

Abstract
NMDA receptor antagonists, such as (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801), potently block glutamate-induced neuronal death in myriad in vitro cell models and effectively attenuate ischemic damage in vivo. In this report, a novel role for MK-801 and other NMDA receptor antagonists in preconditioning neurons to withstand a wide range of subsequent lethal insults is described. A brief 30 min exposure to 0.1 μmMK-801, applied up to 96 hr before a “lethal” insult, protected primary cortical neurons from a diverse group of neurotoxic agents, including NMDA, β-amyloid, staurosporine, etoposide, and oxygen–glucose deprivation. This neuroprotective preconditioning by MK-801 arose from transient NMDA receptor inactivation, because the noncompetitive NMDA receptor antagonists memantine and nylindin and the competitive antagonist AP-5 gave similar effects. MK-801 protection was dependent on new protein synthesis during the first 2 hr, but not from 2 to 5 hr, after MK-801 exposure. The MK-801 transient did not alter the ability of NMDA to trigger normally lethal [Ca2+]i influx 48 hr later, but it did block early downstream signaling events coupled to NMDA neurotoxicity, including PKC inactivation and the activation of calpain. Moreover, MK-801 protected neurons from staurosporine-induced apoptosis, although caspase activation in these cells was unimpeded. It is likely that the stress associated with transient inactivation of NMDA receptors triggered a rapid compensatory survival response that provided long-term protection from a spectrum of insults, inducing apoptotic and nonapoptotic death. The possibility that MK-801 preconditioning blocks an event common to seemingly diverse death mechanisms suggests it will be an important tool for obtaining a clearer understanding of the salient molecular events at work in neuronal death and survival pathways.
Keywords: apoptosis, death signals, lethal injury, MK-801, oxygen–glucose deprivation, stress, preconditioning
It is well established that preconditioning of cardiac and brain tissue with sublethal insults prepares cells to better withstand subsequent injury (Chen and Simon, 1997; Dekker, 1998; Ferdinandy et al., 1998). In nervous tissue,in vivo preconditioning is primarily achieved by brief nonlethal ischemic insults (Kitagawa et al., 1990; Kirino et al., 1991;Liu et al., 1993), although other “stress”-inducing conditions, including hypoxia (Gidday et al., 1994), spreading depression (Kobayashi et al., 1995), and seizure induction (Sasahira et al., 1995), protect the brain from subsequent neurotoxic insults. Both the intensity of the sublethal insult and the latency between preconditioning and the final insult determines the extent of neuroprotection attained (Chen and Simon, 1997). In vitro, mild anoxia–hypoxia (Khaspekov et al., 1998; Tauskela et al., 1999), oxidative phosphorylation inhibition (Kasischke et al., 1996; Riepe et al., 1997), adenosine receptor stimulation (Heurteaux et al., 1995), KCl depolarization (Matsushima et al., 1996; Taga et al., 1997), and heat-shock (Liu et al., 1993) have been reported to spare brain slices or primary neurons from subsequent lethal injury. In general, preconditioning agents–conditions appear to act by inflicting sublethal stresses on neurons that cause a responsive upregulation of intrinsic neuroprotective survival mechanisms (Chen and Simon, 1997).
The cellular mechanisms propagating the initial stress and the subsequent compensatory protective processes in preconditioned neurons are poorly understood. The 24–48 hr necessary to induce ischemic tolerance in brain (Kitagawa et al., 1990; Kato et al., 1991; Kirino et al., 1991) and the observation that protein synthesis inhibitors block ischemic preconditioning (Barone et al., 1998) suggests that new gene expression mediates the response. A role for NMDA receptor signaling in ischemic preconditioning in the CNS is probable because NMDA receptor antagonists attenuate “ischemic tolerance” induced by brief ischemic insults in gerbils (Kato et al., 1992), oxygen–glucose deprivation (OGD) in cortical cultures (Grabb and Choi, 1999), and chemical hypoxia in hippocampal slices (Kasischke et al., 1996). Moreover, sublethal NMDA concentrations protect neurons from lethal death stimuli applied at later times (Bhave et al., 1999).
Competitive and noncompetitive NMDA receptor antagonists inhibit excitotoxic cell death induced by ischemia, trauma, and other insults. (+)-5-Methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) is a potent noncompetitive NMDA receptor antagonist widely used to block glutamate-induced death in a spectrum of in vitro neuronal models, and myriad examples attest to its effectiveness in attenuating ischemic damage in animals (Gill et al., 1992; McCulloch et al., 1993). However, long-term inactivation of NMDA receptor function by MK-801 is lethal to cortical neurons (Hwang et al., 1999), suggesting that a shorter, more controlled MK-801 exposure could possibly evoke a pharmacologically induced preconditioning of cortical cells. In this report, we demonstrate a novel role for MK-801 and other NMDA receptor antagonists as powerful preconditioning agents in primary cortical neurons. The data suggest that a brief, transient inactivation of NMDA receptors triggers a resilient phenotypic change in cortical cells, which confers to them long-lasting protection against a spectrum of insults inducing both apoptotic and nonapoptotic death.
MATERIALS AND METHODS
Reagents. Dulbecco's PBS, Eagle's Minimum Essential Medium (MEM), 5-fluoro-2′-deoxyuridine, glutamate, poly-l-lysine, propidium iodide (PI), trypan blue, and uridine were purchased from Sigma (St. Louis, MO). Heat-inactivated fetal bovine serum (FBS) andl-glutamine were purchased from Life Technologies (Grand Island, NY), and heat-inactivated horse serum was purchased from HyClone (Logan, UT). NMDA, AMPA, MK-801, CNQX, AP-5, glutamate-pyruvate transaminase (GPT), and memantine were purchased from Research Biochemicals Inc. (Natick, MA), and CP101,606 was a generous gift of Pfizer Pharmaceuticals (Groton, CT). Protein reagent was from Bio-Rad (Richmond, CA). Fura-2 AM was purchased from Molecular Probes (Eugene, OR). Staurosporine, etoposide and the cell-permeable calpastatin peptide were from Calbiochem (La Jolla, CA), cycloheximide (CHX) and nylidrin were from Sigma, anti-spectrin antibody was from Chemicon (Temecula, CA), and β-amyloid (1–40) was a generous gift of Neurochem Inc. (Montreal, Quebec, Canada).
Fetal rat cortical neuron isolation. Rat cortical neurons were cultured by a modification of a previously described method (Durkin et al., 1996). Timed-pregnant Sprague Dawley rats were purchased from Charles River Canada (St. Constant, Quebec, Canada). After the mother was killed by cervical dislocation under halothane anesthesia, the fetuses were removed on embryonic day 18 (E18), their brains were removed and placed in ice-cold PBS, and the cortices were dissected. The cortical neurons were dispersed by triturating with a 10 ml pipette, and the cells were centrifuged at 250 × g for 5 min at 4°C. The cells were gently resuspended in plating medium, and viable cells were determined by trypan blue exclusion. The cells were plated at 105 cells/cm2in plating medium consisting of 80% MEM, 10% heat-inactivated FBS, 10% heat-inactivated horse serum, 25 mm glucose, and 2 mml-glutamine. For the determination of neurotoxicity and protein kinase C (PKC) activity, the cells were plated in poly-l-lysine-coated 35 mm tissue culture dishes (DuPont-Life Technologies, Burlington, Ontario, Canada). For the measurement of intracellular free Ca2+ concentrations ([Ca2+]i), cortical neurons were plated on poly-l-lysine-coated 12 mm glass coverslips (Bellco Glass Inc., Vineland, NJ) and incubated in 24-well tissue culture dishes (DuPont-Life Technologies) in 0.5 ml of plating medium at 37°C in an atmosphere of 5% CO2 and 95% air. The cultures were mixed populations containing both neurons and glial cells. Therefore, to minimize glial growth, the cultures were treated with 15 μg/ml 5-fluoro-2′-deoxyuridine and 35 μg/ml uridine on day 4 of culture for 2 d. After exposure to mitotic inhibitors, one-half of the medium was removed and replaced with growth medium consisting of 90% MEM and 10% horse serum. Experiments were performed on cultures after 14–18 d in culture.
Culture treatments. For preconditioning, one-half of the culture conditioned medium (CM) was removed and saved, and the indicated doses of MK-801, memantine, nylidrin, AP-5, or CNQX were added to the cultures for 30 min (2 hr for AP-5). The cells were then washed twice in CSS buffer [(in mm): NaCl 120, KCl 5.4, MgCl2 0.8, CaCl20.8, glucose 15, and buffered with 25 Tris, pH 7.4], and the removed CM was replaced and incubated up to 96 hr before being subjected to NMDA (50 μm or indicated concentrations) for 5 min, AMPA (200 μm) for 15 min, staurosporine (1 μm or indicated concentrations) or etoposide (30 μm) for 4 hr, or Aβ:1–40 (50 μm) for 24 hr, or subjected to hypoxia–hypoglycemia for 4 hr as described previously (Tauskela et al., 1999). Oxygen–glucose deprivation was performed by placing culture dishes in BSS buffer [(in mm): 140 NaCl, 5 KCl, 2 CaCl2, 10 HEPES, and 0.03 glycine, pH 7.4) in a 37°C incubator housed in an anaerobic glove box (Forma Scientific, Marjetta, OH) in 95% N2–5% CO2, producing an O2partial pressure equal to 10–15 torr. After 4 hr, dishes were removed from the anaerobic incubator, and the BSS was replaced with the stored medium of the cell and maintained at normal atmospheric O2 with 5% CO2 at 37°C. In all cases, after cytotoxic treatment, the cultures were returned to CM. Cells were harvested 2 hr after NMDA addition for PKC activity measurements, 6 hr later for caspase 3 activity determinations (Chakravarthy et al., 1999), and 24 hr later for cell viability measurements assessed by propidium iodide exclusion. For “acute” exposure, MK-801 (1 μm) was added during the time of NMDA or staurosporine treatment and was removed along with the cytotoxin. All other additions to cells, whether transient or persistent, were made in CM taken from sister cultures.
Measurement of calpain activation by spectrin hydrolysis in intact neurons. A calpain-specific anti-brain-α-spectrin antibody was prepared as described previously (Roberts-Lewis et al., 1994) that selectively detects the 155 kDa spectrin breakdown product generated by μ-calpain. Spectrin hydrolysis was also measured with a commercially available polyclonal antibody raised against rat erythrocyte spectrin (240/235E; Chemicon) that detects intact spectrin and calpain-generated spectrin fragments (155 and 150 kDa), as well as the 120 kDa fragment generated by the action of caspase 3 (Pike et al., 1998; Wang et al., 1998). Cell lysates containing equal amounts of protein were separated by 10% SDS-PAGE, and the separated proteins were electrotransferred to polyvinylidene difluoride membranes. The membranes were blocked with 1% BSA in TBST solution (10 mm Tris-HCl, 150 mm NaCl, and 0.05% Tween 20, pH 8.0) and probed with the appropriate primary anti-spectrin antibody. The membranes were washed three times in TBST solution, incubated with alkaline phosphatase-conjugated anti-rabbit IgG, and washed an additional five times. The bands were visualized by an alkaline phosphatase developer solution containing 100 mm Tris-HCl, 100 mm NaCl, 5 mm MgCl2, 120 μm nitroblue tetrazoliumchloride, and 540 μm 5-bromo-4-chloro-3-indolylphosphate, pH 9.5 (Amersham Pharmacia Biotech, Arlington Heights, IL).
[Ca2+]imeasurement.[Ca2+]i was determined by measuring the fluorescence signal from the Ca2+-sensitive indicator fura-2. Neurons were loaded with fura-2 by incubating them for 30 min at 37°C in a Mg2+-free normal buffer solution (140 mm NaCl, 5 mm KCl, 2.5 mm CaCl2, 30 μm glycine, 2.6 mmdextrose, and 10 mm HEPES), containing 2.5 μm fura-2 AM. Experiments were conducted at room temperature on single cells or small groups of four to eight cells on glass coverslips in a custom-made coverslip holder fitted to the stage of a Zeiss IM inverted microscope (Carl Zeiss Canada, Don Mills, Ontario, Canada) coupled to a CM3 cation measurement spectrofluorimeter (Spex Inc., Newark, NJ). Measurements were performed using 350 and 380 nm excitation wavelengths alternating at a frequency of 1 Hz. The sample was illuminated through a 40× epifluorescence objective (UVFL40, numerical aperture of 0.85; Olympus Optical, Tokyo, Japan). The emitted light, after collection by this objective, was passed through a 505 nm interference filter (10 nm bandwidth), and its intensity was recorded by a photon counter detector mounted on the microscope. The [Ca2+]i was indicated by the ratios of the fluorescence intensities of fura-2 emission at 505 nm induced by the alternating excitation wavelengths 350 and 380 nm.
Protein kinase C activity determinations. Cells were washed twice with ice-cold PBS, suspended in 0.5 ml of ice-cold hypotonic lysis medium (1 mm NaHCO3, 5 mm MgCl2, and 100 μm phenylmethyl sulfonylfluoride) for 2 min, and lysed by vortexing vigorously for 2 min. All subsequent steps were performed at 4°C. Nuclei and unlysed cells were sedimented at 600 × g for 5 min, and membrane and cytosol fractions were separated by centrifugation at 100,000 × g for 10 min in a Beckman TL-100 ultracentrifuge. PKC activity in isolated cell membranes was measured by the direct PKC assay as described byChakravarthy et al. (1991, 1994). The PKC activity in the membranes was measured using a peptide substrate, Ac-FKKSFKL-NH2, corresponding to the specific PKC-phosphorylation site of the MARCKS protein. This substrate peptide has been shown to be phosphorylated equally by all major PKC isoforms (Heemskerk et al., 1993). The assay reaction mixture contained 20–50 μl of membrane suspension (containing 3–8 μg of protein) in assay buffer [consisting of 50 mm Tris-HCl (H 7.5), 5 mm MgCl2, 1 μm CaCl2, 100 μm sodium vanadate, 100 μm sodium pyrophosphate, 1 mm sodium fluoride, and 100 μm phenylmethylsulfonyl fluoride] and 10 μl of 750 μm PKC substrate peptide (in 50 mm Tris-HCl buffer, pH 7.5). The total volume was adjusted to 90 μl with 50 mm Tris-HCl buffer, pH 7.5. The reaction was started by adding 10 μl of 500 μm [32P]ATP (220 cpm/pmol in Tris buffer; 0.5 mCi/tube). After incubation for 10 min at 25°C, the reaction was stopped by adding 10 μl of 5% acetic acid, and the samples were clarified by centrifugation at 16,000 ×g for 5 min in a microfuge. A 90 μl sample of each supernatant was applied to P81 Whatman paper (2 cm2), and the papers were washed twice in 5% acetic acid (10 ml/cm2 paper) by gentle stirring for 10 min. The radioactivity bound to the washed papers was determined by liquid scintillation counting. To calculate the amount of radioactivity incorporated specifically into the substrate peptide, the nonspecific binding of32P to the P81 papers was determined as above in the absence of the peptide.
Statistical analysis. Statistical analyses were performed with the software package Statistica, using ANOVA ort tests where appropriate.
RESULTS
Primary cortical neurons were pretreated with 1 μmMK-801 for 30 min in CM, washed, and returned to drug-free CM for up to 96 hr before being subjected to a number of cytotoxic conditions. This brief MK-801 preconditioning was found to evoke a powerful, long-lasting and broad-based neuroprotective response in cortical cells. Thus, cultures transiently exposed to MK-801 were protected from the excitotoxic effects of NMDA applied 48 hr later (Fig.1A). Preconditioning with MK-801 offered complete protection against subsequent NMDA, even when the excitotoxin was added 96 hr after the 30 min MK-801 application (data not shown). Significantly, cortical neurons were also protected from the cytotoxic effects of AMPA, a 6 hr treatment with staurosporine or etoposide, or from Aβ (1–40) when subjected to a 30 min MK-801 treatment 48 hr before insult (Fig. 1A). The neuroprotective effects of MK-801 also extended to cortical cells exposed to 2 hr of OGD 48 hr after transient MK-801 application (Fig.1B). In contrast, the addition of BDNF, under conditions that strongly protect cortical cultures from excitotoxicity (Tremblay et al., 1999), had no ability to protect cells from hypoxia-induced damage (Fig. 1B). In all of the above cases, cell viability was assessed 24 hr after the normally lethal insult was applied. Although the extent of kill induced in cortical cells varied with the specific cytotoxic agent–condition used (i.e., ∼85% for NMDA to ∼40% for etoposide), MK-801 pretreatment consistently reduced the degree of kill in these cultures to <20% (Fig. 1A). Moreover, MK-801 pretreatment protected neurons from concentrations of NMDA and staurosporine that were, respectively, at least fourfold and tenfold higher than normally needed to inflict substantial cell death (Fig.2), indicting that protection was not the result of small changes in the effective lethal dose of these cytotoxins. MK-801 protected neurons from NMDA and staurosporine for at least 3 d after insult (data not shown), suggesting that the effects of transient MK-801 were long-lasting and not just delaying cell death as may well be the case with other protectants, such as caspase inhibitors (Lemaire et al., 1998; Grabb and Choi, 1999).
Fig. 1.

Transient (30 min) exposure of primary cortical neurons to MK-801 provides long-term protection against (A) NMDA, AMPA, staurosporine, etoposide, and β-amyloid (1–40) and (B) hypoxia, applied 48 hr later. E18 cortical cells were pretreated with 1 μmMK-801 or vehicle in CM for 30 min and then returned to drug-free CM for 48 hr. Cultures were then exposed to the indicated concentrations of NMDA for 5 min, staurosporine or etoposide for 6 hr, or Aβ (1–40) for 24 hr, all presented to cells in CM. Control cultures were treated with vehicle under identical conditions. For BDNF addition, cultures were exposed continuously to 100 ng/ml neurotrophin [concentrations and conditions shown to effectively block NMDA injury (Durkin et al., 1996] for 24 hr before hypoxia. In all cases after neurotoxic treatment, dishes were washed once in CSS buffer, and the saved conditioned medium was returned to the cultures. Dishes were incubated an additional 24 hr before cell viability was assessed by counting phase-bright cells (live cells) under phase-contrast microscopy and PI-labeled nuclei (dead cells) under fluorescence microscopy. Viability was calculated as the ratio of phase-bright cells to total cells (i.e., phase-bright plus PI-stained). Cell viability was expressed as a percentage using triplicate dishes, and error bars represent the mean ± SEM from at least three independent experiments. In A, *p < 0.05; **p < 0.01 compared with non-MK-801-treated counterparts. In B, *p < 0.05 compared with control.
Fig. 2.

MK-801 preconditioning offers protection against a wide range of (A) NMDA and (B) staurosporine concentrations. Cortical cultures were pretreated with 1 μm MK-801 or vehicle for 30 min and returned to drug-free CM for 48 hr before being exposed to NMDA for 5 min or staurosporine for 6 hr as described in the legend to Figure 1. Dishes were then washed once in CSS buffer, and saved conditioned medium was returned to the cultures for 24 hr, at which time cell viability was assessed by PI exclusion described in the legend to Figure 1. Results are presented as percent viable cell counts in quadruplicate dishes. InA, values are means ± SEM of at least four separate determinations, and values in B are of two separate experiments. *p < 0.01 compared with non-MK-801-treated conditions.
Several lines of evidence indicated that MK-801 preconditioning was mediated by its interaction with, and inhibition of, NMDA receptors: (1) the neuroprotective effect of MK-801 was mimicked by the structurally unrelated noncompetitive NMDA channel blocker memantine (Fig. 3A), the ifenprodil–halperidol site antagonist nylidrin, and the competitive antagonist AP-5 (Fig. 3B); (2) MK-801 protection was not observed in staurosporine- or etoposide-treated human neuroblastoma cells (SH-SY5Y) or primary rat astrocytes (data not shown), cells that do not possess functional NMDA receptors (Backus et al., 1989; Jensen and Chiu, 1990; Porter and McCarthy, 1997); and (3) MK-801 preconditioning protected within the same concentration range (i.e., 0.01–0.1 μm) that the acute administration of MK-801 effectively blocked NMDA toxicity, that is when the drug was added 5 min before and was present during a 5 min NMDA exposure (Fig.3C). In contrast, cortical cells exposed to the AMPA receptor antagonist CNQX under similar conditions were not protected from either NMDA or AMPA applied 48 hr later (Fig. 3D). Interestingly, the enzymatic removal of basal glutamate present in the conditioned culture medium of cortical cultures by a 2 hr exposure to 10 U/ml glutamate-pyruvate transaminase somewhat mimicked the effects of NMDA receptor inactivation in that the cells were partially protected from lethal NMDA applied 48 hr later (Fig. 3B). Increasing the concentration of GPT in the medium beyond 10 U/ml was unable to enhance the neuroprotective effects (data not shown). Thus, briefly blocking NMDA receptor activity by any of a number of NMDA receptor antagonists or conditions caused a protracted cellular response that protected cells from diverse cytotoxic agents.
Fig. 3.

Transient NMDA receptor inactivation in cortical neurons provides long-term protection from excitotoxicity. Transient application of (A) the NMDA receptor channel blocker memantine and (B) the competitive NMDA antagonist AP-5, the ifenprodil–halperidol site antagonist nylidrin, or GPT provides long-term protection against NMDA neurotoxicity.C, MK-801 concentrations that block NMDA receptor activation and NMDA-mediated cell death when added concomitantly (i.e.,t = 0) are the same that provide long-term neuroprotection when added transiently 2 d before NMDA (i.e.,t = −48 hr). D, MK-801, but not the AMPA channel blocker CNQX, provides protection against both NMDA- and AMPA-mediated neurotoxicity. Cortical cultures were pretreated with the indicated concentrations of MK-801, memantine, nylidrin, or vehicle for 30 min or with CNQX, AP-5, GPT, or vehicle for 2 hr in conditioned medium before being washed and returned to drug-free CM for 48 hr. In the case of GPT, 50 μm pyruvate was added along with the enzyme to ensure that the reaction would favor the deamimation of glutamate present in the media. Cells were then exposed to NMDA for 5 min or AMPA for 20 min as described in the legend to Figure 1. In some cases, previously untreated cultures were exposed to the indicated concentrations of MK-801 (B; t = 0) immediately before the 5 min NMDA exposure. After neurotoxin treatment, the dishes were washed with CSS buffer, retained drug-free conditioned medium was returned to the cultures for 24 hr, and cell viability was then assessed by PI exclusion as described in the legend to Figure 1. Results are presented as percent viable cell counts in quadruplicate dishes; values are means ± SEM and are (A) typical of at least two separate determinations. In A, *p < 0.05; **p < 0.01 compared with NMDA alone. B is the average of three separate experiments. In B, **p < 0.01 compared with NMDA alone. C is typical of at least three separate determinations. In C, **p < 0.01 compared with NMDA alone.D is typical of at least three separate determinations. In D, **p < 0.01 compared with respective excitotoxin-treated alone.
Neuroprotection by MK-801 and other NMDA receptor antagonists arose from long-lasting changes induced in cells during and shortly after their application rather than to any effects on NMDA receptors arising from residual drug present at the time of lethal injury. First, long-term neuroprotection afforded by transient NMDA receptor blockage occurred with a spectrum of NMDA receptor antagonists, including agents that would be expected to have rapid washout rates [i.e., the competitive antagonist AP-5 (Fig. 3B)]. In addition, MK-801 preconditioning had no discernible effects on subsequent Ca2+ influx via the NMDA receptor. Control and MK-801 preconditioned neurons were loaded with fura-2 AM 48 hr later and subsequently challenged with NMDA. As shown in Figure4A, NMDA-induced Ca2+ influx was not discernibly different in control and MK-801 pretreated cells, a result inconsistent with the neuroprotective effects of MK-801 arising from residual drug present at the time of the excitotoxic insult. Moreover, although MK-801 preconditioning blocked NMDA-induced cell death, it did not prevent a rapid and transient NMDA-mediated cell swelling (Fig. 4C), a well described early marker of glutamate–NMDA-induced injury (Basavappa and Ellory, 1996; Churchwell et al., 1996; Sakaguchi et al., 1999). More compelling evidence came from the observation that, as reported previously (Prehn et al., 1997), staurosporine-induced neuronal death is independent of glutamate receptors. As shown in Figure 5A, adding MK-801 to cortical cultures immediately before a 6 hr staurosporine exposure had no ameliorative effects on cell death, indicating that NMDA receptor activation was not coupled to staurosporine neurotoxicity in cortical cells. Despite this observation, MK-801 protection became apparent in cultures that were transiently (i.e., 30 min) pretreated with the drug 1–2 hr, or longer, before exposure to staurosporine (Fig.5A). Thus, the transient suppression of NMDA receptor function evoked a neuroprotective response that was not immediate but took time (albeit a relatively short period) to develop and mature. Attempts to perform similar “timing” experiments with NMDA-treated cells were not possible, because unlike staurosporine, the acute addition of MK-801 predictably blocked NMDA toxicity (Fig.3C). However, it was possible to perform such experiments with AP-5 that, as a competitive antagonist, would be expected to exhibit rapid off rates upon its removal from the culture medium. NMDA toxicity was effectively blocked by 100 μm AP-5 present during the 10 min exposure to lethal NMDA (Fig. 5B, compare lanes b, c). In contrast, AP-5 added transiently for 30 min 1 hr before NMDA exposure was not protective (Fig. 5B, lane d), indicating the drug indeed was effectively removed by washing. Nevertheless, almost complete protection was observed in cultures exposed for 2 hr to AP-5 between 150 and 30 min before NMDA (Fig. 5B, lane e). This finding demonstrated that, like the MK-801–staurosporine combination in Figure 5A, blocking NMDA receptor function transiently ∼2 hr before a lethal death signal was sufficient to generate the neuroprotective response. However, once triggered in neurons, this neuroprotective mechanism exhibited remarkable longevity. As shown in Figure 5C, a 2 hr exposure to AP-5 protected cells completely from lethal NMDA added up to 48 hr later. Unlike MK-801, which was still maximally protective in cortical neurons challenged with NMDA 96 hr later (data not shown), the neuroprotective effects of AP-5 gradually declined beyond the 48 hr point (Fig.5C).
Fig. 4.
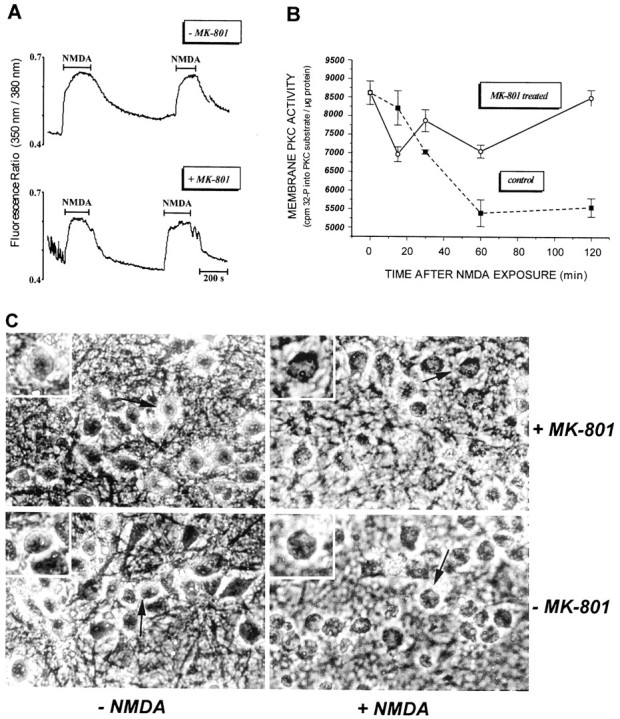
MK-801 preconditioning does not affect (A) Ca2+ influx via the NMDA receptor and (C) NMDA-induced cell swelling but (B) does block the NMDA-induced inactivation of membrane PKC. Cortical cultures were pretreated with 1 μmMK-801 or vehicle for 30 min and returned to drug-free CM for 48 hr before being exposed to NMDA for 5 min. A, For [Ca2+]i determination, cells were preloaded for 30 min with fura-2 AM before being treated with 50 μm NMDA for the indicated periods, and [Ca2+]i was measured as described in Materials and Methods. B, PKC activity was measured in membranes isolated from cortical cells 1 hr after a 5 min NMDA treatment by the method of Chakravarthy et al. (1991) described in Materials and Methods. Values are means ± SEM of three separate experiments each run in triplicate dishes (control and MK-801 values at 120 min are statistically significant; p < 0.001).C, Phase-contrast microphotographs of cortical neurons taken 1 hr after a 5 min exposure to 50 μm NMDA or vehicle, showing that although MK-801 pretreatment protects neurons from the excitotoxin, the characteristic rapid swelling of neurons after NMDA is not attenuated by MK-801 preconditioning.Arrows and insets (higher magnification) indicate representative cells.
Fig. 5.
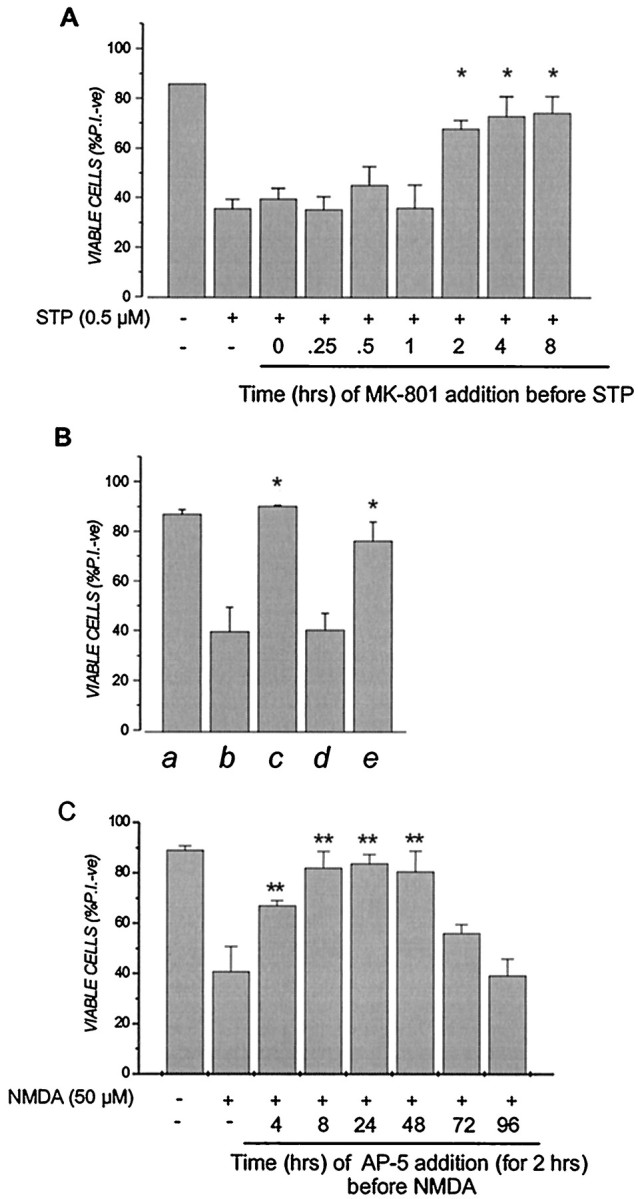
Transient NMDA receptor inactivation induces a rapid but long-lasting neuroprotective response against staurosporine (STP) or NMDA. A, Cortical cultures were pretreated with 1 μm MK-801 or vehicle in conditioned medium for 30 min and returned to drug-free conditioned medium for the indicated times before being exposed to 5 μmstaurosporine for 6 hr as described in the legend to Figure 1. For the 0 and 0.25 hr time points, MK-801 was left in the medium during the 6 hr staurosporine treatment period before being removed. Dishes were then washed once in CSS buffer, and saved conditioned medium was returned to the cultures for 24 hr. *p < 0.05 compared with staurosporine alone. B, Cultures were left untreated (lanes a–c) or pretreated with 100 μm AP-5 for 30 min (lane d) or 2 hr (lane e). All cultures were then washed once in CSS, and saved conditioned media was returned to the culture for 30 min before some cultures (lanes b–e) were exposed to 50 μm NMDA for 5 min. AP-5 was added to lane c during the 5 min NMDA exposure as positive control. Dishes were then washed once in CSS buffer, and saved conditioned medium was returned to the cultures for 24 hr. *p < 0.05 compared with NMDA alone. C, Cultures were pretreated with 100 μm AP-5 for 2 hr and washed once in CSS, and saved conditioned media was returned to the cultures for the indicated time before dishes were exposed to 50 μm NMDA for 5 min. Dishes were then washed once in CSS, and saved conditioned medium was returned to the cultures for 24 hr, at which time cell viability was assessed by PI exclusion, as described in the legend to Figure 1. **p < 0.01 compared with NMDA alone. Results are presented as percent viable cell counts in triplicate dishes; values are means ± SEM and are typical of at least four separate determinations.
Neuroprotective preconditioning afforded by MK-801 was effectively blocked by the protein synthesis inhibitor CHX present during and briefly after MK-801 treatment. Cells were exposed to CHX (10 μg/ml) during and for 90 min after a 30 min MK-801 treatment, or alternatively, from 2 to 5 hr after MK-801 withdrawal. Cultures treated in this manner were subsequently challenged with lethal NMDA (50 μm) 48 hr later. As shown in Figure6, MK-801 neuroprotection was reduced by >50% when CHX was applied during the first 2 hr after MK-801. In contrast, delaying the addition of CHX until 2 hr after the MK-801 transient was far less effective in reversing the preconditioning effect (Fig. 6). At the concentration of CHX used (i.e., 10 μg/ml), the steady-state level of protein synthesis in cortical cultures was inhibited by ∼50% within 30 min (data not shown). These results suggest that MK-801 preconditioning was dependent on new protein synthesis, and likely on new gene expression, during the first few hours after the MK-801 transient.
Fig. 6.

Neuroprotection afforded by MK-801 preconditioning is blocked by the protein synthesis inhibitor cycloheximide (CHX). Cortical cultures were pretreated (1) for 30 min with 10 μg/ml CHX before 1 μm MK-801 was added for 30 min [CHX was maintained in the cultures for an additional 90 min after MK-801 removal, i.e., CHX (0 to 2 hr)], or (2) for 30 min with 1 μm MK-801 before (10 μg/ml CHX was added for 3 hr 90 min after MK-801 removal [i.e., CHX (2 to 5 hr)]. After CHX treatment, cultures were washed in CSS buffer, returned to drug-free CM for 48 hr, and exposed to 50 μm NMDA for 5 min, and cell viability was determined 24 hr later as described in the legend to Figure 1. Results are presented as percent viable cell counts in triplicate dishes; values are means ± SEM and are typical of three separate determinations. *p < 0.05, significantly different from NMDA plus MK-801 group.
Perhaps the most significant aspect of MK-801 preconditioning was its ability to prevent the death of cortical cells by diverse agents acting by seemingly different routes. Thus, both staurosporine and etoposide kill cells, including cortical neurons, by apoptotic mechanisms (Bertrand et al., 1994; Koh et al., 1995; Solovyan et al., 1998; Tronov et al., 1999). In contrast, hypoxia and NMDA can trigger cortical death by necrosis or what has been described as “nonclassical” apoptosis (Ankarcrona et al., 1995; Lesort et al., 1997; Ankarcrona, 1998; Sohn et al., 1998). In our hands, staurosporine, but not NMDA, caused condensed chromatin and cell morphology characteristic of “classical” apoptosis (Fig.7A). This distinction was also evident by changes in apoptosis-associated caspases after such insults. Whereas lethal concentrations of staurosporine and etoposide caused activation of caspase 3 in cortical cells, NMDA did not (Fig.7B). In the case of NMDA, caspase activity was measured 6 hr after the NMDA transient, a point at which ∼30% of the cells were already dead as measured by propidium iodide uptake. Interestingly, caspase 3 activity increased in MK-801 preconditioned cortical cells challenged with staurosporine, despite the fact MK-801 protected the cells from death (Fig. 7B). This staurosporine-induced increase in caspase activation in MK-801 protected cortical cultures was not likely attributable to activation of the protease in glial cells present in the mixed cortical culture because staurosporine did not stimulate caspase in glial-enriched cultures in which the neuronal population was previously removed by chronic NMDA exposure (data not shown). This finding is consistent with a previous study (MacManus et al., 1997) showing that 1 μmstaurosporine is not toxic to glial cells present in mixed cortical cultures. These data suggest that the neuroprotective response evoked by MK-801 acts either downstream, or is independent, of caspases in staurosporine-injured neurons.
Fig. 7.
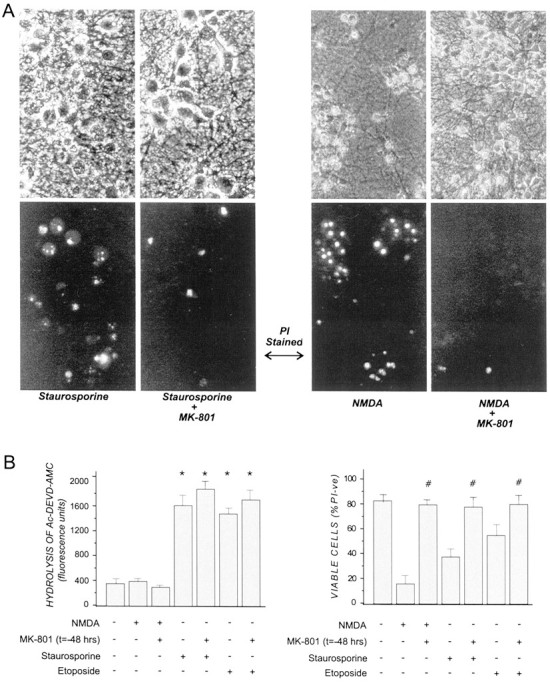
Caspase 3 activation was unaffected by MK-801 preconditioning in staurosporine- and etoposide-treated cortical cultures. Cortical cells were pretreated with 1 μm MK-801 or vehicle in CM for 30 min and returned to drug-free CM for 48 hr before being subjected to 50 μm NMDA for 5 min or to 0.5 μm staurosporine or 5 μm etoposide for 6 hr. Cultures were then either (A) photographed 24 hr later under phase-contrast (top panels) or fluorescence (bottom panels) after the additional of propidium iodide or (B) (1) lysed 6 hr after cytotoxin addition and caspase activity in lysates determined by hydrolysis of the caspase substrate Ac-DEVD-AMC, as described previously (Chakravarthy et al., 1999) or (2) incubated for 24 hr before cell viability was determined by PI exclusion as described in the legend to Figure 1. Results are presented as percent viable cell counts in quadruplicate dishes; values are means ± SEM and are (A) an average of three separate experiments (*p < 0.05 compared with untreated control) and (B) typical of at four separate determinations (#p < 0.05 compared with non-MK-801-treated counterpart).
Another protease linked to death mechanisms in neurons is the Ca2+-dependent, neutral protease calpain (Siman and Noszek, 1988; Wang and Yuen, 1994; Brorson et al., 1995;Rami et al., 1997). We have demonstrated previously that calpain activation is selectively coupled to the influx of Ca2+ via the NMDA receptor. Ca2+ influx triggered by other routes, such as voltage-dependent calcium channels or ionomycin, failed to induce calpain activation or cell death in cortical cells (Hewitt et al., 1998). The activation of calpain appears to be a critical event in the mechanism by which NMDA effects cortical death because concentrations of the calpain inhibitor calpeptin, which effectively block NMDA-induced hydrolysis of endogenous spectrin [a marker of calpain activation (Hewitt et al., 1998)], were also effective in preventing neuronal death by the excitotoxin (Fig.8A). As shown in Figure8B, a 30 min MK-801 transient applied either 48 hr before or concomitant with NMDA strongly ablated calpain activation and cell death effected by the excitotoxin. These results are consistent with those presented in Figure 4 in which the rapid inactivation of PKC activity, one of the cardinal Ca2+-dependent signaling events coupled to NMDA receptor overstimulation and toxicity (Durkin et al., 1996, 1997), was also blocked by MK-801 pretreatment 48 hr earlier (Fig.4B). Thus, both the inactivation of PKC and calpain activation were attenuated in MK-801 protected neurons, although Ca2+ influx via hyperactivated NMDA receptors appeared unaffected (Fig. 4). Collectively, these results suggest that the protracted protection afforded by MK-801 against NMDA may arise at least in part from its ability to block cellular events normally coupling the lethal influx of Ca2+ to a rapid PKC inactivation and calpain activation. However, this possible mechanism for MK-801 neuroprotection under excitotoxic conditions appeared not to translate to cortical neurons challenged with staurosporine. Unlike with NMDA, staurosporine-challenged neurons did not respond by activating calpain (Fig. 8C). In addition, whereas the selective calpain inhibitor calpastatin blocked NMDA neurotoxicity, it had no protective effects in cortical cultures treated with lethal staurosporine (Fig.8C).
Fig. 8.

MK-801 preconditioning prevents calpain activation in NMDA-injured cortical neurons. A, The ability of the calpain inhibitor calpeptin to attenuate spectrin hydrolysis and cell death in neurons treated with NMDA. Cortical cultures were treated with the indicated concentrations of calpeptin for 1 hr before being exposed to 50 μm NMDA for 5 min and then (1) returned to CM for 2 hr before cell lysates were prepared and calpain activity was estimated in Western blots by the extent of spectrin hydrolysis, as described in Materials and Methods or (2) returned to CM for 24 hr before cell viability was determined by PI exclusion as described in the legend to Figure 1. Bars represent the mean ± SD (*p < 0.05, significantly different from NMDA alone). B, NMDA, but not staurosporine, causes calpain activation in cortical cells. Cells were treated with NMDA for 5 min or continuous staurosporine exposure, and calpain activation was determined 3 hr later by the extent of spectrin hydrolysis measured in lysates subjected to Western blot analysis as described above. The calpain inhibitor calpastatin was added to cultures 1 hr before the cytotoxic agents. Cell viability was determined in parallel cultures 24 hr later as described in the legend to Figure 1. *p< 0.01, significantly different from NMDA alone. C, The effect of MK-801 preconditioning on NMDA-mediated calpain activation. Cortical cells were pretreated with 1 μm MK-801 or vehicle for 30 min and returned to drug-free CM for 48 hr before being subjected to 50 μm NMDA for 5 min. Cultures were then subjected to Western blot or cell viability analyses as described above. Viability results are presented as percent viable cell counts in triplicate dishes; values are means ± SEM and are typical of at three separate determinations (*p < 0.01, significantly different from all other groups).
DISCUSSION
Neuroprotective preconditioning evoked by MK-801 in cortical cells was directly mediated by transient inhibition of the NMDA receptor because the effect was also observed with a series of other NMDA receptor antagonists. Because MK-801 and memantine are use-dependent, open-channel NMDA receptor antagonists (Parsons et al., 1995; Palmer et al., 1997), it is likely that transient blockade of basal NMDA receptor activity is responsible for initiating the resultant neuroprotective response. The fact that AP-5, an antagonist that competes with glutamate for its binding site, is as neuroprotective as MK-801 is strong evidence that preconditioning arises from transient blockage of basal NMDA receptor activity. In the mixed cortical cultures used, the levels of glutamate secreted by both glial cells and neurons is likely sufficient to maintain basal glutamate receptor activation. Thus, one explanation for MK-801 neuroprotection is that selective blockade of NMDA receptor function by transient MK-801, in the presence of active AMPA and metabotropic receptors, exerts a nonlethal stress on neuronal cells. The finding that brief exposure to GPT partially mimicked the neuroprotective effects of MK-801 against lethal NMDA 48 hr later is consistent with this concept.
It has been shown recently that a continuous 48 hr exposure to MK-801 is lethal to cortical neurons (Hwang et al., 1999), indicating that long-term inhibition of NMDA receptor activity constitutes a lethal insult. As such, the 30 min MK-801 exposure used in the present study constitutes a “sublethal” stress that is consistent with conventional thinking related to preconditioning mechanisms. As with other preconditioning paradigms (Ferdinandy et al., 1998; de Zeeuw et al., 1999), this imposed stress can stimulate compensatory survival mechanisms that protects neurons from lethal insults at later times. However, to the best of our knowledge, no reported preconditioning agent or condition exhibits such long-lasting protection against such a diverse group of neurotoxic agents as does MK-801. This is in stark contrast to OGD preconditioning, which although effective against subsequent “lethal” OGD, has little capacity to protect neurons from staurosporine or excitotoxins (Grabb and Choi, 1999).
CHX effectively blocked neuroprotection when present during the first 2 hr after the 30 min MK-801 transient but not when added 2 hr after MK-801 withdrawal. Thus, the preconditioning effects of transient NMDA receptor inactivation depends on either new protein synthesis within the initial hours of NMDA receptor blockage or maintaining existing proteins with a high turnover rate. During this critical period, the broad-based neuroprotective response generated by MK-801 remains responsive for at least 48 hr and for 96 hr or longer in the case of NMDA-induced cortical death (data not shown). These findings indicate that a brief 1–2 hr recovery period was sufficient, and necessary, for a MK-801 transient to protect cortical cells from staurosporine (Fig. 5). The finding that a 2 hr exposure to AP-5 removed 30 min before NMDA application was highly neuroprotective supports this contention. Parenthetically, the fact that AP-5 exposure times of <2 hr gave progressively less protection from subsequent NMDA suggests that the degree of neuroprotection seen with a 30 min MK-801 exposure (i.e., Fig. 1) was attributable to slow washout rates, an understatement of the actual time required to exact maximal neuroprotection. Collectively, the data are consistent with neuroprotection arising from an ∼2 hr inactivation of the NMDA receptor complex. Regardless, the neuroprotective response upregulated by NMDA receptor inactivation in cortical cells develops rapidly and is remarkably long-lasting.
It is well known that NGF and BDNF protect neurons from injury under various in vivo and in vitro scenarios (Kirschner et al., 1996; Cheng et al., 1997; Kaplan and Miller, 1997; Miller and Kaplan, 1998). Because NMDA preconditioning in cerebellar neurons has been reported to stimulate BDNF expression (Bhave et al., 1999), it was possible that MK-801 neuroprotection arose, at least in part, from MK801-mediated BDNF production. In fact, the inability of MK-801 preconditioning to affect NMDA-induced Ca2+ influx, but to block the downstream inactivation of PKC that normally arises from lethal Ca2+ through the NMDA receptor (i.e., Fig.4), was similar to that reported for neuroprotection of cortical cells from NMDA by BDNF applied 24 hr before insult (Tremblay et al., 1999). However, several lines of evidence argue against MK-801 preconditioning being mediated solely by BDNF. First, whereas MK-801 protected cortical cells from OGD, BDNF did not (Fig. 1B). Although BDNF protected cortical cells from both NMDA and staurosporine (Tremblay et al., 1999), unlike MK-801, it was unable to protect from Aβ toxicity (our unpublished observations). Moreover, MK-801 protects cortical neurons from staurosporine without blocking caspase 3 activation (Fig. 7B), whereas BDNF blocked staurosporine-mediated caspase 3 activation under similar conditions (data not shown). Thus, MK-801 neuroprotection cannot be accounted for solely by the upregulation of BDNF in neurons or in supporting glia.
The broad-based neuroprotective response evoked by brief MK-801 exposure could have arisen from upregulation of a pleotropic neuroprotective “cassette”-containing elements capable of interceding at pivotal points along the death pathways triggered by NMDA, staurosporine, etoposide, hypoxia, Aβ, and other neurotoxins. Alternatively, the neuroprotective mechanism elicited by MK-801 may be focused on a single event at, or downstream of, a point at which all death pathways converge. Clearly, if the latter were the case, identifying such a “convergence point” would reveal potentially valuable therapeutic targets for blocking neuronal death by myriad processes. However, one line of evidence suggests this may not be the case. MK-801 blocked both the NMDA-induced inactivation of PKC and the activation of calpain, events critical for the excitotoxic process (Durkin et al., 1996, 1997; Hewitt et al., 1998; Tremblay et al., 1999). In fact, the inactivation of PKC appears to be indirectly responsible for calpain activation under these conditions (our unpublished observations). These early steps in the death mechanism take place within the first hour of NMDA addition, well before the first signs of irreversible cell damage. Because calpain inhibitors also protect cortical neurons from NMDA (Fig.8A,C), it is reasonable to suggest that MK-801 protection arises from its ability, direct or indirect, to prevent activation of this protease. However, calpain activation appears not to play a critical role in the death process imparted by staurosporine in these same cells. Staurosporine failed to induce calpain-mediated spectrin hydrolysis, nor was calpain inhibition able to ablate cell death at the hands of staurosporine. Hence, MK-801 appears to interfere with at least two different “death” events that can occur in cortical cells. These data are consistent with transient NMDA receptor inactivation eliciting a broad-based neuroprotective response capable of interceding at different points along the death pathways initiated by various neurotoxic agents.
On the other hand, the effects of MK-801 preconditioning on PKC activity may partially account for the pleotropic neuroprotective effects of the drug. We and others have reported that PKC activity is dramatically reduced in neuronal cells exposed to glutamate (Durkin et al., 1996, 1997)) and stroke (Cardell and Wieloch, 1993), and a recent study indicates that Aβ evokes a rapid loss of PKCα in human fibroblasts (Favit et al., 1998). Similarly, staurosporine is a broad-spectrum kinase inhibitor with a clear ability to block PKC activity in cortical cells. Thus, inactivation of specific PKC isoforms may be a crucial and common step in the death paradigms by which MK-801 preconditioning is efficacious. The data in Figures 4 and 8 indicate that the ability of a MK-801 transient to block NMDA-induced PKC inactivation 48 hr later, as well as the activation of calpain coupled to PKC inhibition, is not attributable to the drug affecting NMDA-induced Ca2+ influx. Because both PKC inactivation and calpain activation are fully dependent on Ca2+ influx through NMDA receptors, MK-801 preconditioning likely prevents signaling steps downstream of Ca2+ influx responsible for inactivating PKC and activating calpain. On a broader scope, these data suggest that a seminal neuroprotective response evoked by the brief MK-801 transient is to provide a long-term suppression of cellular processes responsible for inactivating specific PKC pools that are part of multiple death pathways in neurons.
In summary, we have demonstrated that transient inhibition of basal NMDA receptor activity in cortical neurons causes the upregulation of a powerful and long-lasting survival pathway(s) in cortical neurons that is able to intersect and block death mechanisms coupled to a variety of insults leading to apoptotic and nonapoptotic death. Whether MK-801 protection arises from a multifaceted, broad-spectrum neuroprotective cassette or from a specific response acting at or downstream of a point of convergence for multiple death pathways is not clear. The nature of the signaling events during the first few hours of transient NMDA receptor inactivation that initiate and maintain the protective state within cortical cells is clearly a critical issue that is presently being addressed. As such, MK-801 and other NMDA receptor antagonists should be important tools for garnering a clearer understanding of the salient molecular events at work in neuronal survival pathways, as well as providing information as to how this neuroprotective mechanism impacts and blocks death pathways evoked by a diverse and broad group of neurotoxic agents.
Footnotes
We thank R. E. Williams of this institute for providing the Ac-FKKSFKL-NH2 PKC substrate, D. Song for technical support, and Y. Wang for constructive discussions. This investigation was supported in part by Heart and Stroke Foundation of Ontario Grant T-3338.
Correspondence should be addressed to Dr. Jon P. Durkin, Cellular Neurobiology Group, Institute for Biological Sciences, National Research Council of Canada, Building M-54, Ottawa, Ontario, Canada K1A 0R6. E-mail: jon.durkin@nrc.ca.
REFERENCES
- 1.Ankarcrona M. Glutamate induced cell death: apoptosis or necrosis? Prog Brain Res. 1998;116:265–272. doi: 10.1016/s0079-6123(08)60442-2. [DOI] [PubMed] [Google Scholar]
- 2.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 3.Backus KH, Kettenmann H, Schachner M. Pharmacological characterization of the glutamate receptor in cultured astrocytes. J Neurosci Res. 1989;22:274–282. doi: 10.1002/jnr.490220307. [DOI] [PubMed] [Google Scholar]
- 4.Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, Feuerstein GZ. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke. 1998;29:1937–1950. doi: 10.1161/01.str.29.9.1937. [DOI] [PubMed] [Google Scholar]
- 5.Basavappa S, Ellory JC. The role of swelling-induced anion channels during neuronal volume regulation. Mol Neurobiol. 1996;13:137–153. doi: 10.1007/BF02740638. [DOI] [PubMed] [Google Scholar]
- 6.Bertrand R, Solary E, O'Connor P, Kohn KW, Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- 7.Bhave SV, Ghoda L, Hoffman PL. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J Neurosci. 1999;19:3277–3286. doi: 10.1523/JNEUROSCI.19-09-03277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brorson JR, Marcuccilli CJ, Miller RJ. Delayed antagonism of calpain reduces excitotoxicity in cultured neurons. Stroke. 1995;26:1259–1266. doi: 10.1161/01.str.26.7.1259. [DOI] [PubMed] [Google Scholar]
- 9.Cardell M, Wieloch T. Time course of the translocation and inhibition of protein kinase C during complete cerebral ischemia in the rat. J Neurochem. 1993;61:1308–1314. doi: 10.1111/j.1471-4159.1993.tb13623.x. [DOI] [PubMed] [Google Scholar]
- 10.Chakravarthy BR, Bussey A, Whitfield JF, Sikorska M, Williams RE, Durkin JP. The direct measurement of protein kinase C (PKC) activity in isolated membranes using a selective peptide substrate. Anal Biochem. 1991;196:144–150. doi: 10.1016/0003-2697(91)90130-l. [DOI] [PubMed] [Google Scholar]
- 11.Chakravarthy BR, Whitfield JF, Durkin JP. Inactive membrane protein kinase Cs: a possible target for receptor signalling. Biochem J. 1994;304:809–816. doi: 10.1042/bj3040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakravarthy BR, Walker T, Rasquinha I, Hill IE, MacManus JP. Activation of DNA-dependent protein kinase may play a role in apoptosis of human neuroblastoma cells. J Neurochem. 1999;72:933–942. doi: 10.1046/j.1471-4159.1999.0720933.x. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Simon R. Ischemic tolerance in the brain. Neurology. 1997;48:306–311. doi: 10.1212/wnl.48.2.306. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM. Marked age-dependent neuroprotection by brain-derived neurotrophic factor against neonatal hypoxic-ischemic brain injury. Ann Neurol. 1997;41:521–529. doi: 10.1002/ana.410410416. [DOI] [PubMed] [Google Scholar]
- 15.Churchwell KB, Wright SH, Emma F, Rosenberg PA, Strange K. NMDA receptor activation inhibits neuronal volume regulation after swelling induced by veratridine-stimulated Na+ influx in rat cortical cultures. J Neurosci. 1996;16:7447–7457. doi: 10.1523/JNEUROSCI.16-23-07447.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Zeeuw S, Van den Doel MA, Duncker DJ, Verdouw PD. New insights into cardioprotection by ischemic preconditioning and other forms of stress. Ann NY Acad Sci. 1999;874:178–191. doi: 10.1111/j.1749-6632.1999.tb09235.x. [DOI] [PubMed] [Google Scholar]
- 17.Dekker LR. Toward the heart of ischemic preconditioning [Editorial]. Cardiovasc Res. 1998;37:14–20. doi: 10.1016/s0008-6363(97)00241-1. [DOI] [PubMed] [Google Scholar]
- 18.Durkin JP, Tremblay R, Buchan A, Blosser J, Chakravarthy B, Mealing G, Morley P, Song D. An early loss in membrane protein kinase C activity precedes the excitatory amino acid-induced death of primary cortical neurons. J Neurochem. 1996;66:951–962. doi: 10.1046/j.1471-4159.1996.66030951.x. [DOI] [PubMed] [Google Scholar]
- 19.Durkin JP, Tremblay R, Chakravarthy B, Mealing G, Morley P, Small D, Song D. Evidence that the early loss of membrane protein kinase C is a necessary step in the excitatory amino acid-induced death of primary cortical neurons. J Neurochem. 1997;68:1400–1412. doi: 10.1046/j.1471-4159.1997.68041400.x. [DOI] [PubMed] [Google Scholar]
- 20.Favit A, Grimaldi M, Nelson TJ, Alkon DL. Alzheimer's-specific effects of soluble beta-amyloid on protein kinase C-alpha and -gamma degradation in human fibroblasts. Proc Natl Acad Sci USA. 1998;95:5562–5567. doi: 10.1073/pnas.95.10.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferdinandy P, Szilvassy Z, Baxter GF. Adaptation to myocardial stress in disease states: is preconditioning a healthy heart phenomenon? Trends Pharmacol Sci. 1998;19:223–229. doi: 10.1016/s0165-6147(98)01212-7. [DOI] [PubMed] [Google Scholar]
- 22.Gidday JM, Fitzgibbons JC, Shah AR, Park TS. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett. 1994;168:221–224. doi: 10.1016/0304-3940(94)90455-3. [DOI] [PubMed] [Google Scholar]
- 23.Gill R, Nordholm L, Lodge D. The neuroprotective actions of 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX) in a rat focal ischemia model. Brain Res. 1992;580:35–43. doi: 10.1016/0006-8993(92)90924-x. [DOI] [PubMed] [Google Scholar]
- 24.Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci. 1999;19:1657–1662. doi: 10.1523/JNEUROSCI.19-05-01657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heemskerk FM, Chen HC, Huang FL. Protein kinase C phosphorylates Ser152, Ser156 and Ser163 but not Ser160 of MARCKS in rat brain. Biochem Biophys Res Commun. 1993;190:236–241. doi: 10.1006/bbrc.1993.1036. [DOI] [PubMed] [Google Scholar]
- 26.Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci USA. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hewitt KE, Lesiuk HJ, Tauskela JS, Morley P, Durkin JP. Selective coupling of mu-calpain activation with the NMDA receptor is independent of translocation and autolysis in primary cortical neurons. J Neurosci Res. 1998;54:223–232. doi: 10.1002/(SICI)1097-4547(19981015)54:2<223::AID-JNR10>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Hwang JY, Kim YH, Ahn YH, Wie MB, Koh JY. N-Methyl-d-aspartate receptor blockade induces neuronal apoptosis in cortical culture. Exp Neurol. 1999;159:124–130. doi: 10.1006/exnr.1999.7126. [DOI] [PubMed] [Google Scholar]
- 29.Jensen AM, Chiu SY. Fluorescence measurement of changes in intracellular calcium induced by excitatory amino acids in cultured cortical astrocytes. J Neurosci. 1990;10:1165–1175. doi: 10.1523/JNEUROSCI.10-04-01165.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 31.Kasischke K, Ludolph AC, Riepe MW. NMDA-antagonists reverse increased hypoxic tolerance by preceding chemical hypoxia. Neurosci Lett. 1996;214:175–178. doi: 10.1016/0304-3940(96)12915-3. [DOI] [PubMed] [Google Scholar]
- 32.Kato H, Liu Y, Araki T, Kogure K. Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil: cumulative damage and protective effects. Brain Res. 1991;553:238–242. doi: 10.1016/0006-8993(91)90831-f. [DOI] [PubMed] [Google Scholar]
- 33.Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- 34.Khaspekov L, Shamloo M, Victorov I, Wieloch T. Sublethal in vitro glucose-oxygen deprivation protects cultured hippocampal neurons against a subsequent severe insult. NeuroReport. 1998;9:1273–1276. doi: 10.1097/00001756-199805110-00003. [DOI] [PubMed] [Google Scholar]
- 35.Kirino T, Tsujita Y, Tamura A. Induced tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood Flow Metab. 1991;11:299–307. doi: 10.1038/jcbfm.1991.62. [DOI] [PubMed] [Google Scholar]
- 36.Kirschner PB, Jenkins BG, Schulz JB, Finkelstein SP, Matthews RT, Rosen BR, Beal MF. NGF, BDNF and NT-5, but not NT-3 protect against MPP+ toxicity and oxidative stress in neonatal animals. Brain Res. 1996;713:178–185. doi: 10.1016/0006-8993(95)01513-2. [DOI] [PubMed] [Google Scholar]
- 37.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab. 1995;15:721–727. doi: 10.1038/jcbfm.1995.92. [DOI] [PubMed] [Google Scholar]
- 39.Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- 40.Lemaire C, Andreau K, Souvannavong V, Adam A. Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett. 1998;425:266–270. doi: 10.1016/s0014-5793(98)00252-x. [DOI] [PubMed] [Google Scholar]
- 41.Lesort M, Esclaire F, Yardin C, Hugon J. NMDA induces apoptosis and necrosis in neuronal cultures. Increased APP immunoreactivity is linked to apoptotic cells. Neurosci Lett. 1997;221:213–216. doi: 10.1016/s0304-3940(96)13310-3. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Kato H, Nakata N, Kogure K. Temporal profile of heat shock protein 70 synthesis in ischemic tolerance induced by preconditioning ischemia in rat hippocampus. Neuroscience. 1993;56:921–927. doi: 10.1016/0306-4522(93)90138-6. [DOI] [PubMed] [Google Scholar]
- 43.MacManus JP, Rasquinha I, Black MA, Laferriere NB, Monette R, Walker T, Morley P. Glutamate-treated rat cortical neuronal cultures die in a way different from the classical apoptosis induced by staurosporine. Exp Cell Res. 1997;233:310–320. doi: 10.1006/excr.1997.3558. [DOI] [PubMed] [Google Scholar]
- 44.Matsushima K, Hogan MJ, Hakim AM. Cortical spreading depression protects against subsequent focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1996;16:221–226. doi: 10.1097/00004647-199603000-00006. [DOI] [PubMed] [Google Scholar]
- 45.McCulloch J, Ozyurt E, Park CK, Nehls DG, Teasdale GM, Graham DI. Glutamate receptor antagonists in experimental focal cerebral ischaemia. Acta Neurochir Suppl (Wien) 1993;57:73–79. doi: 10.1007/978-3-7091-9266-5_11. [DOI] [PubMed] [Google Scholar]
- 46.Miller FD, Kaplan DR. Life and death decisions: a biological role for the p75 neurotrophin receptor [Editorial]. Cell Death Differ. 1998;5:343–345. doi: 10.1038/sj.cdd.4400385. [DOI] [PubMed] [Google Scholar]
- 47.Palmer GC, Miller JA, Cregan EF, Gendron P, Peeling J. Low-affinity NMDA receptor antagonists. The neuroprotective potential of ARL 15896AR. Ann NY Acad Sci. 1997;825:220–231. doi: 10.1111/j.1749-6632.1997.tb48432.x. [DOI] [PubMed] [Google Scholar]
- 48.Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34:1239–1258. doi: 10.1016/0028-3908(95)00092-k. [DOI] [PubMed] [Google Scholar]
- 49.Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL. Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. NeuroReport. 1998;9:2437–2442. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- 50.Porter JT, McCarthy KD. Astrocytic neurotransmitter receptors in situ and in vivo. Prog Neurobiol. 1997;51:439–455. doi: 10.1016/s0301-0082(96)00068-8. [DOI] [PubMed] [Google Scholar]
- 51.Prehn JH, Jordan J, Ghadge GD, Preis E, Galindo MF, Roos RP, Krieglstein J, Miller RJ. Ca2+ and reactive oxygen species in staurosporine-induced neuronal apoptosis. J Neurochem. 1997;68:1679–1685. doi: 10.1046/j.1471-4159.1997.68041679.x. [DOI] [PubMed] [Google Scholar]
- 52.Rami A, Ferger D, Krieglstein J. Blockade of calpain proteolytic activity rescues neurons from glutamate excitotoxicity. Neurosci Res. 1997;27:93–97. doi: 10.1016/s0168-0102(96)01123-6. [DOI] [PubMed] [Google Scholar]
- 53.Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, Ludolph AC, Dirnagl U, Hugon J. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning.”. J Cereb Blood Flow Metab. 1997;17:257–264. doi: 10.1097/00004647-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 54.Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sakaguchi T, Kuno M, Kawasaki K. Disparity of cell swelling and rapid neuronal death by excitotoxic insults in rat hippocampal slice cultures. Neurosci Lett. 1999;274:135–138. doi: 10.1016/s0304-3940(99)00702-8. [DOI] [PubMed] [Google Scholar]
- 56.Sasahira M, Lowry T, Simon RP, Greenberg DA. Epileptic tolerance: prior seizures protect against seizure-induced neuronal injury. Neurosci Lett. 1995;185:95–98. doi: 10.1016/0304-3940(94)11233-9. [DOI] [PubMed] [Google Scholar]
- 57.Siman R, Noszek JC. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–287. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 58.Sohn S, Kim EY, Gwag BJ. Glutamate neurotoxicity in mouse cortical neurons: atypical necrosis with DNA ladders and chromatin condensation. Neurosci Lett. 1998;240:147–150. doi: 10.1016/s0304-3940(97)00936-1. [DOI] [PubMed] [Google Scholar]
- 59.Solovyan V, Bezvenyuk Z, Huotari V, Tapiola T, Suuronen T, Salminen A. Distinct mode of apoptosis induced by genotoxic agent etoposide and serum withdrawal in neuroblastoma cells. Brain Res Mol Brain Res. 1998;62:43–55. doi: 10.1016/s0169-328x(98)00234-4. [DOI] [PubMed] [Google Scholar]
- 60.Taga K, Patel PM, Drummond JC, Cole DJ, Kelly PJ. Transient neuronal depolarization induces tolerance to subsequent forebrain ischemia in rats. Anesthesiology. 1997;87:918–925. doi: 10.1097/00000542-199710000-00027. [DOI] [PubMed] [Google Scholar]
- 61.Tauskela JS, Chakravarthy BR, Murray CL, Wang Y, Comas T, Hogan M, Hakim A, Morley P. Evidence from cultured rat cortical neurons of differences in the mechanism of ischemic preconditioning of brain and heart. Brain Res. 1999;827:143–151. doi: 10.1016/s0006-8993(99)01322-0. [DOI] [PubMed] [Google Scholar]
- 62.Tremblay R, Hewitt K, Lesiuk H, Mealing G, Morley P, Durkin JP. Evidence that brain-derived neurotrophic factor neuroprotection is linked to its ability to reverse the NMDA-induced inactivation of protein kinase C in cortical neurons. J Neurochem. 1999;72:102–111. doi: 10.1046/j.1471-4159.1999.0720102.x. [DOI] [PubMed] [Google Scholar]
- 63.Tronov VA, Konoplyannikov MA, Nikolskaya TA, Konstantinov EM. Apoptosis of unstimulated human lymphocytes and DNA strand breaks induced by the topoisomerase II inhibitor etoposide (VP-16). Biochemistry (Mosc) 1999;64:345–352. [PubMed] [Google Scholar]
- 64.Wang KK, Yuen PW. Calpain inhibition: an overview of its therapeutic potential. Trends Pharmacol Sci. 1994;15:412–419. doi: 10.1016/0165-6147(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 65.Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem. 1998;273:22490–22497. doi: 10.1074/jbc.273.35.22490. [DOI] [PubMed] [Google Scholar]