Bothwell, 1992; Wetmore and Olson, 1995; Apfel et al., 1996; Zhou et al., 1998; Brady et al., 1999), can be transported in the central projections of dorsal root ganglion (DRG) neurons (Zhou and Rush, 1996; Tonra, 1999), and is localized to dense-core vesicles within DRG central axon terminals (Michael et al., 1997). Our studies demonstrated that survival of sensory neurons that both express and depend on BDNF can be supported by long-term exposure to elevated potassium, indicating that BDNF can be released under depolarizing conditions in culture (Brady et al., 1999). More recently we found that BDNF acutely inhibits AMPA-mediated currents in second-order sensory relay neurons, indicating that BDNF may modulate glutamatergic primary afferent transmission (Balkowiec et al., 2000). In addition, Kerr et al. (1999) demonstrated that BDNF can potentiate nociceptive spinal reflexes by enhancing NMDA receptor-mediated responses. Despite these findings, little is known about activity-dependent release of endogenous BDNF, either from primary sensory neurons or other neuronal cell types.
Analysis of regulated secretion of endogenous neurotrophins from identified neurons has been hampered by the limited ability of conventional assays to detect the relatively small quantities of these factors released during physiological stimulation. Studies to date have used ELISA to detect neurotrophin release either from tissue slices or following neurotrophin overexpression in transfected cells (Blöchl and Thoenen, 1995, 1996; Goodman et al., 1996; Heymach et al., 1996; Canossa et al., 1997; Krüttgen et al., 1998; Griesbeck et al., 1999). It is unknown, however, whether overexpression to very high concentrations alters normal routes of BDNF trafficking and release. Moreover, most studies of regulated neurotrophin release have stimulated cells using continuous membrane-depolarizing agents, including elevated extracellular potassium, veratridine, or glutamate receptor agonists (Ghosh et al., 1994; Blöchl and Thoenen, 1995;Androutsellis-Theotokis et al., 1996; Goodman et al., 1996; Heymach et al., 1996; Griesbeck et al., 1999). It is well established, however, that release of classical as well as peptide transmitters depends on nerve impulse pattern (Lundberg et al., 1989; Whim and Lloyd, 1994).
To address these issues, the present study compared the effects of continuous chemical depolarization and patterned electrical stimulation on BDNF release from primary sensory neurons, using a modification of conventional ELISA methodology, termed ELISA in situ. This technique, described by Beech et al. (1997) for measuring cytokine release from T-cells, incorporates a substrate-bound monoclonal antibody against the peptide of interest into the cell culture system, so that the released peptide is immediately captured for subsequent detection by colorimetric methods. We found that, using this technique, we can readily detect release of endogenous BDNF from newborn primary sensory neurons following short-term patterned electrical stimulation. Moreover, we found that short-term stimulation with high-frequency bursts is strikingly more effective at releasing BDNF than KCl-induced depolarization over the same time period.
MATERIALS AND METHODS
Cell preparation and culture. Newborn rats (Sprague Dawley strain; Zivic-Miller, Zelienople, PA) were deeply anesthetized by hypothermia and decapitated. Nodose and petrosal ganglia (NPG) were (1) aseptically removed from the animals, (2) digested in 0.1% trypsin (Worthington Biochemical, Lakewood, NJ) with 0.01% deoxyribonuclease I (Sigma, St. Louis, MO) dissolved in Ca2+- and Mg2+-free HBSS (Mediatech, Herndon, VA) for 30 min at 37°C in a humidified atmosphere of 5% CO2 and 95% air, (3) rinsed in 0.1% soybean trypsin inhibitor (Worthington) dissolved in Ca2+- and Mg2+-containing Dulbecco's phosphate-buffered salt solution (Mediatech), (4) transferred to culture medium, and (5) triturated through siliconized, fire-polished Pasteur pipettes. Dissociated NPG neurons were plated in UV-sterilized, 96-well flat-bottom ELISA plates (MaxiSorp; Nalge Nunc International, Naperville, IL) at a density of one NPG per well. Cultures of NPG neurons were grown for 3 d in Neurobasal-A medium supplemented with B-27 serum-free supplement, 0.5 mml-glutamine, 0.025 mm glutamic acid, and 1% penicillin-streptomycin-neomycin antibiotic mixture (Life Technologies, Gaithersburg, MD).
BDNF immunoassays. BDNF protein was measured with both a conventional and a modified sandwich ELISA using the BDNF Emax immunoassay system (Promega, Madison, WI) according to the protocol of the manufacturer, except that the concentrations of the anti-BDNF monoclonal antibody and anti-human BDNF polyclonal antibody were 5 and 2 μg/ml, respectively, and the dilution of the anti-IgY-HRP antibody was 1:1000. All reagents used prior to cell plating were sterilized with 0.2 μm Acrodisc syringe filters (Pall, Ann Arbor, MI).
Conventional BDNF ELISA. NPG cells were grown in uncoated 96-well ELISA plates. In some control experiments, wells were precoated with an irrelevant monoclonal antibody (anti-NGF; Promega) to rule out any potential influence of antibody presence on BDNF release. These wells were treated prior to cell plating as described below for anti-BDNF monoclonal antibody. On the day of the assay, a standard curve was generated for each plate using BDNF diluted in the same medium used for cell culture. Standards (in duplicate) and undiluted fresh samples of cell-conditioned culture medium (in duplicate or triplicate) were incubated in ELISA plates precoated with anti-BDNF monoclonal antibody, according to the manufacturer's protocol. Following the incubation and washing steps, anti-human BDNF polyclonal antibody was applied (see below).
BDNF ELISA in situ. Ninety-six-well ELISA plates were UV-sterilized for 30 min and coated with anti-BDNF monoclonal antibody at 4°C for 16.5 hr. Next, plates were washed and blocked, followed by two 1 hr incubations with culture medium to remove any residue of the ELISA washing solution. Then the NPG neurons were prepared as described above, plated in anti-BDNF-coated wells, and grown for 3 d under various experimental conditions (see Results). BDNF samples used to generate the standard curves were incubated in the same plate as the cells. At the end of the culture period, plates were extensively washed to remove all cells and cell debris, and the anti-human BDNF polyclonal antibody was applied, followed by subsequent steps according to the manufacturer's protocol. In experiments designed to compare the conventional BDNF ELISA with BDNF ELISA in situ, all steps of the protocol, beginning with the application of the anti-human BDNF antibody, were performed simultaneously for both assays. Absorbance values were read at 450 nm in a plate reader (Vmax; Molecular Devices, Sunnyvale, CA). For control wells in which anti-BDNF monoclonal antibody was omitted, absorbance values were not significantly different from the absorbance of blank wells.
Electrical field stimulation of NPG neurons. NPG cultures were prepared as described above for BDNF ELISA in situ. After an initial 3-d incubation, three adjacent culture wells were connected to each other in series through thin strips of 1% agarose gel permeated with culture medium, and to the stimulator (MultiStim System; Digitimer) through Ag:AgCl stimulating electrodes (modified from the methods of Brevet et al., 1976; McDonough et al., 1994). Three additional wells were also connected to each other by agarose bridges but were not connected to the stimulator and served as controls. The plate was put back to the incubator, and the neurons were stimulated for 30 or 60 min with biphasic rectangular pulses delivered at various patterns (see Results). In experiments comparing the effects of patterned electrical stimulation with potassium-induced continuous depolarization, KCl was added to three additional wells, to a final concentration of 40 mm, at the beginning of the stimulation period. In addition, BDNF standards were prepared in the same plate, also at the beginning of the stimulation period. After stimulation, all wells were vigorously washed prior to the ELISA steps described above.
Calculations and statistical analysis. BDNF levels were calculated from the standard curve prepared for each plate, using SOFTmax PRO version 3.0 software (Molecular Devices). The standard curves were linear within the range used (0–500 pg/ml), and the quantities of BDNF in experimental samples were always within the linear range of the standard curve. Data are expressed as mean ± SE. Samples were compared using ANOVA followed by Duncan's multiple-comparison procedure, and p < 0.05 was considered significant.
Immunocytochemistry. Cultures for immunocytochemical staining were fixed with 4% paraformaldehyde in 0.1 msodium phosphate buffer, pH 7.4, for 30 min at room temperature. Protein gene product (PGP) 9.5 immunostaining was performed as previously described (Brady et al., 1999). The number of neurons in each culture was evaluated by counting all PGP9.5-immunoreactive cells per well. Experiments were performed three times with three cultures per experimental group. Values were compared using ANOVA followed by Duncan's multiple-comparison procedure, and p < 0.05 was considered significant. TrkB and phospho-cAMP response element-binding protein (P-CREB) immunostaining were performed as previously described (Brady et al., 1999), using rabbit polyclonal anti-TrkB (Chemicon, Temecula, CA) or rabbit anti-phospho-CREB IgG (Upstate Biotechnology, Lake Placid, NY) and goat anti-rabbit biotinylated IgG (Vector Laboratories, Burlingame, CA). Control cultures, in which primary antibody was omitted, were completely devoid of staining.
Anti-TrkB IgG1 (clone 47; Transduction Laboratories, Lexington, KY; catalog #T16020, special order, without additives) was used to inhibit binding of BDNF to TrkB receptors (see Results). We established that 5 μg/ml anti-TrkB was sufficient to inhibit survival of BDNF-dependent embryonic day 16.5 NPG neurons (Erickson et al., 1996) grown in the presence of 10 ng/ml BDNF. Survival of newborn NPG neurons, which are not BDNF-dependent (Brady et al., 1999), was not affected by treatment with 10 μg/ml antibody (1197.56 ± 102.78 neurons per well in control and 1025.5 ± 80.49 neurons per well with anti-TrkB; n = 9; p = 0.37578).
RESULTS
Initial studies sought to compare extracellular levels of BDNF in cultures of newborn NPG neurons grown for 72 hr in the absence (control) or presence of depolarizing concentrations of KCl (40 mm; Brosenitsch et al., 1998; Brady et al., 1999) using a conventional BDNF ELISA protocol. Using this approach, we were able to detect only very low levels of BDNF in control cultures and saw no significant change in BDNF concentration in KCl-treated groups compared with controls (Fig. 1; control, 2.88 ± 0.84 pg/ml; n = 23; KCl-treated, 4.92 ± 0.81 pg/ml; n = 21; p = 0.88256). This result was at odds with previous findings from our laboratory that endogenous BDNF can support survival of NPG neurons in culture following chronic exposure to elevated KCl (Brady et al., 1999). We therefore sought to improve detectability of BDNF in our culture system using a modification of the conventional ELISA, termed ELISA in situ, in which cells are grown in wells precoated with a monoclonal antibody against the peptide of interest, which is thus immobilized and subsequently detected using standard colorimetric methods (Beech et al., 1997). Indeed, using BDNF ELISA in situ, we were able not only to measure higher levels of BDNF in control cultures but also to detect significant release of BDNF following chronic depolarization. Specifically, the concentration of BDNF averaged 65.34 ± 5.29 pg/ml (n = 23) in 72 hr control NPG cultures and 228.16 ± 19.47 pg/ml (n = 21) following 72 hr treatment with elevated KCl (p = 0.00011; Fig. 1). The increase in BDNF levels detected in KCl-treated cultures was not attributable to increased neuronal survival (1234.3 ± 71.05 neurons per well in control cultures vs 1429.6 ± 91.22 neurons per well in KCl-treated cultures; n = 9; p = 0.53294). Similarly, survival was not significantly increased by the presence of the BDNF antibody during the cell culture period (1234.3 ± 71.05 neurons per well in the presence of anti-BDNF vs 1197.56 ± 102.78 neurons per well in the absence of anti-BDNF;n = 9; p = 0.70989). To determine whether the presence of the capture antibody by itself stimulated BDNF release, sister cultures were grown in wells precoated with an irrelevant anti-NGF monoclonal antibody, in the presence or absence of elevated KCl, and extracellular BDNF levels were measured using conventional BDNF ELISA. The presence of the monoclonal antibody had no effect on BDNF levels, either in control cultures (0.67 ± 0.45 pg/ml in the presence of anti-NGF vs 0.73 ± 0.26 pg/ml in the absence of anti-NGF; n = 8; p = 0.97377) or KCl-treated cultures (5.02 ± 2.28 pg/ml in the presence of anti-NGF vs 4.76 ± 1.46 pg/ml in the absence of anti-NGF; n = 8; p = 0.89129). However, we cannot exclude the possibility that the presence of the antibody stimulated release of low levels of BDNF, below the limits of detectability by conventional ELISA. To rule out the possibility that the observed increase in BDNF release was due simply to the increased osmolarity of the KCl-supplemented medium, we compared cultures grown in control medium with cultures supplemented with 40 mm NaCl and found that BDNF levels, measured using ELISA in situ, were unchanged in the presence of elevated NaCl (92.38 ± 13.24 vs 76.76 ± 7.38 pg/ml in control groups; n = 9; p = 0.46864).
Fig. 1.

Long-term exposure to elevated extracellular potassium induces BDNF release from newborn NPG neurons in culture. Mean BDNF levels were measured with standard ELISA and ELISA in situ in sister cultures grown for 72 hr in the absence (Control, gray bars) or presence of 40 mmpotassium (KCl, black bars). n = 21; ***p < 0.001; n.s., not significant.
These findings demonstrated that markedly higher levels of BDNF were detected in control and KCl-treated NPG cultures using ELISA in situ compared with conventional ELISA, and that this difference could not be attributed to increased neuronal survival or other nonspecific effects. We hypothesized, therefore, that the in situ assay protocol increased BDNF detectability by rapidly capturing and immobilizing released BDNF and thereby protecting the peptide from binding to cells and/or degradation. To test these possibilities, 500 pg/ml exogenous human recombinant BDNF (Promega) was added to culture wells containing either medium alone or NPG dissociate cultures. Following 72 hr of incubation, BDNF levels were compared in both groups using conventional ELISA and ELISA in situ. No significant differences were found between the levels of BDNF detected by standard ELISA (310.83 ± 39.31 pg/ml; n = 17) and ELISA in situ (304.64 ± 27.13 pg/ml;n = 10; p = 0.88643; Fig.2) in wells containing culture medium alone plus BDNF. This experiment demonstrated that there are no intrinsic differences in the sensitivity of the two assays. However, when compared among wells containing NPG neurons plus BDNF, there was a highly significant (p = 0.00047) difference between the levels of BDNF detected with the two assays. Specifically, using standard ELISA, the BDNF concentration was only 32.12 ± 5.42 pg/ml (n = 19) after 72 hr, a value that was not significantly different from BDNF levels in medium from control NPG cultures grown without added BDNF (p = 0.30941; Fig. 2). This result indicates that, in the presence of NPG cells, BDNF is lost over time from the culture medium, perhaps through degradation or binding to the high-affinity BDNF receptor TrkB, which is expressed by newborn NPG neurons (Zhuo and Helke, 1996; present study, Fig.3A). In fact, treatment of cultures with a function blocking anti-TrkB antibody (Transduction Laboratories; for details, see Materials and Methods) significantly increased the ability of conventional ELISA to detect BDNF added to standard NPG cultures. Specifically, addition of the anti-TrkB antibody increased detection of BDNF by conventional ELISA by nearly threefold compared with control cultures grown without anti-TrkB (with anti-TrkB, 211.58 ± 62.71 pg/ml; n = 5; vs controls, 68.36 ± 11.44 pg/ml; n = 13; p = 0.00056; Fig. 3B). These data suggest that the relative inability of conventional ELISA to detect BDNF release in NPG cultures is attributable, in large part, to binding of BDNF to TrkB on the cultured cells.
Fig. 2.

Detectability of exogenous BDNF by standard ELISAversus ELISA in situ. BDNF (500 pg/ml) was added at plating to newborn NPG cultures (hatched gray bars) and to culture medium (Med.) alone (hatched white bars) and incubated for 72 hr in the absence (Standard ELISA) or presence of anti-BDNF monoclonal capture antibody (ELISA in situ). BDNF levels were also measured in control cultures (solid gray bars) to which exogenous BDNF was not added. ***p < 0.001; n.s., not significant.
Fig. 3.
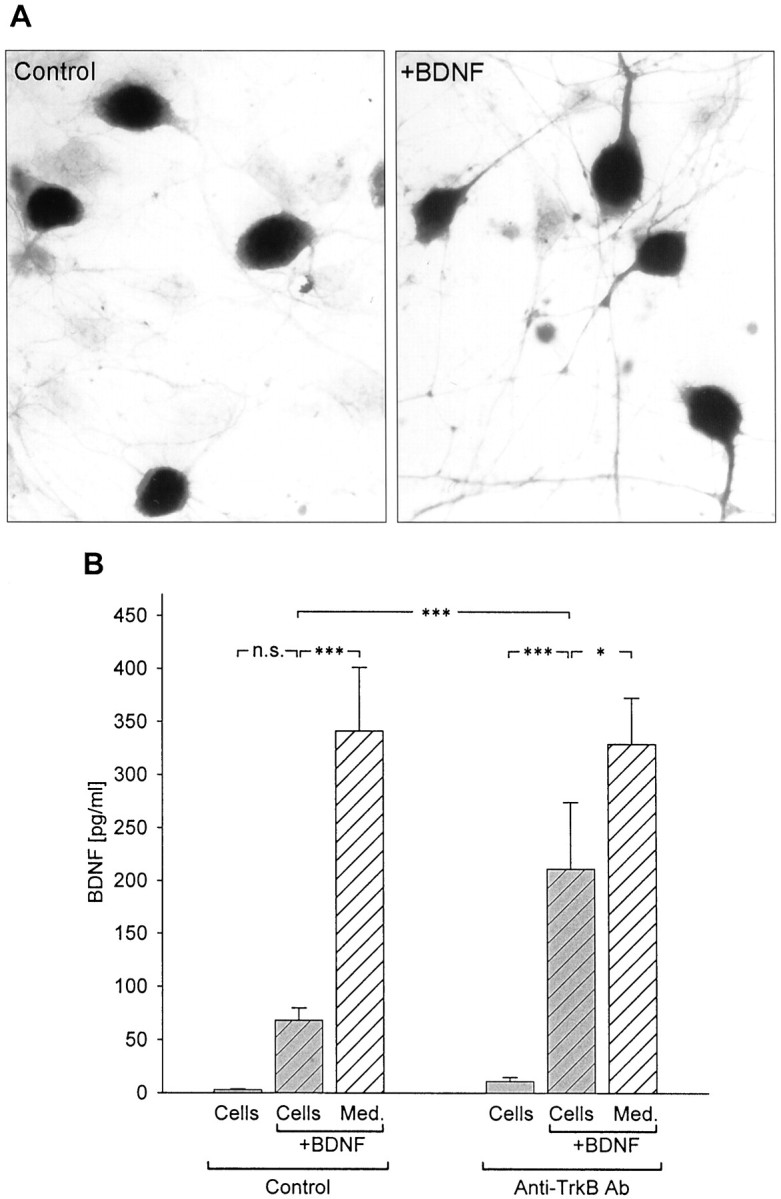
Inhibition of BDNF binding to cells increases BDNF detectability by standard ELISA. A, Immunostaining with an antibody against the extracellular domain of TrkB (Chemicon) in newborn NPG cultures grown for 3 d in the absence (Control) or presence of exogenous BDNF (+BDNF, 500 pg/ml). B, Mean levels of BDNF, detected with standard ELISA, in the absence (Control) or presence of anti-TrkB antibody (Anti-TrkB Ab, 10 μg/ml) in NPG cultures (hatched gray bars) and culture medium alone (hatched white bars) 48 hr after addition of 500 pg/ml BDNF. BDNF levels were also measured in control cultures (solid gray bars) to which exogenous BDNF was not added. ***p < 0.001;*p < 0.05;n.s., not significant.
In contrast to the results obtained with standard ELISA, ELISA in situ detected 337.33 ± 25.93 pg/ml BDNF (n = 18) after 72 hr, a level not significantly different from that in wells to which BDNF was added in the absence of cells (p = 0.92444; Fig. 2). These data demonstrate that the substrate-bound anti-BDNF, which is present throughout the culture period in the in situ paradigm, successfully competes with BDNF binding to TrkB on cells, thereby enhancing detectability of BDNF in the culture medium.
Previous studies of activity-dependent neurotransmitter release demonstrated that chronic depolarization is markedly less effective than high-frequency electrical stimulation at releasing both classical transmitters and peptide co-transmitters (Belai et al., 1987; Agoston et al., 1988). To examine whether BDNF release is similarly regulated, we compared the effects of patterned electrical field stimulation for 30 min (50 biphasic rectangular pulses of 25 msec, at 20 Hz, every 5 sec) with 30 min of continuous depolarization by 40 mm KCl, on BDNF release from newborn NPG neurons, using ELISA in situ. To determine whether these stimulation protocols were effective at activating NPG neurons, we performed immunostaining with an antibody against the phosphorylated form of CREB, a marker of neuronal depolarization (Ghosh et al., 1994; Moore et al., 1996). Both KCl treatment and patterned electrical field stimulation led to marked increases in P-CREB staining in the vast majority of cells (Fig.4A), indicating that both protocols were effective at activating neurons in these cultures.
Fig. 4.

Patterned electrical stimulation is markedly more effective at releasing BDNF from newborn NPG neurons than KCl-induced continuous depolarization. A, P-CREB immunostaining of newborn NPG cultures after 30 min electrical field stimulation (20 Hz;Electrical stimulation) or 30 min continuous depolarization (40mm KCl) compared with unstimulated controls. B, Mean levels of BDNF released in sister cultures of newborn NPG neurons during 30 min of control conditions (no stimulation), electrical field stimulation (50 biphasic rectangular pulses of 25 msec, at 20 Hz, every 5 sec), or continuous depolarization with 40 mm KCl. Each value represents the difference between the BDNF level measured after stimulation and the level measured in sister cultures at the beginning of the stimulus period. ***p < 0.001;n.s., not significant.
BDNF levels were compared following 30 min of either control conditions, patterned electrical stimulation, or KCl-induced chronic depolarization. Patterned electrical stimulation at 20 Hz resulted in a highly significant increase in BDNF release from NPG neurons (62.95 ± 4.19 pg/ml vs 2.87 ± 1.11 pg/ml in control; n = 16; p = 0.00011; Fig. 4B). In contrast, KCl-induced chronic depolarization over the same time period was completely ineffective at increasing detectable BDNF release (−1.35 ± 3.58 pg/ml; n = 20; p = 0.87134; Fig. 4B). The release of BDNF induced by electrical stimulation was abolished by treatment of cultures with 1.5 μm tetrodotoxin (TTX), an inhibitor of voltage-dependent Na+ channels, before stimulation (1.50 ± 0.70 pg/ml with TTX vs 35.12 ± 0.94 pg/ml without TTX; n = 4; p = 0.0000001), indicating that activation of voltage-gated sodium channels is required for this release. To rule out the possibility that the enhanced BDNF release was due to damage of cells by electrical activation, we compared cell survival between control cultures and cultures stimulated for 30 min with bursts of 50 biphasic rectangular pulses of 25 msec, at 20 Hz, delivered every 5 sec. Twenty-four hours after stimulation, there was no significant difference in the number of cells in control and stimulated cultures (per well: control, 1575 ± 60.35; stimulated, 1480 ± 57.15; n = 9;p = 0.28485).
It is well established that peptide neurotransmitter release can be differentially regulated by distinct patterns of neuronal activity (Lundberg et al., 1986, 1989; Whim and Lloyd, 1994; Vilim et al., 1996). To examine the effect of stimulus pattern on the release of BDNF from NPG neurons, we used a paradigm in which the overall number of pulses, and consequently, average frequency, as well as the number of pulses in individual bursts, remained constant, whereas intraburst frequency and interburst interval were varied. Specifically, BDNF levels were compared following 60 min of either control conditions or electrical field stimulation with 50 biphasic rectangular pulses of 10 msec, delivered at 5, 10, 20, and 50 Hz, with interburst intervals, respectively, of 0 (tonic stimulation), 10, 15, and 18 sec (Fig.5A). BDNF release was significantly higher during stimulation with high-frequency bursts (20 Hz, 34.95 ± 4.98 pg/ml; p = 0.0144; 50 Hz, 48.07 ± 7.18 pg/ml; p = 0.0005) compared with tonic stimulation at 5 Hz (18.09 ± 2.79 pg/ml; n= 10; Fig. 5B). When compared among different bursting patterns, stimulation with 2 sec 50 Hz bursts delivered every 20 sec was most effective, despite the short burst duration and long interburst interval characteristic of this pattern (Fig. 5).
Fig. 5.

Activity-dependent release of BDNF is regulated by the pattern of stimulation. A, Schematic representation of the stimulation patterns applied to each group of cultures.B, Mean levels of BDNF released from newborn NPG neurons during 60 min of electrical field stimulation with 50 biphasic rectangular pulses of 10 msec, delivered at 5, 10, 20, and 50 Hz, with interburst intervals, respectively, of 0, 10, 15, and 18 sec as shown in A. ***p < 0.001; *p < 0.05; n.s., not significant.
DISCUSSION
The present study demonstrates that primary sensory neurons can release BDNF in an activity-dependent manner. The amount of released BDNF is regulated by both stimulus frequency and pattern, and high-frequency bursts are markedly more effective at evoking release than either continuous depolarization with elevated extracellular KCl or tonic electrical stimulation. Moreover, our results demonstrate that the detectability of released BDNF by ELISA in situ is greatly enhanced compared with conventional ELISA. Thus, we are able to quantify release of endogenous BDNF from dissociated neurons, without the need to enhance peptide levels by genetic overexpression, as in other studies (Blöchl and Thoenen, 1995,1996; Goodman et al., 1996; Heymach et al., 1996; Canossa et al., 1997;Krüttgen et al., 1998; Griesbeck et al., 1999).
Previous analyses of neurotrophin release have usedcontinuous depolarization, induced by elevated potassium, veratridine, or glutamate agonists to activate cells (Ghosh et al., 1994; Blöchl and Thoenen, 1995, 1996; Androutsellis-Theotokis et al., 1996; Goodman et al., 1996; Heymach et al., 1996; Griesbeck et al., 1999). Indeed, continuous depolarization is highly effective at inducing calcium influx and activation of intracellular signaling pathways required for both genomic and nongenomic responses (Sheng et al., 1990; Ginty et al., 1991; Bito, 1998; Brosenitsch et al., 1998;Tao et al., 1998). In the present study, for example, short-term exposure to 40 mm KCl was sufficient to increase CREB phosphorylation, a marker of neuronal depolarization and activation of multiple intracellular signaling cascades (Sheng et al., 1990; Davis et al., 1996; Deisseroth et al., 1996; Moore et al., 1996). Despite this, 30 min of KCl depolarization was completely ineffective at evoking detectable release of BDNF. Similarly, Griesbeck et al. (1999) reported that short-term KCl-induced depolarization was ineffective at releasing BDNF from primary cultures of hippocampal neurons. In contrast, we found that 30 min of patterned electrical stimulation led to a marked 20-fold rise in the concentration of extracellular BDNF. These data suggest that, rather than depolarization per se, activation of specific signaling pathways by patterned stimulation is required to evoke detectable BDNF release (see alsoBuonanno and Fields, 1999).
We did find that exposure to KCl for 3 d evokes detectable release of BDNF from NPG neurons, albeit much less than only 30 min of high-frequency electrical stimulation. However, such release likely reflects multiple sequelae of long-term continuous depolarization, including increased BDNF expression (Shieh et al., 1998; Shieh and Ghosh, 1999), and is therefore probably not a useful model for elucidating mechanisms that govern release of preexisting BDNF pools.
Our results demonstrate for the first time that the amount of BDNF release depends on stimulus pattern, indicating that BDNF can encode temporal features of presynaptic neuronal activity. This finding may be of particular significance in light of the proposed role of BDNF in activity-dependent mechanisms of neuronal development and function (Cabelli et al., 1995; Thoenen, 1995; Acheson and Lindsay, 1996;Bonhoeffer, 1996; Galuske et al., 1996; Katz and Shatz, 1996;McAllister et al., 1996; Snider and Lichtman, 1996; Stoop and Poo, 1996; Cabelli et al., 1997; Marty et al., 1997; Black, 1999; Lu and Chow, 1999; McAllister et al., 1999), including homeostatic regulation of synaptic strength (Rutherford et al., 1998; Turrigiano, 1999). For example, by encoding afferent firing patterns, BDNF could provide a mechanism for distinguishing among competing inputs during activity-dependent refinement of synaptic connections. Once released from presynaptic terminals, BDNF could act directly or, alternatively, by modulating responses to classical neurotransmitters. We recently found, for example, that BDNF, acting through TrkB, strongly inhibits AMPA responses of developing sensory relay neurons (Balkowiec et al., 2000).
Studies of other peptide transmitter systems, such as neuropeptide Y, vasoactive intestinal polypeptide, or the small cardioactive peptide, all indicate that the pattern of nerve impulses is critical for coding peptide release (Lundberg et al., 1986; Agoston et al., 1988; Lundberg et al., 1989; Pernow et al., 1989; Whim and Lloyd, 1994). Specifically, high-frequency stimulation releases larger amounts of neuropeptides compared with low-frequency stimulation. Moreover, KCl depolarization is significantly less effective, or even completely ineffective, at stimulating the release of vasoactive intestinal polypeptide compared with high-frequency electrical impulses (Belai et al., 1987; Agoston et al., 1988). Therefore, in this regard, activity-dependent BDNF release resembles that of other peptide neurotransmitters. In addition, studies of the intracellular distribution of BDNF have shown that the peptide is localized to dense-core vesicles in sensory axon terminals (Michael et al., 1997), as is typical of other sensory neuropeptides (Zupanc, 1996), and to vesicles of the regulated secretory pathway in cortical neurons (Fawcett et al., 1997; Haubensak et al., 1998). Thus, our current findings provide functional data, consistent with the subcellular distribution of BDNF, that support its role as a peptide neuromodulator at sensory synapses (Kerr et al., 1999; Balkowiec et al., 2000) as well as other synapses (Lohof et al., 1993; Lessmann et al., 1994; Kang and Schuman, 1995; Lessmann and Heumann, 1998; Levine et al., 1998). Moreover, the stimulus frequencies applied in the current study to evoke BDNF release from NPG neurons are within the physiological range for these cells (Jaffe and Sampson, 1976; Thoren, 1976; Coleridge et al., 1987).
In conclusion, the present study shows that primary sensory neurons can release endogenous BDNF in an activity-dependent manner, and that the magnitude of release depends on the pattern and frequency of stimulation. Given that transient, repetitive electrical stimulation resembles patterns of nerve activity in vivo more closely than continuous depolarization, we believe that this model provides new opportunities for defining physiological mechanisms of BDNF release and, consequently, BDNF roles in synaptic development and function.
Footnotes
This work was supported by US Public Health Service National Heart, Lung, and Blood Institute grants to D.M.K.
Correspondence should be addressed to Dr. David M. Katz, Department of Neurosciences, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106. E-mail: dmk4@po.cwru.edu.
REFERENCES
- 1.Acheson A, Lindsay RM. Non target-derived roles of the neurotrophins. Philos Trans R Soc Lond B Biol Sci. 1996;351:417–422. doi: 10.1098/rstb.1996.0037. [DOI] [PubMed] [Google Scholar]
- 2.Agoston DV, Conlon JM, Whittaker VP. Selective depletion of the acetylcholine and vasoactive intestinal polypeptide of the guinea-pig myenteric plexus by differential mobilization of distinct transmitter pools. Exp Brain Res. 1988;72:535–542. doi: 10.1007/BF00250599. [DOI] [PubMed] [Google Scholar]
- 3.Androutsellis-Theotokis A, McCormack WJ, Bradford HF, Stern GM, Pliego-Rivero FB. The depolarisation-induced release of [125I]BDNF from brain tissue. Brain Res. 1996;743:40–48. doi: 10.1016/s0006-8993(96)00981-x. [DOI] [PubMed] [Google Scholar]
- 4.Apfel SC, Wright DE, Wiideman AM, Dormia C, Snider WD, Kessler JA. Nerve growth factor regulates the expression of brain-derived neurotrophic factor mRNA in the peripheral nervous system. Mol Cell Neurosci. 1996;7:134–142. doi: 10.1006/mcne.1996.0010. [DOI] [PubMed] [Google Scholar]
- 5.Balkowiec A, Kunze DL, Katz DM. Brain-derived neurotrophic factor acutely inhibits AMPA-mediated currents in developing sensory relay neurons. J Neurosci. 2000;20:1904–1911. doi: 10.1523/JNEUROSCI.20-05-01904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beech JT, Bainbridge T, Thompson SJ. Incorporation of cells into an ELISA system enhances antigen-driven lymphokine detection. J Immunol Methods. 1997;205:163–168. doi: 10.1016/s0022-1759(97)00072-0. [DOI] [PubMed] [Google Scholar]
- 7.Belai A, Ralevic V, Burnstock G. VIP release from enteric nerves is independent of extracellular calcium. Regul Pept. 1987;19:79–89. doi: 10.1016/0167-0115(87)90077-2. [DOI] [PubMed] [Google Scholar]
- 8.Bito H. The role of calcium in activity-dependent neuronal gene regulation. Cell Calcium. 1998;23:143–150. doi: 10.1016/s0143-4160(98)90113-0. [DOI] [PubMed] [Google Scholar]
- 9.Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41:108–118. [PubMed] [Google Scholar]
- 10.Blöchl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 11.Blöchl A, Thoenen H. Localization of cellular storage compartments and sites of constitutive and activity-dependent release of nerve growth factor (NGF) in primary cultures of hippocampal neurons. Mol Cell Neurosci. 1996;7:173–190. doi: 10.1006/mcne.1996.0014. [DOI] [PubMed] [Google Scholar]
- 12.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 13.Brady R, Zaidi SIA, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brevet A, Pinto E, Peacock J, Stockdale FE. Myosin synthesis increased by electrical stimulation of skeletal muscle cell cultures. Science. 1976;193:1152–1154. doi: 10.1126/science.959833. [DOI] [PubMed] [Google Scholar]
- 15.Brosenitsch TA, Salgado-Commissariat D, Kunze DL, Katz DM. A role for L-type calcium channels in developmental regulation of transmitter phenotype in primary sensory neurons. J Neurosci. 1998;18:1047–1055. doi: 10.1523/JNEUROSCI.18-03-01047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buonanno A, Fields RD. Gene regulation by patterned electrical activity during neural and skeletal muscle development. Curr Opin Neurobiol. 1999;9:110–120. doi: 10.1016/s0959-4388(99)80014-2. [DOI] [PubMed] [Google Scholar]
- 17.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 18.Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- 19.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci USA. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleridge HM, Coleridge JC, Schultz HD. Characteristics of C fibre baroreceptors in the carotid sinus of dogs. J Physiol (Lond) 1987;394:291–313. doi: 10.1113/jphysiol.1987.sp016871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis GW, Schuster CM, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron. 1996;17:669–679. doi: 10.1016/s0896-6273(00)80199-3. [DOI] [PubMed] [Google Scholar]
- 22.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 23.Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J Neurosci. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- 25.Galuske RA, Kim DS, Castrén E, Thoenen H, Singer W. Brain-derived neurotrophic factor reverses experience-dependent synaptic modifications in kitten visual cortex. Eur J Neurosci. 1996;8:1554–1559. doi: 10.1111/j.1460-9568.1996.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 27.Ginty DD, Glowacka D, Bader DS, Hidaka H, Wagner JA. Induction of immediate early genes by Ca2+ influx requires cAMP-dependent protein kinase in PC12 cells. J Biol Chem. 1991;266:17454–17458. [PubMed] [Google Scholar]
- 28.Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- 29.Griesbeck O, Canossa M, Campana G, Gärtner A, Hoener MC, Nawa H, Kolbeck R, Thoenen H. Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc Res Tech. 1999;45:262–275. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<262::AID-JEMT10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 30.Haubensak W, Narz F, Heumann R, Lessmann V. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci. 1998;111:1483–1493. doi: 10.1242/jcs.111.11.1483. [DOI] [PubMed] [Google Scholar]
- 31.Heymach JV, Krüttgen A, Suter U, Shooter EM. The regulated secretion and vectorial targeting of neurotrophins in neuroendocrine and epithelial cells. J Biol Chem. 1996;271:25430–25437. doi: 10.1074/jbc.271.41.25430. [DOI] [PubMed] [Google Scholar]
- 32.Jaffe RA, Sampson SR. Analysis of passive and active electrophysiologic properties of neurons in mammalian nodose ganglia maintained in vitro. J Neurophysiol. 1976;39:802–815. doi: 10.1152/jn.1976.39.4.802. [DOI] [PubMed] [Google Scholar]
- 33.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 34.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 35.Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krüttgen A, Möller JC, Heymach JV, Shooter EM. Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc Natl Acad Sci USA. 1998;95:9614–9619. doi: 10.1073/pnas.95.16.9614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 38.Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- 39.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lohof AM, Ip NY, Poo M-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 41.Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58:76–87. [PubMed] [Google Scholar]
- 42.Lundberg JM, Rudehill A, Sollevi A, Fried G, Wallin G. Co-release of neuropeptide Y and noradrenaline from pig spleen in vivo: importance of subcellular storage, nerve impulse frequency and pattern, feedback regulation and resupply by axonal transport. Neuroscience. 1989;28:475–486. doi: 10.1016/0306-4522(89)90193-0. [DOI] [PubMed] [Google Scholar]
- 43.Lundberg JM, Rudehill A, Sollevi A, Theodorsson-Norheim E, Hamberger B. Frequency- and reserpine-dependent chemical coding of sympathetic transmission: differential release of noradrenaline and neuropeptide Y from pig spleen. Neurosci Lett. 1986;63:96–100. doi: 10.1016/0304-3940(86)90020-0. [DOI] [PubMed] [Google Scholar]
- 44.Marty S, Berzaghi MP, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci. 1997;20:198–202. doi: 10.1016/s0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- 45.McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 46.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 47.McDonough PM, Stella SL, Glembotski CC. Involvement of cytoplasmic calcium and protein kinases in the regulation of atrial natriuretic factor secretion by contraction rate and endothelin. J Biol Chem. 1994;269:9466–9472. [PubMed] [Google Scholar]
- 48.Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moore AN, Waxham MN, Dash PK. Neuronal activity increases the phosphorylation of the transcription factor cAMP response element-binding protein (CREB) in rat hippocampus and cortex. J Biol Chem. 1996;271:14214–14220. doi: 10.1074/jbc.271.24.14214. [DOI] [PubMed] [Google Scholar]
- 50.Pernow J, Schwieler J, Kahan T, Hjemdahl P, Oberle J, Wallin BG, Lundberg JM. Influence of sympathetic discharge pattern on norepinephrine and neuropeptide Y release. Am J Physiol. 1989;257:H866–H872. doi: 10.1152/ajpheart.1989.257.3.H866. [DOI] [PubMed] [Google Scholar]
- 51.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 52.Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- 53.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 54.Shieh PB, Ghosh A. Molecular mechanisms underlying activity-dependent regulation of BDNF expression. J Neurobiol. 1999;41:127–134. [PubMed] [Google Scholar]
- 55.Shieh PB, Hu S-C, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 56.Snider WD, Lichtman JW. Are neurotrophins synaptotrophins? Mol Cell Neurosci. 1996;7:433–442. doi: 10.1006/mcne.1996.0031. [DOI] [PubMed] [Google Scholar]
- 57.Stoop R, Poo M-M. Synaptic modulation by neurotrophic factors. Prog Brain Res. 1996;109:359–364. doi: 10.1016/s0079-6123(08)62118-4. [DOI] [PubMed] [Google Scholar]
- 58.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 59.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 60.Thoren PN. Atrial receptors with nonmedullated vagal afferents in the cat. Discharge frequency and pattern in relation to atrial pressure. Circ Res. 1976;38:357–362. doi: 10.1161/01.res.38.5.357. [DOI] [PubMed] [Google Scholar]
- 61.Tonra JR. Classical and novel directions in neurotrophin transport and research: anterograde transport of brain-derived neurotrophic factor by sensory neurons. Microsc Res Tech. 1999;45:225–232. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<225::AID-JEMT6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 62.Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci. 1999;22:221–227. doi: 10.1016/s0166-2236(98)01341-1. [DOI] [PubMed] [Google Scholar]
- 63.Vilim FS, Cropper EC, Price DA, Kupfermann I, Weiss KR. Release of peptide cotransmitters in Aplysia: regulation and functional implications. J Neurosci. 1996;16:8105–8114. doi: 10.1523/JNEUROSCI.16-24-08105.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wetmore C, Olson L. Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. J Comp Neurol. 1995;353:143–159. doi: 10.1002/cne.903530113. [DOI] [PubMed] [Google Scholar]
- 65.Whim MD, Lloyd PE. Differential regulation of the release of the same peptide transmitters from individual identified motor neurons in culture. J Neurosci. 1994;14:4244–4251. doi: 10.1523/JNEUROSCI.14-07-04244.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou X-F, Chie ET, Rush RA. Distribution of brain-derived neurotrophic factor in cranial and spinal ganglia. Exp Neurol. 1998;149:237–242. doi: 10.1006/exnr.1997.6716. [DOI] [PubMed] [Google Scholar]
- 67.Zhou X-F, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–951. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]
- 68.Zhuo H, Helke CJ. Presence and localization of neurotrophin receptor tyrosine kinase (TrkA, TrkB, TrkC) mRNAs in visceral afferent neurons of the nodose and petrosal ganglia. Brain Res Mol Brain Res. 1996;38:63–70. doi: 10.1016/0169-328x(95)00313-h. [DOI] [PubMed] [Google Scholar]
- 69.Zupanc GK. Peptidergic transmission: from morphological correlates to functional implications. Micron. 1996;27:35–91. doi: 10.1016/0968-4328(95)00028-3. [DOI] [PubMed] [Google Scholar]