

Abstract
We report here that activation of the caspase-3 apoptotic cascade in spinal cord injury is regulated, in part, by calcineurin-mediated BAD dephosphorylation. BAD, a proapoptotic member of the bcl-2 gene family, is rapidly dephosphorylated after injury, dissociates from 14-3-3 in the cytosol, and translocates to the mitochondria of neurons where it binds to Bcl-xL. Pretreatment of animals with FK506, a potent inhibitor of calcineurin activity, or MK801, an NMDA glutamate receptor antagonist, blocked BAD dephosphorylation and abolished activation of the caspase-3 apoptotic cascade. These findings extend previous in vitroobservations and are the first to implicate the involvement of glutamate-mediated calcineurin activation and BAD dephosphorylation as upstream, premitochondrial signaling events leading to caspase-3 activation in traumatic spinal cord injury.
Keywords: calcineurin, caspase-3, FK506, glutamate receptors, NMDA, Bcl-xL, BAD translocation
Traumatic injury to the spinal cord results in widespread apoptotic cell death affecting several neuronal and glial cell populations (Crowe et al., 1997; Liu et al., 1997;Shuman et al., 1997). The loss of these cells contributes to long-term neurological dysfunction involving voluntary motor control and sensory deficits below the site of injury. Recent studies examining human and rat spinal cord suggest activation of the cytochromec-dependent capsase-3 apoptotic cascade in neuronal and oligodendroglial cell death after injury (Emery et al., 1998; Springer et al., 1999). The involvement of caspase-3 activation also has been implicated in other CNS injuries, including brain trauma (Yakovlev et al., 1997; Pike et al., 1998; Clark et al., 1999; Zhang et al., 1999a) and cerebral ischemia (Hara et al., 1997; Endres et al., 1998; Fink et al., 1998; Namura et al., 1998). However, the extracellular signals and upstream intracellular events responsible for activation of the cytochrome c-dependent caspase-3 cascade in spinal cord injury are not well understood.
Cytochrome c release can be regulated by members of the Bcl-2 gene family, including Bcl-2 and Bcl-xL, which are anchored to the mitochondrial outer membrane (Adachi et al., 1997; Kluck et al., 1997; Yang et al., 1997; Jurgensmeier et al., 1998). BAD is a proapoptotic member of the bcl-2 gene family that promotes apoptosis by binding to and inhibiting the actions of the antiapoptotic proteins Bcl-2 and Bcl-xL (Yang et al., 1995; Hsu et al., 1997; Zha et al., 1997). Under nonapoptotic conditions, BAD is maintained in an inactive phosphorylated state by serine-threonine kinases, including Akt (Datta et al., 1997; del Peso et al., 1997; Kennedy et al., 1999; Scheid et al., 1999), which has been shown to inhibit cytochrome c release (Kennedy et al., 1999). In a phosphorylated state, BAD is normally sequestered to the cytosol by 14-3-3 protein (Zha et al., 1996; Hsu et al., 1997; Zhang et al., 1999b). However, in the presence of an apoptotic stimulus, BAD is dephosphorylated, dissociates from 14-3-3, and translocates to the mitochondria where it binds to Bcl-2 and Bcl-xL.
A recent study provided evidence that the Ca2+-dependent protein phosphatase calcineurin is responsible for BAD dephosphorylation and subsequent apoptosis of primary hippocampal neurons and neuronal cell lines exposed to glutamate (Wang et al., 1999). Previous in vitrostudies implicate a role for glutamate-mediated calcineurin activation in neuronal apoptosis (Ankarcrona et al., 1996; Bochelen et al., 1999;Morioka et al., 1999). However, studies linking calcineurin-mediated BAD dephosphorylation to caspase-3 activation in vivo after CNS injury are lacking. It is well known that the extracellular levels of glutamate increase dramatically in spinal cord within minutes after injury (Faden et al., 1989; Panter et al., 1990; Farooque et al., 1996), followed hours later by the appearance of apoptotic features within the injury site (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997; Springer et al., 1999). Therefore, in the present study, we examined whether the dephosphorylation of BAD by glutamate-mediated calcineurin activation observed in vitro (Wang et al., 1999) also occurs in vivo after spinal cord injury. In addition, we determined whether these premitochondrial events are linked to activation of the caspase-3 apoptotic cascade reported previously (Emery et al., 1998; Springer et al., 1999).
MATERIALS AND METHODS
Spinal cord injury and drug treatments. Spinal cord contusion injuries were performed using the NYU impactor device. Animals receiving injury were anesthetized with pentobarbital (40 mg/kg), and a dorsal laminectomy was performed to expose the spinal cord at thoracic level T10. The vertebral column was stabilized by clamping the column at vertebra T8 and T11, and the 10 gm weight impactor probe was dropped from a distance of 25 mm. Control animals received a dorsal laminectomy only. At different times after injury, animals were killed by decapitation (for immunoblotting and caspase-3 activity assay experiments) or by perfusion with saline followed by 4% paraformaldehyde (immunohistofluorescence experiments). Independent groups of animals were pretreated with either FK506 (5 mg/kg, i.p.), a potent inhibitor of calcineurin activity (Fruman et al., 1992; Wiederrecht et al., 1993; Sewell et al., 1994), or vehicle. A separate group of animals were pretreated with MK801 (1 mg/kg, i.p.), an NMDA glutamate receptor antagonist, or saline vehicle. Animals receiving FK506 or MK801 were killed 1 hr after injury. All procedures used followed the guidelines established in the US Public Health Service Policy on Humane Care and Use of Laboratory Animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Kentucky Institutional Animal Care and Use Committee.
Immunoblotting. A 5.0 mm segment of freshly dissected spinal cord (containing the impact site in injured spinal cords) was homogenized in 0.5 ml of homogenization buffer, pH 7.4, containing 10 mm HEPES, 250 mm sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonicacid, 1 mm EDTA, 5 mm dithiothreitol, and 10 μl each of pepstatin (1 mg/ml), leupeptin (2.5 mg/ml), aprotinin (2 mg/ml), and phenylmethylsulfonyl fluoride (0.2 m). The samples were centrifuged at 40,000 × g for 30 min, and protein levels of the postmitochondrial supernatant fraction and resuspended pellet were determined using the BCA assay (Pierce, Rockford, IL). Samples of spinal cord protein (75–100 μg) were diluted in 4× SDS-PAGE running buffer, loaded onto 15% Tris-glycine BioRad (Hercules, CA) Ready gels, and electrophoresed at 4°C for 90 min at 100 V. Proteins were transferred to nitrocellulose blots (Amersham Pharmacia Biotech, Arlington Heights, IL), and blots were probed with polyclonal antibodies to Bcl-xL(1:1000; Santa Cruz Biotechnologies, Santa Cruz, CA), 14-3-3 (1:1000; Santa Cruz Biotechnologies), calcineurin A (1:1000; Chemicon, Temecula, CA), phosphorylation state-specific (Ser-112) BAD (1:1000; New England Biolabs, Beverly, MA), University of Texas Southwestern Medical Center, Dallas, TX/DNA fragmentation factor 45 (DFF45, 1:2500; a gift from X. Wang) and caspase-9 (1:5,000, a gift from X. Wang), and a monoclonal antibody to phosphorylation state-independent BAD (1:1000; Transduction Laboratories, Lexington, KY). Protein bands were visualized using peroxidase-conjugated secondary antibodies (adsorbed to rat serum proteins; Jackson ImmunoResearch, West Grove, PA) and enhanced chemiluminescence with ECL Hyperfilm (Amersham). The specificity of the immunoblotting signal was verified by omitting the primary antibody. Semiquantitative analysis of bands of interest corresponding to levels of phosphorylated BAD, activated caspase-9, and caspase-3-like DFF45 cleavage was performed using laser-scanned densitometric analysis.
Co-immunoprecipitation experiments. A phosphorylation state-independent BAD antibody (New England Biolabs) was cross-linked to protein G-Sepharose beads (Sigma, St. Louis, MO) using 20 mm dimethylpimelimidate. Postmitochondrial supernatant fractions (14-3-3 and calcineurin) or pellet fractions (Bcl-xL) of control and injured spinal cord (n = 4 laminectomy control; n = 4 injured animals per group) were incubated with the immobilized BAD antibody overnight at 4°C on a rocker table. After repeated washing in 0.1 m PBS, proteins bound to BAD were eluted away from the protein G-Sepharose–BAD antibody complex with 100 mm glycine, pH 2.7, and the eluant was neutralized with 1 m Tris, pH 9.0. Eluted samples were then separated by SDS-PAGE, and immunoblotting was used as above to analyze 14-3-3, calcineurin A, and Bcl-xLbound to BAD.
Immunohistofluorescence. At 1 hr after injury, control or injured animals (n = 6 per group) were perfused with saline followed by 4% paraformaldehyde. Spinal cords were cryoprotected in 25% sucrose and sectioned in a longitudinal manner on a freezing microtome at a thickness of 30 μm. Sections were incubated overnight at room temperature with the monoclonal antibody to phosphorylation state-independent BAD and a polyclonal antibody recognizing the mitochondrial heat shock protein 60 (HSP60, 1:250; Santa Cruz Biotechnologies). Detection was performed using a goat anti-mouse antibody (1:600) conjugated to AlexaFlour 488 (Molecular Probes, Eugene, OR) for BAD and a goat anti-rabbit antibody (1:400) conjugated to Cy3 (Jackson ImmunoResearch) for HSP60. Some sections were co-stained with the BAD antibody and a polyclonal antibody to the activated form of caspase-3 (1:500; R & D Systems, Minneapolis, MN) to identify cells exhibiting BAD translocation and undergoing early stages of apoptosis. Immunoreactivity was examined using a Leica (Nussloch, Germany) confocal laser scanning microscope. The specificity of the immunoreactive signal was verified by omitting the primary antibody in adjacent sections.
Caspase-3 enzyme activity assay. Postmitochondrial supernatant fractions (100 μg) from control or injured spinal cords (n = 4 animals per group) were added to 400 μl of caspase homogenization buffer containing 15.0 μm Z-DEVD-7-amino-4-trifluoromethyl coumarin (Enzyme Systems Products, Livermore, CA). Samples were incubated at room temperature for 5 min, and relative fluorescence (excitation at 380 nm and emission at 460 nm) was measured for 30 min. The specific activity of the samples was calculated relative to a standard curve using recombinant caspase-3 (Upstate Biotechnology, Lake Placid, NY).
Statistical analysis. The caspase-3 enzyme activity assay data and immunoblotting experiments were analyzed by a one-way ANOVA followed by Scheffe's post hoc analysis using the Statview 4.5 software program (Abacus Concepts, Berkeley, CA).
RESULTS
In our initial set of experiments, immunoblotting was used to examine potential changes in BAD, 14-3-3, Bcl-xL, and calcineurin protein levels at different time points after a contusion spinal cord injury. The time points examined (30 min and 1, 4, and 24 hr; n = 5 animals per time point) were chosen on the basis of our previous observation that caspase-3 activation can be detected as early as 1 hr after injury (Springer et al., 1999). These experiments indicate that the overall levels of BAD, 14-3-3, Bcl-xL, and calcineurin in extracts of injured spinal cord are no different from levels obtained from laminectomy control spinal cord at any of the time points examined (Fig.1). However, when blots were probed with a phosphorylation state-specific antibody to BAD, the levels of phosphorylated BAD in the supernatant fraction were found to be significantly reduced (p < 0.01) as early as 30 min after injury and were maintained at significantly low levels for as long as 24 hr (p < 0.01) after injury (Fig.1B). Omitting the primary antibodies on separate blots resulted in a complete loss of immunoreactive signal (data not shown).
Fig. 1.
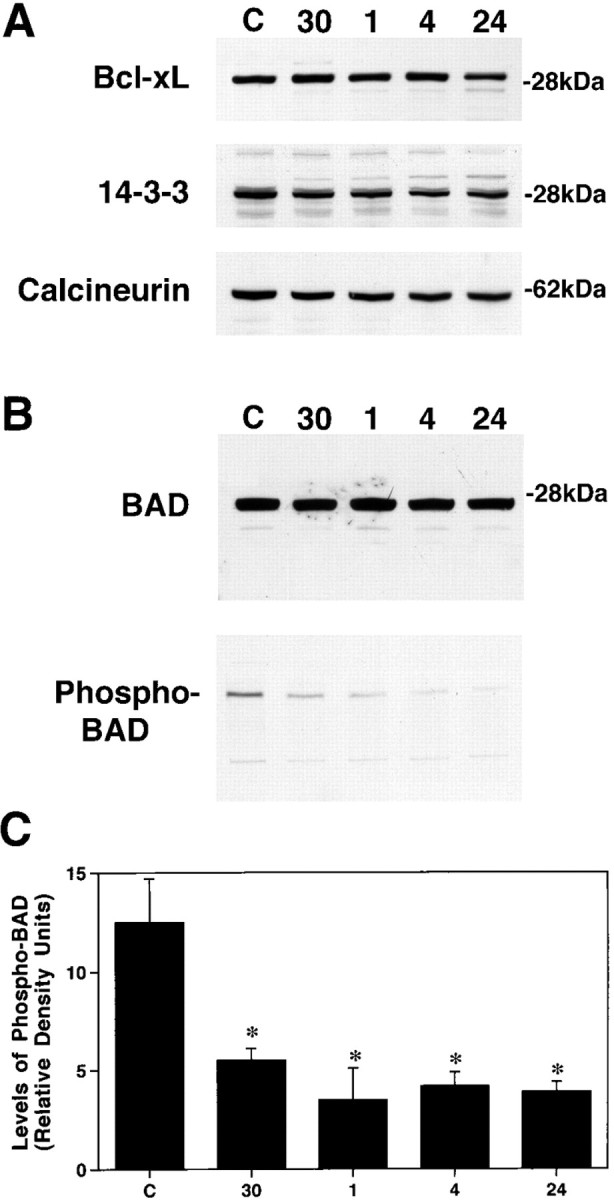
Representative immunoblots demonstrating that spinal cord injury results in rapid BAD dephosphorylation.A, Immunoblotting experiments demonstrate that spinal cord injury has no effect on overall levels of Bcl-xL, 14-3-3, or calcineurin A over 24 hr.B, BAD levels also are not affected over this period; however, the levels of phosphorylated BAD rapidly decline as early as 30 min after injury. C, Semiquantitative analysis reveals that the levels of phosphorylated BAD are significantly reduced at all time points examined after injury. *p < 0.01 (Scheffe's post hoc analysis).
Calcineurin-mediated dephosphorylation of BAD will result in its dissociation from the 14-3-3–calcineurin complex and the subsequent binding to Bcl-xL (Wang et al., 1999). Given that the levels of phosphorylated BAD decrease after spinal cord injury, we predicted that a similar event (dissociation of BAD from the 14-3-3–calcineurin complex) would occur in our injury model. Co-immunoprecipitation experiments were conducted on the supernatant and pellet fractions of laminectomy control and injured spinal cords (n = 4 animals per group). We found that 14-3-3 and calcineurin, but not Bcl-xL, co-immunoprecipitated with BAD in the supernatant fraction of laminectomy control animals (Fig. 2). In contrast, there is clear evidence that BAD rapidly dissociates from this complex as early as 30 min after injury (Fig. 2). At the same time point, the binding of BAD to Bcl-xL increases dramatically in the pellet fraction (Fig. 2), suggesting a translocation of BAD from the cytosol to the mitochondrial outer membrane where Bcl-xL is localized.
Fig. 2.

Representative immunoblots demonstrating that spinal cord injury results in the rapid dissociation of BAD from 14-3-3 and calcineurin A followed by binding to Bcl-xL. A phosphorylation state-independent BAD antibody (New England Biolabs) was cross-linked to protein G-Sepharose beads (Sigma). Postmitochondrial supernatant fractions (14-3-3 and calcineurin) or resuspended pellet fractions (Bcl-xL) of control or injured spinal cord were incubated overnight at 4°C with the immobilized BAD antibody. Proteins bound to BAD were eluted and separated by SDS-PAGE, and immunoblotting was used to analyze 14-3-3, calcineurin A and Bcl-xL bound to BAD. The data are representative of experiments from four control laminectomy and four spinal cord-injured animals. CTL, Control;30′, 30 min after injury.
To provide additional evidence that BAD redistributes to the mitochondria after injury, immunofluorescence histochemistry was used to examine the cellular staining pattern of phospho-independent BAD and activated capsase-3 in sections from control and injured spinal cord (n = 6 per group). BAD staining was weak and diffuse in neurons of dorsal and ventral gray spinal cord from laminectomy control animals (Fig. 3A). At 1 hr after injury, a localized punctate staining pattern indicative of organelle labeling was observed in numerous neurons in both the dorsal and ventral spinal cord (Fig. 3B). This staining pattern was not restricted to any particular neuronal cell size in sections examined from the injury epicenter; however, only the small and medium-sized neurons continued to show mitochondrial staining for BAD in sections examined 2.0 mm rostral and caudal to the injury epicenter. Double labeling of sections with an antibody to HSP60 (Fig.3C,D) supports the hypothesis that BAD has translocated to the mitochondria where it can bind to and inhibit Bcl-xL. These findings are consistent with the immunoprecipitation experiments described above.
Fig. 3.
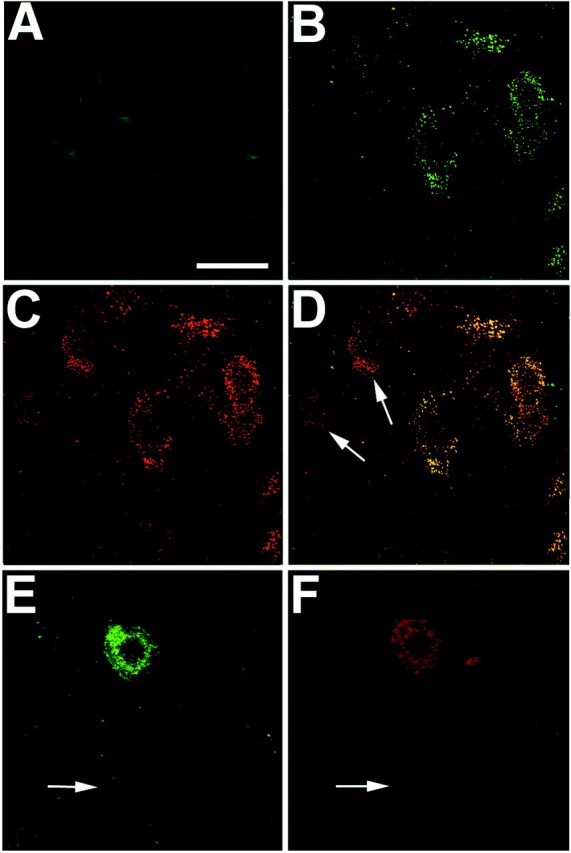
Representative laser-scanned confocal images demonstrating rapid translocation of BAD to the mitochondria and caspase-3 activation after spinal cord injury. Immunofluorescence histochemistry was performed on longitudinal spinal cord sections containing the lesion epicenter. A, Control section demonstrating weak diffuse immunoreactivity for BAD in uninjured gray matter neurons. B, Punctate BAD immunoreactivity in ventral horn neurons in the injury epicenter 1 hr after injury.C, HSP60 immunoreactivity double labeling of the same section as in B. D, Merged images fromB and C providing evidence that BAD immunoreactivity is associated with mitochondria. (Note the absence of colocalization in neurons denoted by arrows.) E, F, Double-labeling experiments indicate that caspase-3 activation (F) occurs in cells exhibiting BAD translocation (E). Arrows inE and F denote the absence of BAD or activated capsase-3 staining in a large α motor neuron. These photomicrographs are from 30-μm-thick spinal cord sections corresponding to 2.0 mm rostral to the injury epicenter. Scale bar, 25 μm.
In all sections analyzed, this mitochondrial staining pattern was restricted to neurons located in gray matter and was not observed in glial cell types as identified using markers for astrocytes (glial fibrillary acid protein) or oligodendroglia (CC1) (data not shown). The translocation of BAD to the mitochondria in neurons located within and near the injury epicenter suggests that early stages of the caspase-3 apoptotic process have been activated in these cells. To test this directly, sections were double-labeled with an antibody that recognizes the activated form of caspase-3. Examination of these sections revealed that the activated form of capsase-3 was observed in those neurons exhibiting mitochondrial staining for BAD (Fig.3E,F). As described above, the large α motor neurons distant to the injury epicenter (2.0 mm rostral or caudal) did not exhibit any evidence of BAD translocation or capsase-3 activation (Fig. 3E,F, arrows). Omitting the primary antibody from adjacent sections resulted in a complete loss of immunoreactive signal (data not shown).
The recent observations that glutamate stimulation can lead to Ca2+-mediated calcineurin activation and BAD dephosphorylation (Wang et al., 1999) and the findings that glutamate levels rise to excitotoxic levels within minutes after spinal cord injury (Faden et al., 1989; Panter et al., 1990; Farooque et al., 1996) suggest that glutamate-mediated calcineurin activation is responsible for BAD dephosphorylation after injury. Therefore, we examined whether glutamate-mediated calcineurin activation is an upstream event leading to BAD dephosphorylation and whether both contribute to the activation of the caspase-3 apoptotic cascade observed after injury (Springer et al., 1999). To accomplish this, animals (n = 5 animals per group) were pretreated with MK801, an antagonist of the Ca2+-permeable NMDA glutamate receptor subtype, FK506, an immunosuppressant that is a potent inhibitor of calcineurin activity (Dawson et al., 1994; Ide et al., 1996; Bochelen et al., 1999), or vehicle. Relative to vehicle-treated injured animals, pretreatment with either MK801 or FK506 had no effect on overall BAD levels but dramatically increased the levels of phosphorylated BAD (Fig.4A).
Fig. 4.
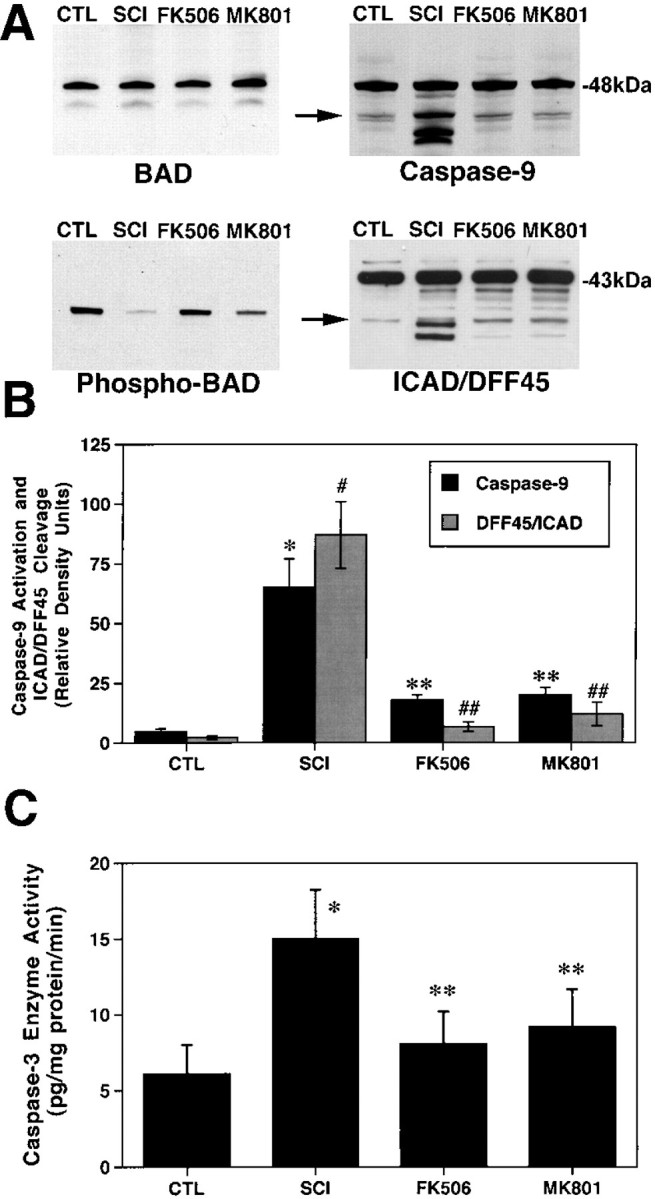
FK506 and MK801 treatments inhibit BAD dephosphorylation and caspase-3 activation. Animals were pretreated with FK506 (10 mg/kg, i.p., 1 hr before injury) or MK801 (0.3 mg/kg, i.p., 30 min before injury), and spinal cords were obtained 1 hr after injury. A, Representative immunoblots demonstrating that FK506 and MK801 treatments inhibited BAD dephosphorylation, caspase-9 activation, and ICAD/DFF45 cleavage. Postmitochondrial supernatant fractions were analyzed by immunoblotting using antibodies to phosphorylation state-independent and -dependent BAD, caspase-9, and ICAD/DFF45. B, Semiquantitative analysis of bands corresponding to activated caspase-9 and caspase-3-like ICAD/DFF45 cleavage (arrows) revealed a significant effect of FK506 and MK801. *p < 0.01 for caspase-9;#p < 0.01 for ICAD/DFF45 (Scheffe'spost hoc analysis). C, The caspase fluorogenic assay demonstrated that both FK506 and MK801 significantly reduced caspase-3 enzyme activity after spinal cord injury compared with vehicle treatments. *p < 0.01 compared with laminectomy controls; **p < 0.01 compared with spinal cord injury (Scheffe's post hoc analysis).CTL, Control; SCI, spinal cord injury.
BAD is normally phosphorylated by the serine-threonine kinase Akt when the latter is phosphorylated by phosphatidylinositol-3-kinase. A recent study reported that exposure of cerebellar granule cells to glutamate resulted in Akt dephosphorylation and a loss of kinase activity (Chalecka-Franaszek and Chuang, 1999). Therefore, it is possible that the dephosphorylation of BAD observed in the present study is attributable to a loss of Akt kinase activity and not an increase in calcineurin phosphatase activity. Although we did not measure Akt kinase activity in the present study, immunoblotting experiments with phosphorylation-specific Akt antibodies revealed that the phosphorylation state of Akt was unchanged in our injured spinal cord samples (data not shown).
If BAD dephosphorylation is directly linked to activation of the caspase-3 apoptotic cascade, then the same treatments should block the downstream steps involved in this cell death cascade. Therefore, we determined whether MK801 or FK506 treatment also affected caspase-9 and caspase-3 activation. Protein bands corresponding to caspase-9 activation are clearly evident in spinal cord extracts of vehicle-treated animals at 1 hr after injury (Fig.4A). However, extracts from the MK801- and FK506-treated animals show no indication of caspase-9 activation. Spinal cord extracts probed with an antibody to the caspase-3 substrate ICAD/DFF45 also indicate that caspase-3 activation is inhibited by FK506 or MK801 pretreatment. As previously reported, caspase-3-like ICAD/DFF45 cleavage is detectable 1 hr after injury (Springer et al., 1999). However, the relative absence of ICAD/DFF45 cleavage products in extracts from FK506- or MK801-treated animals suggests that these two treatments blocked caspase-3 activation. Semiquantitative analysis of bands corresponding to activated caspase-9 and caspase-3-like ICAD/DFF45 cleavage revealed a significant effect of FK506 and MK801 (Fig. 4B). Specifically, both treatments significantly reduced (p < 0.01) caspase-9 activation and the appearance of the caspase-3-mediated cleavage product of ICAD/DFF45. An enzyme activity assay was used to directly test whether FK506 or MK801 pretreatment suppressed caspase-3 activation in a different set of animals (n = 5 animals per group), as suggested by the immunoblotting experiments. As shown in Figure 4B, caspase-3 enzyme activity was significantly elevated 1 hr after spinal cord injury, which replicates the results of our previous study (Springer et al., 1999). However, caspase-3 enzyme activity levels were significantly reduced in animals pretreated with FK506 and MK801 (Fig. 4B), which is consistent with the observation that both treatments reduced the cleavage of ICAD/DFF45 (Fig. 4A).
DISCUSSION
There is clear evidence that apoptotic cell death occurs in several neuronal and glial cell types after traumatic spinal cord injury (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997). Moreover, it is apparent that manifestation of this cell death process involves activation of the caspase-3 apoptotic cascade (Emery et al., 1998; Springer et al., 1999). However, the extracellular and intracellular events contributing to caspase-3 activation in spinal cord injury are not well understood and need to be identified to develop appropriate therapeutic approaches targeting this cell death process. The findings of the present study are consistent with previous studies using cell lines (Zha et al., 1996; Harada et al., 1999;Kennedy et al., 1999; Wang et al., 1999) but provide the first evidence linking calcineurin-mediated BAD dephosphorylation to the rapid activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Specifically, spinal cord neurons within and in close proximity to the injury epicenter were found to exhibit mitochondrial staining for BAD within 1 hr after injury. These studies also demonstrate that spinal cord neurons exhibiting mitochondrial translocation of BAD contain evidence of caspase-3 activation. This early time point after injury corresponds to times when the levels of phosphorylated BAD first decrease and the binding of BAD to Bcl-xL is first observed. In addition, the translocation of BAD correlates with the time point when caspase-3 activation is first detected using biochemical measures (Springer et al., 1999). These observations indicate that, as might be predicted, BAD translocation and caspase-3 activation occur before the appearance of the classic morphological features associated with an apoptotic phenotype (Crowe et al., 1997;Liu et al., 1997; Shuman et al., 1997).
An interesting observation in the present study is that, within the injury epicenter, BAD translocation and caspase-3 activation are present in numerous small neurons scattered throughout the dorsal horn, as well as medium- and large-diameter (α motor neurons) neurons of the ventral horn. However, this staining pattern was absent in the large α motor neurons of sections distant to the injury epicenter. These findings are consistent with our previous observation that events associated with caspase-3 activation occur primarily in α motor neurons in relatively close proximity to the injury epicenter (Springer et al., 1999). The reason for the lack of BAD translocation and caspase-3 activation in α motor neurons distant to the injury epicenter is not clear at this time but may be related to the time points examined, the differential response of the α motor neurons to the actions of glutamate (Urca and Urca, 1990; Regan, 1996; Pizzi et al., 2000), or possibly the presence of high levels of a naturally occurring inhibitor of apoptosis in the spinal cord α motor neuron population (Xu et al., 1997; Pari et al., 2000). Regardless, these findings provide additional evidence supporting the hypothesis that caspase-3 activation is rapidly occurring in neurons exhibiting BAD translocation.
The results of this and previous studies suggest that calcineurin and caspase-3 can be activated, in part, by the actions of glutamate on the Ca2+-permeable NMDA glutamate receptor subtype (Lieberman and Mody, 1994; Ankarcrona et al., 1996; Wood and Bristow, 1998; Wang et al., 1999). Specifically, pretreatment of animals with FK506, an immunosuppressant that inhibits calcineurin, or the NMDA receptor antagonist NMDA inhibited BAD dephosphorylation and subsequent caspase-3 activation. However, it cannot be ruled out that glutamate could also be acting through other receptor subtypes, including AMPA/kainate and metabotropic receptors. In addition, it is unclear whether FK506 pretreatment inhibits or simply delays activation of the apoptotic process. There is evidence that FK506 levels remain elevated in the CNS for up to 72 hr after a peripheral administration (Butcher et al., 1997), suggesting that the actions of FK506 may persist for several days. Therefore, studies examining the long-term consequences of FK506 treatment on injury-related tissue loss and functional recovery are warranted, and these experiments are currently being conducted. Regardless, the results of the present experiments using FK506 pretreatment as a tool support our hypothesis that calcineurin activation is involved in rapid activation of the caspase-3 apoptotic cascade (Springer et al., 1999).
Although glutamate-mediated neuronal apoptosis has been suggested to occur under certain conditions (Ankarcrona et al., 1995; Nicotera et al., 1997), the present findings demonstrate the first mechanism by which glutamate activates the caspase-3 apoptotic cascade after spinal cord injury. Taken together, we postulate that the release of glutamate soon after injury (<1 hr) results in the influx of Ca2+ through NMDA receptors, leading to calcineurin activation and BAD dephosphorylation. After dephosphorylation, BAD dissociates from the 14-3-3–calcineurin complex and then translocates to the mitochondria where it binds and inhibits Bcl-xL. The binding of proapoptotic proteins such as BAD to 14-3-3 proteins has been shown to promote cell survival by suppressing apoptotic signaling events (Wang et al., 1999; Zhang et al., 1999b). Therefore, our results demonstrating the dissociation of BAD from 14-3-3 after injury indicate that this step is an early event in the apoptotic process. Inhibiting the actions of Bcl-xL could result in the release of cytochromec from the mitochondria, which leads to activation of the postmitochondrial caspase-3 apoptotic cascade (Adachi et al., 1997;Kluck et al., 1997; Yang et al., 1997; Jurgensmeier et al., 1998). Our recent study demonstrating rapid activation of caspase-3 after spinal cord injury is consistent with this hypothesis (Springer et al., 1999).
It should be pointed out that the results of our study do not imply that glutamate-mediated excitotoxic events are not involved in cell death after spinal cord injury. Rather, these findings suggest that glutamate can result in cell death through apoptotic as well as necrotic mechanisms. Regardless, understanding the biochemical pathways involved in the apoptotic process is essential for identifying potential treatment strategies to examine the involvement of necrotic versus apoptotic neuronal cell death. Interestingly, a recent study reported that inhibitors of caspase-3 and MK801 can act synergistically to reduce infarct size after transient focal cerebral ischemia (Ma et al., 1998). In summary, the results of the present study provide strong evidence that glutamate release after spinal cord injury results in calcineurin-mediated BAD dephosphorylation, which is an upstream step leading to caspase-3 activation. Future studies targeting these signaling events will be of fundamental importance for determining the contribution of this cell death process to neurological dysfunction after spinal cord injury.
Footnotes
This work was supported by National Institutes of Health Grant NS40015 and a grant from the Kentucky Spinal Cord and Head Injury Research Trust. We thank Dr. Bruce Maley of the Electron Microscopy and Imaging Facility for assistance with the confocal images.
Correspondence should be addressed to Dr. Joe E. Springer, Department of Anatomy and Neurobiology, University of Kentucky Medical Center, 800 Rose Street, Lexington, KY 40536-0084. E-mail: jspring@pop.uky.edu.
REFERENCES
- 1.Adachi S, Cross AR, Babior BM, Gottlieb RA. Bcl-2 and the outer mitochondrial membrane in the inactivation of cytochrome c during Fas-mediated apoptosis. J Biol Chem. 1997;272:21878–21882. doi: 10.1074/jbc.272.35.21878. [DOI] [PubMed] [Google Scholar]
- 2.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 3.Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- 4.Bochelen D, Rudin M, Sauter A. Calcineurin inhibitors FK506 and SDZ ASM 981 alleviate the outcome of focal cerebral ischemic/reperfusion injury. J Pharmacol Exp Ther. 1999;288:653–659. [PubMed] [Google Scholar]
- 5.Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase akt-1 and suppresses glutamate-induced inhibition of akt-1 activity in neurons (in process citation). Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, Hamilton RL, Loeffert JE, Graham SH. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813–821. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- 8.Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 9.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 10.Dawson TM, Steiner JP, Lyons WE, Fotuhi M, Blue M, Snyder SH. The immunophilins, FK506 binding protein and cyclophilin, are discretely localized in the brain: relationship to calcineurin. Neuroscience. 1994;62:569–580. doi: 10.1016/0306-4522(94)90389-1. [DOI] [PubMed] [Google Scholar]
- 11.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 12.Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD. Apoptosis after traumatic human spinal cord injury. J Neurosurg. 1998;89:911–920. doi: 10.3171/jns.1998.89.6.0911. [DOI] [PubMed] [Google Scholar]
- 13.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 15.Farooque M, Hillered L, Holtz A, Olsson Y. Changes of extracellular levels of amino acids after graded compression trauma to the spinal cord: an experimental study in the rat using microdialysis. J Neurotrauma. 1996;13:537–548. doi: 10.1089/neu.1996.13.537. [DOI] [PubMed] [Google Scholar]
- 16.Fink K, Zhu J, Namura S, Shimizu-Sasamata M, Endres M, Ma J, Dalkara T, Yuan J, Moskowitz MA. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J Cereb Blood Flow Metab. 1998;18:1071–1076. doi: 10.1097/00004647-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA. 1992;89:3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 20.Hsu SY, Kaipia A, Zhu L, Hsueh AJ. Interference of BAD (Bcl-xL/Bcl-2-associated death promoter)-induced apoptosis in mammalian cells by 14-3-3 isoforms and P11. Mol Endocrinol. 1997;11:1858–1867. doi: 10.1210/mend.11.12.0023. [DOI] [PubMed] [Google Scholar]
- 21.Ide T, Morikawa E, Kirino T. An immunosuppressant, FK506, protects hippocampal neurons from forebrain ischemia in the mongolian gerbil. Neurosci Lett. 1996;204:157–160. doi: 10.1016/0304-3940(96)12352-1. [DOI] [PubMed] [Google Scholar]
- 22.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 25.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 26.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma J, Endres M, Moskowitz MA. Synergistic effects of caspase inhibitors and MK-801 in brain injury after transient focal cerebral ischaemia in mice. Br J Pharmacol. 1998;124:756–762. doi: 10.1038/sj.bjp.0701871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morioka M, Hamada J, Ushio Y, Miyamoto E. Potential role of calcineurin for brain ischemia and traumatic injury. Prog Neurobiol. 1999;58:1–30. doi: 10.1016/s0301-0082(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 29.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicotera P, Ankarcrona M, Bonfoco E, Orrenius S, Lipton SA. Neuronal necrosis and apoptosis: two distinct events induced by exposure to glutamate or oxidative stress. Adv Neurol. 1997;72:95–101. [PubMed] [Google Scholar]
- 31.Panter SS, Yum SW, Faden AI. Alteration in extracellular amino acids after traumatic spinal cord injury. Ann Neurol. 1990;27:96–99. doi: 10.1002/ana.410270115. [DOI] [PubMed] [Google Scholar]
- 32.Pari G, Berrada F, Verge G, Karpati G, Nalbantoglu J. Immunolocalization of NAIP in the human brain and spinal cord. NeuroReport. 2000;11:9–14. doi: 10.1097/00001756-200001170-00003. [DOI] [PubMed] [Google Scholar]
- 33.Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL. Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. NeuroReport. 1998;9:2437–2442. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- 34.Pizzi M, Benarese M, Boroni F, Goffi F, Valerio A, Spano PF. Neuroprotection by metabotropic glutamate receptor agonists on kainate-induced degeneration of motor neurons in spinal cord slices from adult rat (in process citation). Neuropharmacology. 2000;39:903–910. doi: 10.1016/s0028-3908(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 35.Regan RF. The vulnerability of spinal cord neurons to excitotoxic injury: comparison with cortical neurons. Neurosci Lett. 1996;213:9–12. doi: 10.1016/0304-3940(96)12823-8. [DOI] [PubMed] [Google Scholar]
- 36.Scheid MP, Schubert KM, Duronio V. Regulation of Bad phosphorylation and association with Bcl-xL by the MAPK/Erk kinase. J Biol Chem. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- 37.Sewell TJ, Lam E, Martin MM, Leszyk J, Weidner J, Calaycay J, Griffin P, Williams H, Hung S, Cryan J. Inhibition of calcineurin by a novel FK-506-binding protein. J Biol Chem. 1994;269:21094–21102. [PubMed] [Google Scholar]
- 38.Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res. 1997;50:798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 39.Springer J, Azbill R, Knapp P. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 40.Urca G, Urca R. Neurotoxic effects of excitatory amino acids in the mouse spinal cord: quisqualate and kainate but not N-methyl-d-aspartate induce permanent neural damage. Brain Res. 1990;529:7–15. doi: 10.1016/0006-8993(90)90805-l. [DOI] [PubMed] [Google Scholar]
- 41.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 42.Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of FK-506 and cyclosporin A. Ann NY Acad Sci. 1993;696:9–19. doi: 10.1111/j.1749-6632.1993.tb17137.x. [DOI] [PubMed] [Google Scholar]
- 43.Wood AM, Bristow DR. N-Methyl-d-aspartate receptor desensitisation is neuroprotective by inhibiting glutamate-induced apoptotic-like death. J Neurochem. 1998;70:677–687. doi: 10.1046/j.1471-4159.1998.70020677.x. [DOI] [PubMed] [Google Scholar]
- 44.Xu DG, Korneluk RG, Tamai K, Wigle N, Hakim A, Mackenzie A, Robertson GS. Distribution of neuronal apoptosis inhibitory protein-like immunoreactivity in the rat central nervous system. J Comp Neurol. 1997;382:247–259. [PubMed] [Google Scholar]
- 45.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 48.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 49.Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- 50.Zhang C, Raghupathi R, Saatman K, LaPlaca M, McIntosh T. Regional and temporal alterations in DNA fragmentation factor (DFF)-like proteins following experimental brain trauma in the rat. J Neurochem. 1999a;73:1650–1659. doi: 10.1046/j.1471-4159.1999.0731650.x. [DOI] [PubMed] [Google Scholar]
- 51.Zhang L, Chen J, Fu H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci USA. 1999b;96:8511–8515. doi: 10.1073/pnas.96.15.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]