

Abstract
Neurotrophin (NT) signals may be moved from axon terminals to neuron cell bodies via signaling endosomes—organelles in which NTs continue to be bound to their activated receptors. Suggesting that clathrin-coated membranes serve as one source of signaling endosomes, in earlier studies we showed that nerve growth factor (NGF) treatment increased clathrin at the plasma membrane and resulted in colocalization of clathrin with TrkA, the receptor tyrosine kinase for NGF. Strikingly, however, we also noted that most clathrin puncta at the surface of NGF-treated cells did not colocalize with TrkA, raising the possibility that NGF induces a general increase in clathrin-coated membrane formation. To explore this possibility further, we examined the distribution of clathrin in NGF- and BDNF-treated cells. NGF signaling in PC12 cells robustly redistributed the adaptor protein AP2 and the clathrin heavy chain (CHC) to surface membranes. Using confocal and epifluorescence microscopy, as well as biochemical assays, we showed the redistribution of clathrin to be attributable to the activation of TrkA. Significantly, NGF signaled through TrkA to induce an increase in clathrin-mediated membrane trafficking, as revealed in the increased endocytosis of transferrin. In that BDNF treatment increased AP2 and clathrin at the surface membranes of hippocampal neurons, these findings may represent a physiologically significant response to NTs. We conclude that NT signaling increases clathrin-coated membrane formation and clathrin-mediated membrane trafficking and speculate that this effect contributes to their trophic actions via the increased internalization of receptors and other proteins that are present in clathrin-coated membranes.
Keywords: NGF, BDNF, neurotrophin, signaling, TrkA, clathrin, transferrin, endocytosis
The neurotrophins (NTs) regulate the trophic state of neurons (Yuen et al., 1996; Kaplan and Miller, 1997;Casaccia-Bonnefil et al., 1999). An interesting question is how signals generated at the terminals of axons are communicated retrogradely to neuronal cell bodies. That such communication occurs is strongly supported by studies showing that NTs are produced in target tissues and that this source of NTs is critical for the survival of responsive neurons (Snider, 1994; Li et al., 1995; Silos-Santiago et al., 1995;Francis et al., 1999). We (Beattie et al., 1996; Grimes et al., 1996,1997) and others (Ehlers et al., 1995; Bhattacharyya et al., 1997;Riccio et al., 1997; Senger and Campenot, 1997; Tsui-Pierchala and Ginty, 1999; Watson et al., 1999) have provided evidence that retrograde NT signals are transmitted through the formation of signaling endosomes, organelles that arise via the endocytosis of complexes in which NTs are bound to their Trk receptors.
Earlier findings in this laboratory suggested that clathrin-coated membranes may be used to move NTs and their receptors into signaling endosomes. Nerve growth factor (NGF) treatment of PC12 cells resulted in a >10-fold increase in the colocalization of TrkA, the receptor tyrosine kinase for NGF, with the clathrin heavy chain (CHC) at or near the cell surface (Beattie et al., 1996; Grimes et al., 1996, 1997). CHC is a constituent of clathrin that is used to mark the presence of clathrin-coated membranes (Nathke et al., 1992; Schmid, 1997; Marsh and McMahon, 1999). Thus, as is the case for a number of other cell surface receptors [e.g., the transferrin receptor (TfnR) and the epidermal growth factor receptor (EGFR) (Schmid, 1997)], clathrin-coated membranes may mediate the endocytosis of Trk receptors. In the same experiments we noted that NGF treatment markedly increased clathrin-immunostained puncta at or near the plasma membrane (Grimes et al., 1996). Although consistent with earlier, as well as more recent, observations on the effect of NGF, EGF, and insulin (Connolly et al., 1981, 1984; Corvera, 1990; Wilde et al., 1999), the extent of the change and its rapidity were impressive. Quite unexpectedly, we also found that most (∼80%) clathrin puncta near the surface of NGF-treated cells failed to stain for TrkA, suggesting that clathrin was recruited to membranes containing little or no TrkA. Our observations gave evidence that NGF signaling regulates clathrin-coated membrane formation. They predicted that NGF increases endocytic trafficking of TrkA and other proteins that are found in these membranes.
Now we have tested the suggested link between NGF signaling and clathrin-mediated membrane trafficking. NGF signaled through TrkA to increase the formation of clathrin-coated membranes. Significantly, TrkA activation also increased clathrin-mediated membrane endocytic traffic, as revealed by the increased uptake of transferrin (Tfn). These findings may reflect a physiological action of NTs because BDNF increased clathrin association with the surface membranes of hippocampal neurons. We speculate that enhanced clathrin-mediated membrane trafficking may be a common feature of NT actions that supports their trophic properties.
MATERIALS AND METHODS
Reagents. X22, a mouse monoclonal antibody against CHC (Brodsky, 1985), was used for immunoprecipitation and immunostaining. TD.1, another mouse monoclonal antibody to CHC (Nathke et al., 1992), was used for probing Western blots. AP.6 (Chin et al., 1989), a monoclonal antibody to the clathrin adaptor protein AP2 (Schmid, 1997), was used in the immunostaining experiments. Immunoprecipitation of tyrosine-phosphorylated proteins was accomplished by using mouse monoclonal antibody 4G10 as an agarose conjugate (Upstate Biotechnology, Lake Placid, NY). Blotting for tyrosine-phosphorylated proteins also used 4G10. To detect the presence of mouse antibody binding to the blots, we used HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA). For immunofluorescence studies the goat-anti-mouse IgG antibodies conjugated to FITC or to rhodamine were obtained from Cappel (Costa Mesa, CA). The membrane marker DiI C-7000 was from Molecular Probes (Eugene, OR).
PC12 nnr5 cells and nnr5 derivatives stably transfected with wild-type human TrkA or with the TrkA mutants 22.7 (activation loop mutant, YY674/675FF) or M1 (kinase inactive, K538N) were obtained from Robert Kupta in the Louis Reichardt laboratory (University of California at San Francisco). The mutant constructs were created by David Kaplan and colleagues (Ferrari et al., 1995; Cunningham et al., 1997) and were used with permission. For PC12 cells we used KB PC12 cells (Grimes et al., 1996). Parental 3T3 cells and 3T3 cells expressing either TrkA or p75NTR were supplied by Chin-shiou Huang and maintained as indicated (Huang et al., 1999).
NGF was isolated from the mouse submaxillary gland (Mobley et al., 1976). BDNF was a gift of Regeneron (Tarrytown, NY). Papain was from Worthington Biochemical (Lakewood, NJ). Serum extender and poly-d-lysine were from Collaborative Research (Bedford, MA). Collagen was obtained from Cohesion (Palo Alto, CA). Normal goat serum was from Jackson ImmunoResearch (West Grove, PA). Geneticin, Neurobasal medium, and B-27 serum-free supplement were from Life Technologies (Rockville, MD). FITC-dextran was from Molecular Probes. Tfn, K252a, saponin, and Triton X-100 were from Sigma (St. Louis, MO). Unless otherwise stated, all other reagents were from Sigma. Tissue culture media and media additions were supplied by the University of California at San Francisco Cell Culture Facility.
Preparation of cultured hippocampal neurons. A modification of a previously described procedure was used (Lester et al., 1989). The hippocampi of six to nine postnatal day 0 (P0) Sprague Dawley rats were removed and placed in a dissecting solution [containing (in mm) 161 NaCl, 5.0 KCl, 2.9 CaCl2, 5.0 HEPES, and 5.5 glucose, plus 530 μmMgSO4 and 5.6 μm phenol red, pH 7.4]. The dentate gyri were dissected and discarded. The remaining tissue was treated with papain (20 U/ml) in 10 ml of the same solution containing (in mm) 1.7 cysteine, 1 CaCl2, and 0.5 EDTA for 45 min at 37°C. The digestion was stopped by decanting the solution and by adding 10 ml of Complete medium [MEM with Earle's salts withoutl-glutamine and with 20 mm glucose, serum extender (1:1000), and 5% heat-inactivated fetal calf serum] containing 25 mg of BSA and 25 mg of trypsin inhibitor type 3–0. Then the tissue was triturated in a small volume of this solution with a fire-polished Pasteur pipette. Using coverslips coated with poly-d-lysine (0.1 mg/ml) and collagen (0.06 mg/ml), we plated the cells overnight in Neurobasal medium containing the additives B-27 serum-free supplement (1×) andl-alanyl–l-glutamine (2 mm). One-half of the medium was replaced with the Neurobasal medium plus additives the next day. Cultures were refed by replacing one-half of the volume of medium at weekly intervals. At week one they were refed with Complete medium containing B-27. At week two and beyond they were fed with the Neurobasal medium plus additives. Astrocyte growth was inhibited at day 12 by adding 5-fluoro-2′-deoxyuridine (0.3 mm) plus uridine (0.7 mm). Cultures were used for experiments between weeks three and four.
Immunofluorescence studies. All cells were grown and maintained at 37°C with 5% CO2. PC12 cells, PC12 nnr5 cells, and nnr5 variants were cultured in DME-H21, 10% horse serum, and 5% fetal calf serum on collagen-coated plates. The medium used for maintaining TrkA variant-expressing nnr5 PC12 cell lines included 100 μg/ml of Geneticin. Priming of PC12 cells was accomplished by adding NGF (2 nm) for 7 d. 3T3 cells were grown on plastic in DME-H21 in 10% horse serum.
In preparation for the immunostaining experiments on primed PC12 cells, the cells were washed at 37°C with serum-free medium (minus NGF) in three changes (30 min each). Then they were treated with NGF or the vehicle in serum-free medium, as indicated below. For experiments on unprimed PC12 cells, PC12 nnr5 cells, and 3T3 cells the cells were incubated first in DME-H21 containing 1% horse serum overnight before treatment in the same medium. Hippocampal neurons were maintained and treated in the medium described above. To examine the effects of NGF treatment, usually we first incubated the cells with NGF under conditions in which NGF would bind to its receptors without inducing membrane-trafficking events. Thus, the cells were incubated at 4°C in medium with NGF (2 nm) or with the vehicle (0.2% acetic acid in the same small volume as used to add NGF) for 1 hr before warming at 37°C. In some experiments NGF (2 nm) or BDNF (2 nm) was added to the cells at 37°C. After NGF treatment the cells were chilled quickly to 4°C, fixed with 4% paraformaldehyde in PBS for 20 min at 4°C, permeabilized in PBS containing saponin (1 μg/ml), and blocked in 10% normal goat serum. To visualize clathrin (i.e., CHC), we incubated X22 (10 μg/ml) with the cells overnight at 4°C and developed the signal with either rhodamine- or FITC-conjugated goat anti-mouse IgG antibody. To stain AP2, we incubated AP.6 (6 μg/ml) with cells that used the same protocol and developed the signal with rhodamine-conjugated goat anti-mouse IgG. For plasma membrane demarcation DiI (0.5 μg/ml in PBS) was applied to fixed and immunostained cells for 30 min at room temperature in the dark, followed by a brief wash with PBS.
Confocal microscopic analysis of clathrin distribution was accomplished with a MRC 1000 laser scanning confocal microscope (Bio-Rad, Hercules, CA) equipped with a krypton/argon laser and attached to a ZeissAxiovert microscope. Care was taken to ensure that data were collected at a point midway between the substrate-attached plasma membrane and the top of the cell. Immunostained puncta were located near the apparent margin of all of the cells that were stained for CHC or AP2. Although puncta were fewer in number and less intense in vehicle-treated cells, the staining was adequate to delineate the cell margin. The surface of cells was defined as a line that linked the outermost puncta. Figure 2 shows that this method defined the margins of both NGF- and vehicle-treated cells. The length of the line that marked the cell surface was measured by the measurement analysis tool provided with the public domain National Institutes of Health Image program (developed at National Institutes of Health and available on the Internet at http://rsb.info.NIH.gov/National Institutes of Health-image/), and this value was used as the cell perimeter. Immunostained puncta were counted as described (Grimes et al., 1996). Briefly, after defining the edge of the cell, we drew a second line 0.5 μm interior to the first, and we counted all immunostained puncta between the lines. The results were expressed as the number of puncta per micrometer of cell surface or the number per cell.
Fig. 2.
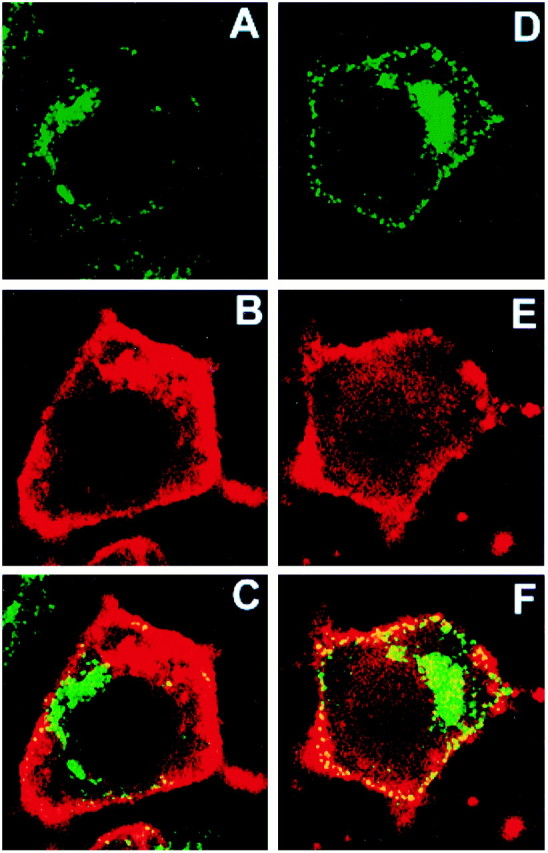
NGF treatment resulted in increased movement of clathrin to the plasma membrane. Confocal microscopy was used to show that CHC immunostaining colocalized with that for the membrane marker DiI. PC12 cells were chilled to 4°C and incubated with the vehicle (A–C) or with NGF (2 nm;d–f). Then the cells were warmed to 37°C for 2 min, chilled, fixed, prepared for CHC immunostaining with X22 (A, D), and stained with DiI (B, E). The merged images for a control (C) and an NGF-treated cell (F) show that CHC colocalized with DiI at the surface of both cells and that the extent of colocalization was much greater in the NGF-treated cell. The width of each panel is 55 μm.
Epifluorescence microscopy was performed with a Nikon Diaphot 300 inverted microscope with a PlanApo 60 Nikon objective. Images were collected and processed with a Princeton Instruments Micro Max CCD camera (Trenton, NJ) and IP Lab Spectrum Image Processing software from Signal Analytics (Vienna, VA). To define the margins of cells and the number of puncta at or near the cell surface, we used the same methods as for confocal microscopy.
Clathrin membrane association studies. For biochemical analyses all of the cells were cultured and prepared for experiments as described above. However, in some cases serum deprivation for 4 hr replaced overnight incubation in 1% horse serum. We used two methods to measure membrane-associated clathrin. In each, gentle conditions were used that favored maintaining the association of clathrin with membranes (Wilde and Brodsky, 1996). The first method produces a cell ghost depleted of cytosol and internal membranes; importantly, however, the plasma membrane remains associated with the cell ghost (Grimes et al., 1996). Cells (5 × 107 per condition) were harvested at 37°C in calcium- and magnesium-free (CMF) PBS. They were pelleted at 1000 × g and resuspended in binding buffer (PBS containing 1 mg/ml of glucose, 1 mg/ml of BSA, and 10 mm HEPES, pH 7.4). Then the cells were treated with NGF (2 nm) or vehicle for 2 min at 37°C. The suspensions were chilled rapidly to 4°C in an ice water bath, pelleted at 1000 × g at 4°C, and resuspended in 1 ml of cold (4°C) MES buffer [containing (in mm) 100 MES, pH 6.8, 0.5 MgCl2, 0.2 DTT, 1 Na-Orthovandate, and 1 PMSF plus 0.1 μg/ml each of leupeptin and aprotinin]. Cells then were disrupted gently by a ball homogenizer, as described previously (Grimes et al., 1996). To separate the cell ghost from the cytosol, we centrifuged the preparation at 8000 × g for 35 min, a procedure that also pelleted the heaviest membranes released from the cell ghost. After washing the pellet once in MES buffer and repelleting at 8000 × g for 35 min, we lysed the pellet with lysis buffer [containing (in mm) 20 Tris, pH 8.0, 137 NaCl, 1 Na-Orthovandate, and 1 PMSF plus 1% NP-40, 0.5% DOC, 10% glycerol, and 1 μg/ml each of leupeptin and aprotinin]. The samples, which represented equivalent numbers of cells, were immunoprecipitated for CHC with X22 (10 μg/ml). Immunoprecipitates were processed by SDS-PAGE and transferred to nitrocellulose as described (Grimes et al., 1996). The blot was probed with TD.1 (3 μg/ml). After incubation with HRP-conjugated goat anti-mouse IgG, the signal was visualized by ECL phosphorescence (Amersham, Buckinghamshire, England). Data were quantified by National Institutes of Health Image software.
In the second method we disrupted the cells more thoroughly in an attempt to eliminate any possible contamination of membrane fractions by cytosol. One 15 cm plate (5 × 107cells) per condition was harvested with CMF-PBS. The cells were pelleted at 1000 × g for 5 min and then resuspended in 5 ml of cold (4°C) DME containing 25 mm HEPES buffer and either NGF (2 nm) or the vehicle. The cell suspensions, in 15 ml conical tubes, were rotated for 1 hr at 4°C and then warmed for the time indicated in a 37°C water bath with periodic gentle mixing. Samples were chilled in an ice bath for 3 min, and the cells were pelleted at 1000 × g for 5 min. Next they were washed once with cold PBS (4°C) and resuspended in 1 ml of cold (4°C) MES buffer. Membranes were disrupted by three cycles of freezing, followed by thawing, after each of which a 25 gauge needle was used to disrupt the material further. Samples then were spun at 1000 × g for 5 min to remove nuclei and intact cells, and the supernatant was centrifuged at 100,000 ×g for 40 min, essentially as described (Grimes et al., 1996). On the basis of results from earlier studies (Grimes et al., 1996), the resulting pellet (P2′) was predicted to contain small and large fragments of the plasma membrane, internal membranes derived from the plasma membranes, other cellular membranes, and organelles such as mitochondria and ribosomes; S2′ contained the cytosol. P2′ was resuspended in the lysis buffer. The supernatant (S2′) was diluted 1:2 in a solution of 0.5 m Tris buffer containing 1% Triton X-100. Lysed P2′ fractions were equalized for protein, as were S2′ fractions, and they were subjected to SDS-PAGE, transferred to nitrocellulose, and immunoblotted for clathrin (i.e., CHC) with TD.1 (3 μg/ml) as described (Grimes et al., 1996). A modification of this method was used to examine PC12 nnr5 cells and PC12nrr5 cells expressing wild-type TrkA and mutant TrkA receptors. After the cells were disrupted as indicated above, they were centrifuged at 10,000 × g for 10 min at 4°C. This produced a pellet containing cell ghosts, cell ghost fragments, and heavy membranes that was similar to that collected with the first method. The pellet was suspended in the lysis buffer and processed as above, except that the samples were slot-blotted to nitrocellulose and immunoblotted for CHC with the same antibody. After the blots were incubated with HRP-conjugated goat anti-mouse IgG, the signal was detected by ECL phosphorescence and quantified with National Institutes of Health Image software. In control studies we verified that protein loads and exposures produced signals in the linear range.
CHC phosphorylation assay. PC12 cells were used in the clathrin phosphorylation assay. Cells (3 × 107 per condition) were treated with NGF (2 nm) or the vehicle at 37°C for 2 min. The medium was removed, and cold (4°C) PBS was used to wash the cells while the plates were transferred onto ice. Then the cells were lysed in the lysis buffer and immunoprecipitated with X22 (10 μg/ml). The immunoprecipitates were subjected to SDS-PAGE and transfer, and they were immunoblotted as described (Grimes et al., 1996). Antibodies against CHC (TD.1; 3 μg/ml) and phosphorylated tyrosine (4G10; 1:2000) were used to probe the blot sequentially. To prepare for reprobing, we acid-stripped the blot by using TBS, pH 2.0, for 30 min at room temperature. After the blots were incubated with HRP-conjugated goat anti-mouse IgG, the signal was detected by ECL phosphorescence and quantified with National Institutes of Health Image software.
FITC-dextran and 125I-transferrin uptake assays. In FITC-dextran uptake studies the PC12 cells (5 × 107 cells per condition) were removed from culture plates in warm (37°C) CMF-PBS and incubated, rotating, for 30 min in serum-free PBS-HEPES (10 mm, pH 7.4) at 37°C. Cells were pelleted for 2 min at 1000 × g and resuspended in 1 ml of HEPES-PBS. FITC-dextran (1 nm), with or without NGF (2 nm), then was added to the suspension, and the cells were incubated, rotating, at 37°C for 5 or 10 min. Cells were chilled (4°C) and washed three times with ice-cold PBS. Next they were lysed in the lysis buffer. After nuclei and insoluble debris were sedimented at 1000 × g, absorbance was measured at 490 nm.
In Tfn uptake time course experiments, PC12 cells from four 15 cm plates (20 × 107 cells) were pooled and suspended in 16 ml of CMF-PBS. Aliquots of 1 ml were incubated at 4°C for 30 min in PBS-HEPES. 125I-Tfn (10 ng/ml), with or without NGF (2 nm), was added to the cells at 4°C, and then the mixtures were incubated at 37°C for the time intervals indicated. Cells were chilled and spun down (1000 × g for 1 min) before acid stripping, as described (Zhou et al., 1995), for 10 min at 4°C. Cells then were pelleted quickly and washed once with cold (4°C) PBS. Radioactivity of the cell pellet was measured in a Beckman gamma counter. In samples that were treated with125I-Tfn or with125I-Tfn and NGF, but not warmed, the counts in the pellet after stripping were <20% of the warmed values and did not differ between NGF-treated and vehicle-treated samples. In experiments testing the role of TrkA kinase activity on125I-Tfn uptake, PC12 cells or PC12nnr5 cells were treated for 5 min with 125I-Tfn or with 125I-Tfn and NGF, essentially as described above. For K252a experiments the cells were pretreated at 37°C in suspension with 200 nm K252a for 30 min and chilled to 4°C. Then they were treated with125I-Tfn or125I-Tfn plus NGF for 5 min, as above.
RESULTS
NGF signaling recruited clathrin to the plasma membrane
To investigate further the NGF effect on clathrin-coated membrane formation, we examined the cellular localization of clathrin in PC12 cells incubated with NGF (2 nm) or the vehicle for 1 hr at 4°C, followed by warming for 2 min at 37°C. After treatment the cells were chilled quickly and then washed, fixed, and permeabilized before being immunostained for CHC. Confocal microscopy of cells that were not treated with NGF showed that CHC staining was seen in small puncta distributed diffusely throughout cells, with most staining in the cytosol (Fig. 1). Staining was noted in the perinuclear region, but there was relatively little associated with the plasma membrane. After NGF treatment there was a redistribution of staining with a marked increase at or near the plasma membrane. There was also a consistent increase in staining in the perinuclear region. The puncta near the cell surface consistently demonstrated a “picket fence” pattern in which they appeared to line up near the edge of the cell. We quantified the increase in puncta within 0.5 μm of the cell surface after NGF treatment. In NGF-treated cells the average number of these puncta was 0.76/μm. With an average cell perimeter of 56 μm, the average number of puncta per cell was 43. The number of puncta per micrometer in NGF-treated cells was 244% of the vehicle-treated control (± 6.8% SEM; n = 15 cells in two separate experiments), a result that was significant (p< 0.01). We conclude that NGF acted to induce redistribution of clathrin.
Fig. 1.

NGF treatment caused clathrin and AP2 redistribution in PC12 cells. A, B, PC12 cells were cultured (i.e., primed) in the presence of NGF (2 nm) for 7 d. After being washed three times with fresh serum-free medium without NGF, the cells were chilled to 4°C and incubated for 1 hr in either the absence (A; i.e., with vehicle alone) or presence (B) of 2 nm NGF in serum-free medium. Then the cells were warmed at 37°C for 2 min, quickly chilled (4°C), fixed, and processed for CHC immunostaining with X22. The panels show confocal micrographs. The width of each panel is 45 μm. C, D, The localization of the adaptor protein AP2 was examined by epifluorescence microscopy. Unprimed PC12 cells were treated with vehicle (C) or NGF (2 nm; D) at 37°C for 2 min. Then they were chilled, fixed, and processed for immunostaining for AP2 with AP.6. NGF increased AP2 near the plasma membrane. The width of each panel is 45 μm.
The change in CHC staining suggested that NGF redistributed clathrin to the plasma membrane. To better define the locus of clathrin near the cell surface, we asked whether CHC staining would colocalize with a lipophilic membrane marker, DiI. Figure2, B and E, shows that DiI effectively marked surface membranes in both vehicle-treated and NGF-treated cells. Permeabilization was required to immunostain for CHC. As a result, DiI sometimes also marked membranes near the cell surface. The redistribution of CHC staining that followed NGF treatment (Fig. 2, compare A, D) resulted in increased colocalization of CHC with DiI at the cell surface. Note the marked increase in the number of CHC puncta colocalized with DiI (yellow denotes colocalization) after NGF treatment (Fig. 2, compare C, F). Because brief (3 min) NGF treatment of PC12 cells has been shown not to increase plasma membrane surface area (Connolly et al., 1984), the NGF-induced increase in CHC immunostaining reflects an increase in clathrin at the plasma membrane.
The change in CHC staining that followed NGF treatment was consistent with an increase in membrane-associated clathrin. To confirm this prediction, we used two methods. In each case the cells were treated with NGF for 2 min. In the first method we gently disrupted cells with a ball homogenizer (Grimes et al., 1996). This method depletes cells of cytosol, internal membranes, and organelles under conditions that do not fragment plasma membrane and that favor the continued association of clathrin with membranes. By harvesting the 8000 × gpellet, we separated clathrin in the cell ghost from cytoplasmic clathrin and from clathrin associated with all but the heaviest membranes released from the cells. CHC in the pellet was examined by SDS-PAGE and transferred to nitrocellulose, followed by immunoblotting. In NGF-treated cells CHC was increased to 158% of untreated controls (Fig. 3A).
Fig. 3.
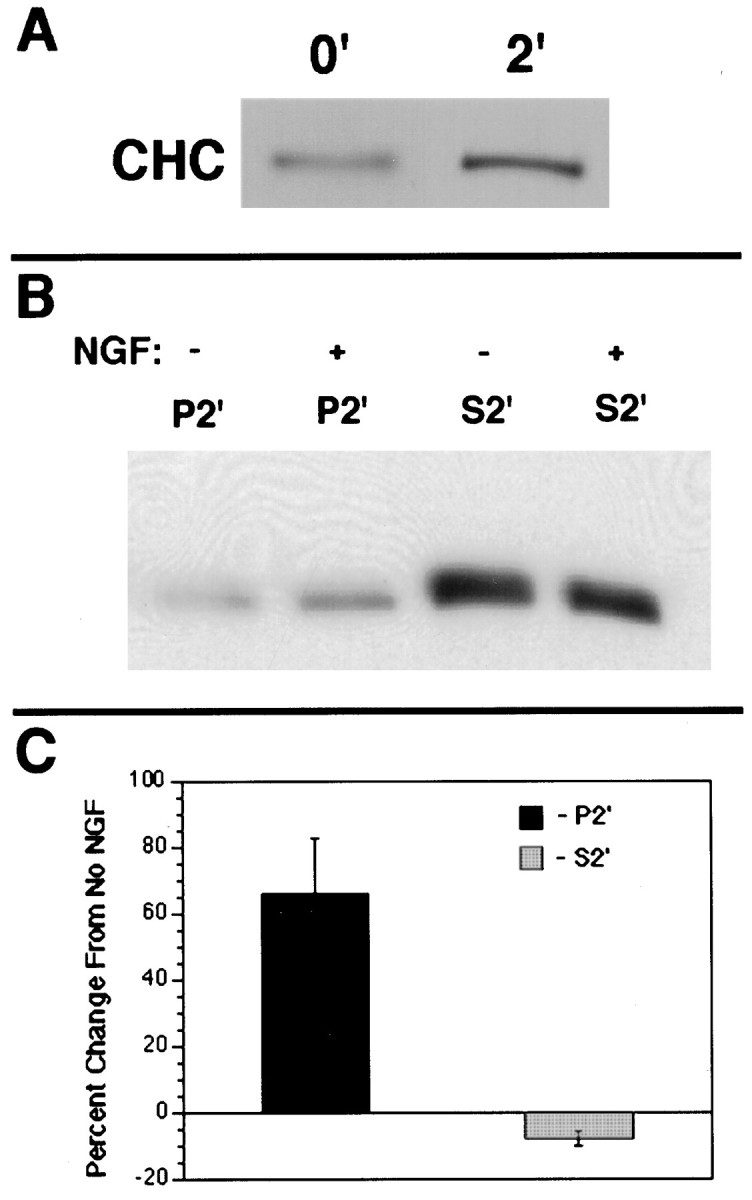
NGF induced an increase in membrane-associated clathrin. To quantify the amount of clathrin that was associated with membranes, we examined CHC in membrane and cytosolic fractions via two methods. A, In the first method equal numbers of PC12 cells were treated with either 2 nm NGF or with the vehicle for 2 min at 37°C; the ghost of gently disrupted cells was separated from the cytosol by pelleting at 8000 × g. CHC was immunoprecipitated with X22 and submitted to SDS-PAGE, followed by transfer to nitrocellulose and immunoblotting with TD.1. NGF treatment caused a 158 ± 9% increase (n = 4;p < 0.01) in membrane-associated clathrin.B, In the second method the cells were disrupted more thoroughly by three cycles of free/thaw. Using samples normalized for protein from the P2′ (membrane-associated) fraction or from the S2′ (cytosolic) fraction, we immunoprecipitated CHC with X22, submitted it to SDS-PAGE, transferred the CHC to nitrocellulose, and immunoblotted it with TD.1. NGF treatment caused a significant increase in CHC in P2′ (166 ± 18% of the vehicle-treated control; n= 3; p < 0.05). There was a concomitant small decrease in CHC in S2′ (92 ± 3.5% of the vehicle-treated control; n = 3; p = 0.06).C, The bands developed in B were quantified by National Institutes of Health Image program; the data from three separate experiments are shown. Error bars represent SEM.
In the second method we used three cycles of freezing and thawing to disrupt the cells more thoroughly to ensure that the trapping of cytosolic clathrin in cell ghosts could not contribute to the findings for membrane-associated clathrin. After pelleting the remaining cell ghosts, we quantified the amount of CHC associated with fragments of the plasma membrane and with membranes released from disrupted cells. These membranes (P2′) were separated from cytosol (S2′) with a 100,000 × g spin. CHC was present in both fractions in both untreated and treated cells. After NGF the amount of CHC in P2′ increased while the amount in S2′ decreased (Fig. 3B,C). In NGF-treated cells the amount of CHC in P2′ was 166% of the vehicle-treated control. There was a small decrease in CHC in S2′.
These observations show that NGF induced the movement of clathrin to membranes, with changes that were comparable via the two methods. In that plasma membrane was a major constituent of the membrane fractions produced by both methods, the findings point to an NGF-induced increase in clathrin at the plasma membrane. The change in CHC immunostaining at the surface of NGF-treated cells is consistent with this view, as is an earlier EM study showing an NGF effect on clathrin-coated membranes (Connolly et al., 1984). The possibility exists that NGF induced an increase in clathrin association with other membranes, including those derived from the plasma membrane.
NGF signaled through TrkA to increase the amount of clathrin at the plasma membrane
NGF signals through two receptors, TrkA and p75NTR. To ask which NGF receptor or receptors were responsible for the redistribution of clathrin, we performed experiments in a number of different cell types. To examine a contribution by TrkA in the absence of p75NTR, we tested 3T3 cells that express TrkA (3T3-TrkA cells). In the absence of NGF treatment CHC was distributed in the pattern seen in untreated PC12 cells (Fig.4A). The addition of NGF to these cultures resulted in the redistribution of CHC such that the number of CHC puncta at or near the plasma membrane was significantly greater (Fig. 4B). Quantification of puncta within 0.5 μm of the cell surface showed that NGF treatment for 2 min resulted in a value that was 250% of the vehicle-treated control (n = 10 cells; p < 0.01). Corresponding to this increase, we found that in 3T3-TrkA cells that were treated with NGF the amount of membrane-associated CHC in the P2′ fraction was increased (at 1 min, 170%; at 2 min, 136%; at 10 min, 137% of the vehicle-treated controls). Very similar results for clathrin redistribution were obtained when 3T3-TrkB cells were treated with BDNF (data not shown). In the 3T3 parental cells the number of puncta at the plasma membrane was unchanged after NGF treatment (3% below vehicle-treated control; n = 10 cells;p = 0.38) (Fig. 4E,F). NGF did not increase CHC-positive puncta near the plasma membrane in 3T3 cells expressing p75NTR (Fig. 4C,D). Indeed, NGF actually decreased the number of such puncta (30% below vehicle-treated control; n = 10 cells;p < 0.05). Further studies are needed to characterize p75NTR effects on CHC distribution. We conclude that the NGF increased clathrin-coated membrane formation through TrkA.
Fig. 4.

NGF induced redistribution of clathrin to the surface of 3T3 cells expressing TrkA, but not p75NTR. National Institutes of Health 3T3 fibroblasts were examined. Parental cells (i.e., cells without Trk or p75NTR) are shown (E, F), as are cells transfected with p75NTR (C, D) or with TrkA (A, B). Cells were treated with NGF (2 nm; B, D, F) or vehicle (A, C, E) for 2 min at 37°C. Then they were chilled at 4°C, fixed, and processed to show the distribution of clathrin by CHC immunostaining. The panels shown are confocal micrographs, and their width is 65 μm. Only the TrkA-expressing cell line displayed an increase in clathrin at the plasma membrane (B).
We tested further the role of TrkA signaling in the clathrin redistribution by performing studies in normal PC12 cells and in a series of PC12 cell variants (i.e., nnr5 PC12 cells) carrying wild-type and mutant TrkA receptors. In all untreated cells clathrin was distributed diffusely and in a cytosolic pattern. In PC12 cells, as expected, NGF signaling resulted in an increase in clathrin staining at or near the plasma membrane (Fig.5A,B). In PC12nnr5 cells, which have extremely low levels of TrkA but normal levels of p75NTR (Loeb and Greene, 1993), there was no evident change in the distribution of clathrin with NGF treatment (Fig. 5E,F). Redistribution of CHC staining was seen in a variant of PC12 nnr5 cells transfected with the wild-type TrkA receptor (Fig. 5C,D). When these cells were treated with NGF, many brightly stained puncta were found at the cell surface. We also examined PC12nnr5 cells stably transfected with the M1 TrkA mutant, in which substituting N for K at residue 538 inactivates the kinase domain, and with the 22.7 mutant, in which two activation loop tyrosines (Y674 and 675) are replaced with phenylalanine. Earlier studies documented markedly decreased TrkA signaling in cells expressing these mutants (Ferrari et al., 1995; Cunningham et al., 1997). We found that NGF treatment failed to induce clathrin redistribution in cells expressing the mutant TrkA receptors (Fig.5G–J). Consistent with these findings, although CHC association with membranes was induced by NGF in the nnr5 TrkA cells (% of vehicle = 113 ± 1.9%; n = 3;p < 0.05), there was no increase in the 22.7 mutant cells (% of vehicle = 96.2 ± 2.1; n = 3;p = 0.26). In the M1 cells NGF treatment caused a decrease that was not significant (% of vehicle = 82 ± 8;n = 3; p = 0.18). Taken together, the data provide strong evidence that NGF signaled via the activation of TrkA kinase to induce the formation of clathrin-coated membranes.
Fig. 5.

NGF induced clathrin redistribution in PC12 cells expressing wild-type TrkA. PC12 cells, PC12 nnr5 cells, and nnr5 variants were chilled (4°C) and then incubated with vehicle or 2 nm NGF for 1 hr before being warmed at 37°C for 2 min. After treatment the cells were chilled quickly, fixed, and processed to determine the distribution of clathrin by immunostaining for CHC with X22. The cell lines that were examined were KB PC12 cells expressing endogenous wild-type TrkA (KB; A, B), nnr5 cells transfected with wild-type TrkA (TrkA nnr5;C, D), nnr5 parental cells (nnr5;E, F), nnr5 cells transfected with kinase-inactivated TrkA (M1 nnr5; G, H), and nnr5 cells transfected with activation loop-mutated TrkA (22.7 nnr5; I, J). Confocal microscopy was used to assess the distribution of clathrin in the vehicle-treated (top row) and NGF-treated (bottom row) conditions. Only cells with wild-type TrkA (B, D) responded to NGF with an increase in clathrin near the plasma membrane (arrowheads). The width of each panel is 55 μm.
NTs signaled to redistribute AP2 and clathrin in PC12 cells and in hippocampal neurons
The marked effect of NGF on clathrin at the plasma membrane suggested that other components of clathrin-coated membranes would be recruited also. We tested this by examining the distribution of AP2, a major constituent of clathrin coats at the plasma membrane (Beck et al., 1992; Wilde and Brodsky, 1996; Marsh and McMahon, 1999). By epifluorescence microscopy, AP2 was distributed in a cytosolic pattern in untreated PC12 cells (see Fig. 1C). After NGF treatment much more AP2 was present at or near the plasma membrane (see Fig.1D). The pattern of staining was essentially identical to that seen for CHC. Surface puncta were counted, as described above, in epifluorescence micrographs. In NGF-treated cells the number of puncta per micrometer was 225 ± 15% (n = 12; p < 0.01) of the vehicle-treated control. Thus, as was true for clathrin, NGF signaling recruited AP2 to the plasma membrane of PC12 cells.
To ask whether NTs influence the distribution of clathrin and AP2 in neurons, we performed studies in hippocampal primary cultures. Hippocampal neurons express little if any TrkA, but they do express TrkB and respond to BDNF (Ip et al., 1993a; Minichiello et al., 1999). In vehicle-treated cells AP2 staining was distributed more or less uniformly in the cytosol (Fig.6A, confocal image;B, epifluorescent image). There was no apparent change in the distribution of AP2 with NGF (data not shown). However, with BDNF treatment there was a clear increase in staining near the plasma membrane (Fig. 6C,D). The surface membranes of both cell bodies and processes showed the change. The change in AP2 staining was quantified in epifluorescence micrographs. In BDNF-treated cells the number of surface puncta was 306 ± 23% (n = 5;p < 0.01) of the vehicle-treated control. To show whether clathrin also was redistributed by BDNF treatment, we examined hippocampal neurons after staining for CHC. In vehicle-treated cells there was a diffuse cytosolic pattern of staining (Fig.6E, confocal image;F,I, epifluorescent images). BDNF treatment for 2 min resulted in marked redistribution of CHC staining to the plasma membrane (Fig. 6G, confocal;H,J, epifluorescent images); the number of puncta was 241 ± 25% (n = 5;p < 0.01) of that in vehicle-treated cells. The redistribution of AP2 and CHC induced by BDNF acting on hippocampal neurons suggests that increased movement of clathrin to surface membranes may represent a physiological response to NT signaling.
Fig. 6.

BDNF induced an increase in AP2 and clathrin at the surface of hippocampal neurons. To show whether NT treatment induced an increase in AP2 and clathrin associated with surface membranes in primary neurons, we used BDNF to treat rat hippocampal neurons. The distribution of AP2 was assessed by confocal (A, C) and epifluorescence (B, D) microscopy of neurons immunostained with AP.6. Clathrin distribution was assessed with confocal (E, G) and epifluorescence (F, H–J) microscopy of neurons immunostained for CHC with X22. BDNF (2 nm;C, D, G, H, J) or vehicle (A, B, E, F, I) was applied to cultured neurons for 2 min at 37°C before they were chilled, fixed, and processed for immunostaining. BDNF increased staining for AP2 (C, D) and CHC (G, H) at the plasma membrane (arrowheads). I and J show sections of neuronal processes and indicate that the BDNF effect also was registered here (J). The width of all panels is 55 μm.
NGF increases phosphorylation of CHC
Recently, we showed that the redistribution of clathrin seen with EGF signaling was associated with the phosphorylation of CHC (Wilde et al., 1999). To determine whether NGF signaling also induced an increase in the phosphorylation of CHC, we quantified tyrosine-phosphorylated CHC in immunoprecipitates from NGF-treated and untreated PC12 cells. Figure 7 shows that phosphorylated CHC was present in untreated cells. The amount was increased significantly after NGF treatment for 2 min; the value averaged 223 ± 19% (n = 3; p < 0.01) of the vehicle-treated control. Similar results were obtained in experiments in which the 4G10 anti-phosphotyrosine antibody was used for immunoprecipitation, followed by probing with the antibody for CHC (data not shown). To evaluate further the effect of NGF on CHC phosphorylation, we performed time course studies. NGF treatment increased CHC phosphorylation as early as 1 min (NGF-treated: 141 ± 9% of control; n = 3; p < 0.01). The effect was maximal at 2 min (see above) and lasted through 15 min (at 5 min: 174 ± 14%, n = 3; at 15 min: 143 ± 5%, n = 3; for both, p < 0.01). These data show that NGF actions on CHC phosphorylation are rapid and robust and that they were correlated in time with clathrin redistribution.
Fig. 7.

NGF treatment increased the phosphorylation of CHC. PC12 cells were treated with NGF or vehicle for 2 min at 37°C. The cells were chilled quickly (4°C) and lysed in lysis buffer. Samples equalized for protein were immunoprecipitated with X22 and subjected to SDS-PAGE, transferred to nitrocellulose, and immunoblotted (IB) with the anti-phosphotyrosine antibody 4G10. Thep-tyrrow shows that CHC phosphorylation was increased by NGF. The CHC row confirms that equal amounts of CHC were present in the NGF and in vehicle-treated samples.
NGF increases endocytosis and trafficking through clathrin-coated membranes
The NGF-induced increase in AP2 and clathrin at the plasma membrane suggested that NGF could signal to induce increased endocytosis through clathrin-coated membranes. To test this idea, we examined NGF effects on two markers of endocytosis. FITC-dextran provides a marker of fluid phase endocytosis. PC12 cells were incubated in the presence of FITC-dextran for 0–10 min at 37°C. After 5 min of NGF treatment the uptake was increased by 20% over baseline; by 10 min the increase was nearly 60% (Fig.8A). These findings show that NGF increases the uptake of solutes that enter the cell by bulk flow.
Fig. 8.

NGF enhanced the uptake of FITC-dextran and of125I-Tfn in PC12 cells. A, PC12 cells were incubated with FITC-dextran for 0–10 min at 37°C in the absence or presence of 2 nm NGF. The amount of internalized FITC-dextran was determined by measuring the absorbance at 490 nm of the lysates of the washed cell pellets. The values are expressed as a percentage of the vehicle-treated samples that were warmed for 10 min. NGF increased the uptake of FITC-dextran by 20 ± 3% (n = 3; p = 0.02) at 5 min and by 60 ± 6% (n = 3; p = 0.01) at 10 min. The increase at 10 min resulted in a value that was 157% of the vehicle-treated control. The error bars represent SEM.B, PC12 cells were incubated with125I-Tfn in the absence or presence of 2 nm NGF for 2, 5, 15, or 30 min at 37°C. Then they were chilled (4°C) and quickly pelleted before acid stripping of the surface-bound Tfn. Cell-associated counts represent internalized125I-Tfn. The values are expressed as a percentage of the vehicle-treated samples at 30 min. NGF treatment increased the uptake of 125I-Tfn by 35 ± 10% (n = 3;p = 0.001) at 5 min to a value that was approximately twice that of the control. By 15 min the increase was 13 ± 1% (n = 3; p = 0.001). Error bars represent SEM. C, TrkA activation was required for the NGF effect on increased endocytosis of125I-Tfn. Cells were incubated with 125I-Tfn in the absence or presence of 2 nm NGF for 5 min at 37°C. Although NGF induced an increased endocytosis of 125I-Tfn in KB PC12 cells (177 ± 6% of the vehicle-treated;n = 3; p = 0.001), it had no significant effect in KB PC12 cells pretreated with 200 nmK252a (110 ± 3%; n = 3;p = 0.08) or in PC12 nnr5 cells (96 ± 3%;n = 3; p = 0.38).
To test the idea that NGF influences trafficking via clathrin-coated membranes, we examined NGF actions on the uptake of Tfn. This ligand is internalized through clathrin-coated pits after binding to TfnR (Schmid, 1997). To measure the uptake of Tfn, we treated PC12 cells with radiolabeled Tfn in either the presence or absence of NGF. With NGF treatment there was a marked increase in Tfn endocytosis over vehicle-treated controls. The increase was marked by 5 min, measuring approximately twofold. The increase in uptake persisted through 15 min (Fig. 8B). By 30 min the level of Tfn uptake in NGF-treated cells was the same as in vehicle-treated cells. The increase in endocytosis was not seen after NGF treatment of PC12 nnr5 cells or in PC12 cells pretreated with K252a, an inhibitor of Trk activation (Koizumi et al., 1988) (Fig. 8C). These findings show that TrkA activation was required to induce endocytosis. That NGF-treated cells more rapidly reached the same plateau for Tfn endocytosis as vehicle-treated cells suggests that NGF acted to allow stimulated cells to reach an equilibrium more quickly with respect to Tfn trafficking. Whether the plateau for Tfn endocytosis reflects the presence of a limiting number of TfnRs or is attributable to an NGF effect on TfnR recycling is unknown. In either case these data are evidence that NGF increases trafficking via clathrin-coated membranes.
DISCUSSION
Endocytosis plays a critical role in cellular functions ranging from nutrient acquisition to synaptic transmission. It is important to elucidate the mechanisms that underlie endocytosis and their regulation (Marsh and McMahon, 1999). The current study shows that NT signaling increased the formation of clathrin-coated membranes. NGF acted via its receptor tyrosine kinase TrkA to increase clathrin on the surface membranes of PC12 cells. In concert, there was increased activity of the clathrin-coated pit pathway, as evidenced by enhanced endocytosis of Tfn. In that BDNF induced clathrin recruitment to the surface membranes of hippocampal neurons, our findings suggest that NTs may act normally to regulate clathrin-coated membrane formation and to increase clathrin-mediated membrane traffic.
A number of protein–protein and protein–lipid interactions underlie the assembly of the clathrin-based endocytic machine. In addition to clathrin (i.e., CHC and light chain chains), AP-2, and, in neurons, AP180 (Schmid, 1997), the proteins include dynamin and amphiphysin (Damke et al., 1994; McMahon et al., 1997). Accessory cytosolic proteins include synaptojanin I, an inositol 5-phosphatase, and Eps15 (Benmerah et al., 1995; Tebar et al., 1996; Haffner et al., 1997; van Delft et al., 1997). In what appears to be a critical step for vesicle formation, dynamin binds and possibly activates endophilin I, a lysophosphatidic acid acyl transferase that catalyzes the conversion of lysophosphatidic acid to phosphatidic acid (Schmidt et al., 1999). How the interaction of these components is regulated is of considerable interest. It has been known for some time that polypeptide growth factor signals influence the formation of coated membranes (Connolly et al., 1981, 1984). Greene and colleagues showed in PC12 cells treated with NGF that the number of clathrin-coated plasma membrane densities increased two- to threefold within 30 sec of NGF addition (Connolly et al., 1981). Similar results were seen in sympathetic neurons treated with NGF and in PC12 cells treated with EGF (Connolly et al., 1984). In related studies, insulin treatment of adipocytes caused a threefold increase in the amount of CHC associated with plasma membrane (Corvera, 1990). Recently, we showed that EGFR activation resulted in a dramatic redistribution of clathrin to the plasma membrane of A431 cells, as judged by confocal microscopy and by quantification of membrane-associated CHC (Wilde et al., 1999). The current study extends these observations by showing that NGF signaled through TrkA to regulate robustly the clathrin coating of the plasma membrane. NGF treatment resulted in a prominent increase in membrane-associated CHC and AP2. This was revealed by immunostaining studies of these proteins and in biochemical studies in which we measured the amount of CHC in membrane fractions.
Our findings, and those for EGF and insulin (Connolly et al., 1984;Corvera, 1990; Wilde et al., 1999), suggest that a common mechanism may link the activation of receptor tyrosine kinases to the induction of clathrin-coated membrane formation. However, beyond the requirement for TrkA kinase activation, the mechanism by which TrkA signaling induces increased clathrin-coated membranes is yet to be defined. Of note, several of the proteins that make up clathrin-coated membranes are subject to phosphorylation and dephosphorylation, and it has been shown that such modifications contribute significantly to the regulation of endocytic function (Slepnev et al., 1998). As was seen with EGF (Wilde et al., 1999), NGF effects on clathrin redistribution were associated with changes in the phosphorylation of CHC. Whether, as is the case for EGFR, pp60src is required downstream of TrkA activation to increase clathrin-coated membrane formation and CHC phosphorylation is yet to be determined. Furthermore, it is uncertain as to whether the increase in clathrin at surface membranes was attributable to local changes in signaling (i.e., because of local recruitment of clathrin and AP-2 by activated TrkA receptors) or to signaling events not spatially contiguous with activated TrkA receptors. However, we have seen TrkA in complex with CHC and AP-2 in PC12 cells, and the amount of such complexes increases with NGF treatment (C. Howe, E. Beattie, and W. Mobley, unpublished observations). This suggests that some membrane-associated clathrin is complexed with activated Trks
To demonstrate the physiological relevance of our findings, we asked whether BDNF would influence the distribution of clathrin in hippocampal neurons. The changes induced by BDNF were identical to those seen for NGF, in that both AP-2 and clathrin were recruited rapidly to surface membranes. Surface membranes were decorated prominently with immunostained puncta. Remarkably, the changes seen with BDNF were registered on cell bodies and processes. These findings show that BDNF signaling induces widespread effects on clathrin-coated membrane formation and suggest that much of the surface of neurons is responsive to this aspect of BDNF actions. The similarity of the findings for BDNF and NGF suggests that each of the NTs will be shown to act via Trk receptors to increase the production of clathrin-coated membranes, a suggestion that is consistent with the existence of some signaling mechanisms that have been shown to be common for the Trk receptors (Ip et al., 1993b; Kaplan and Miller, 1997; Yuen and Mobley, 1999).
Endocytic trafficking of cell surface receptors through clathrin-coated membranes follows from their concentration in clathrin-coated membranes either on a constitutive basis [e.g., the TfnR, the low density lipoprotein receptor (LDL-R)] or in response to ligand binding (e.g., EGFR; Schmid, 1997). Our findings for NGF and EGF suggest that regulated formation of clathrin-coated membranes also contributes to endocytosis. One consequence would be increased endocytosis of the receptors for NGF and EGF. Evidence that this is the case is data showing that, when EGF signaling through pp60src was inhibited, there was an inhibition of clathrin redistribution and a delay in EGF endocytosis (Wilde et al., 1999). In studies on PC12 cells we found that NGF treatment increased endocytosis of TrkA and that TrkA at or near the surface of treated cells was colocalized with clathrin (Grimes et al., 1996). Additional recent findings also support the view that endocytosis of TrkA occurs via clathrin-coated membranes (C. Howe and W. Mobley, unpublished observations). Thus, it is likely that NGF acts to enhance the endocytosis of TrkA receptors via clathrin-coated membranes. We speculate that clathrin-coated vesicles may serve as a source of signaling endosomes.
We entertained the novel possibility that NGF signaling effects on clathrin-mediated membrane formation would result in a general increase in clathrin-mediated endocytosis, augmenting the uptake of markers unrelated to NGF or its receptors. This possibility was suggested, in part, by the finding that many of the clathrin-positive puncta appearing near the surface of NGF-treated cells did not stain for TrkA. Indeed, we discovered that, although NGF induced a >10-fold increase in the number of TrkA puncta that colocalized with CHC, TrkA was detected in only ∼20% of CHC puncta (Grimes et al., 1996). In the current study we showed that NGF treatment increased the endocytosis of FITC-dextran and Tfn. The data for Tfn are especially important; they show that NGF acted via TrkA activation to cause increased clathrin-mediated membrane trafficking of a receptor unrelated to the NGF receptors and for which the endocytosis is constitutive. The endocytosis of other receptors, including those for which the internalization normally is induced by ligand binding, also may be regulated by NGF. Indeed, in preliminary studies the endocytosis of EGFR has been shown to increase after NGF treatment (Howe and Mobley, unpublished observations). Our findings suggest that NGF may regulate the endocytosis of many receptors that are present in clathrin-coated membranes. If so, increased clathrin-coated membrane formation and trafficking may play an important role in mediating the trophic effects of NGF and other NTs. One such effect might involve increased delivery of receptors carrying nutrients, thus rapidly supplying neurons with substrates that are important for growth and differentiation. The enhanced uptake of Tfn shown here may supply the iron that is needed for the function of iron-containing proteins under conditions of NGF stimulation. Through increased endocytosis of plasma membrane receptors for neurotransmitters and growth factors, NTs may impact the ability of a neuron to respond to such influences. The interesting possibility arises that NTs could exert indirect but important influences on signaling through non-NT signaling pathways. Finally, it is tempting to speculate that NT signaling might enhance the uptake of synaptic vesicle proteins through clathrin-coated membranes, an action that could support neurotransmission directly (Berninger et al., 1999;Schinder et al., 2000). It will be important to explore further the significance for neuronal function of the NT-induced increase in clathrin-mediated membrane trafficking.
Footnotes
This work was supported by the Adler Foundation, the McGowan Charitable Fund, gifts from the Powell and Wright Foundations, a Howard Hughes Medial Institute predoctoral fellowship (C.L.H.), and National Institutes of Health Grants GM38093 (F.M.B.) and NS24054 (W.C.M.). We thank Drs. Mark von Zastrow and Nigel Bunnett for the use of epifluorescence and confocal microscopes and for helpful discussions. We thank Janice Valletta for technical assistance. Mark Bunin was most helpful in preparing the hippocampal cultures.
E.C.B. and C.L.H. contributed equally to this work.
Correspondence should be addressed to Dr. William C. Mobley, Department of Neurology and Neurological Sciences, Department of Pediatrics, and the Program in Neuroscience, Stanford University, MSLS P211, 1201 Welch Road, Stanford, CA 94305. E-mail: NGFV1@leland.stanford.edu.
Dr. Beattie's present address: Langley Porter Psychiatric Institute, Department of Psychiatry, University of California at San Francisco, 401 Parnassus Avenue, San Francisco, CA 94143.
Dr. Howe's present address: Department of Neurology and Neurological Sciences, Stanford University, 1201 Welch Road, Stanford, CA 94305.
Dr. Wilde's present address: Carnegie Institution of Washington, Department of Embryology, 115 West University Parkway, Baltimore, MD 21210.
REFERENCES
- 1.Beattie EC, Zhou J, Grimes ML, Bunnett NW, Howe CL, Mobley WC. A signaling endosome hypothesis to explain NGF actions: potential implications for neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:389–406. [PubMed] [Google Scholar]
- 2.Beck KA, Chang M, Brodsky FM, Keen JH. Clathrin assembly protein AP-2 induces aggregation of membrane vesicles: a possible role for AP-2 in endosome formation. J Cell Biol. 1992;119:787–796. doi: 10.1083/jcb.119.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benmerah A, Gagnon J, Bègue B, Mégarbané B, Dautry-Varsat A, Cerf-Bensussan N. The tyrosine kinase substrate Eps15 is constitutively associated with the plasma membrane adaptor AP-2. J Cell Biol. 1995;131:1831–1838. doi: 10.1083/jcb.131.6.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berninger B, Schinder AF, Poo MM. Synaptic reliability correlates with reduced susceptibility to synaptic potentiation by brain-derived neurotrophic factor. Learn Mem. 1999;6:232–242. [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharyya A, Watson FL, Bradlee TA, Pomeroy SL, Stiles CD, Segal RA. Trk receptors function as rapid retrograde signal carriers in the adult nervous system. J Neurosci. 1997;17:7007–7016. doi: 10.1523/JNEUROSCI.17-18-07007.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodsky FM. Clathrin structure characterized with monoclonal antibodies. I. Analysis of multiple antigenic sites. J Cell Biol. 1985;101:2047–2054. doi: 10.1083/jcb.101.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casaccia-Bonnefil P, Gu C, Chao MV. Neurotrophins in cell survival/death decisions. Adv Exp Med Biol. 1999;468:275–282. doi: 10.1007/978-1-4615-4685-6_22. [DOI] [PubMed] [Google Scholar]
- 8.Chin DJ, Straubinger RM, Acton S, Nathke I, Brodsky FM. 100 kDa polypeptides in peripheral clathrin-coated vesicles are required for receptor-mediated endocytosis. Proc Natl Acad Sci USA. 1989;86:9289–9293. doi: 10.1073/pnas.86.23.9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly JL, Green SA, Greene LA. Pit formation and rapid changes in surface morphology of sympathetic neurons in response to nerve growth factor. J Cell Biol. 1981;90:176–180. doi: 10.1083/jcb.90.1.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly JL, Green SA, Greene LA. Comparison of rapid changes in surface morphology and coated pit formation of PC12 cells in response to nerve growth factor, epidermal growth factor, and dibutyryl cyclic AMP. J Cell Biol. 1984;98:457–465. doi: 10.1083/jcb.98.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corvera S. Insulin stimulates the assembly of cytosolic clathrin onto adipocyte plasma membranes. J Biol Chem. 1990;265:2413–2416. [PubMed] [Google Scholar]
- 12.Cunningham ME, Stephens RM, Kaplan DR, Greene LA. Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. J Biol Chem. 1997;272:10957–10967. doi: 10.1074/jbc.272.16.10957. [DOI] [PubMed] [Google Scholar]
- 13.Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of TrkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrari G, Anderson BL, Stephens RM, Kaplan DR, Greene LA. Prevention of apoptotic neuronal death by GM1 ganglioside. Involvement of Trk neurotrophin receptors. J Biol Chem. 1995;270:3074–3080. doi: 10.1074/jbc.270.7.3074. [DOI] [PubMed] [Google Scholar]
- 16.Francis N, Farinas I, Brennan C, Rivas-Plata K, Backus C, Reichardt L, Landis S. NT-3, like NGF, is required for survival of sympathetic neurons, but not their precursors. Dev Biol. 1999;210:411–427. doi: 10.1006/dbio.1999.9269. [DOI] [PubMed] [Google Scholar]
- 17.Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grimes ML, Beattie E, Mobley WC. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proc Natl Acad Sci USA. 1997;94:9909–9914. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haffner C, Takei K, Chen H, Ringstad N, Hudson A, Butler MH, Salcini AE, Di Fiore PP, De Camilli P. Synaptojanin 1: localization on coated endocytic intermediates in nerve terminals and interaction of its 170 kDa isoform with Eps15. FEBS Lett. 1997;419:175–180. doi: 10.1016/s0014-5793(97)01451-8. [DOI] [PubMed] [Google Scholar]
- 20.Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. Nerve growth factor signaling in caveolae-like domains at the plasma membrane. J Biol Chem. 1999;51:36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- 21.Ip NY, Li Y, Yancopoulos GD, Lindsay RM. Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci. 1993a;13:3394–3405. doi: 10.1523/JNEUROSCI.13-08-03394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and non-neuronal cells. Neuron. 1993b;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 24.Koizumi S, Contreras ML, Matsuda Y, Hama T, Lazarovici P, Guroff G. K-252a: a specific inhibitor of the action of nerve growth factor on PC12 cells. J Neurosci. 1988;8:715–721. doi: 10.1523/JNEUROSCI.08-02-00715.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lester RA, Quarum ML, Parker JD, Weber E, Jahr CE. Interaction of 6-cyano-7-nitroquinoxaline-2,3-dione with the N-methyl-d-aspartate receptor-associated glycine binding site. Mol Pharmacol. 1989;35:565–570. [PubMed] [Google Scholar]
- 26.Li YW, Holtzman DM, Kromer LF, Kaplan DR, Chuacouzens J, Clary DO, Knusel B, Mobley WC. Regulation of TrkA and ChAT expression in developing rat basal forebrain—evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loeb DM, Greene LA. Transfection with Trk restores slow NGF binding, efficient NGF uptake, and multiple NGF responses to NGF-nonresponsive PC12 cell mutants. J Neurosci. 1993;13:2919–2929. doi: 10.1523/JNEUROSCI.13-07-02919.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsh M, McMahon HT. The structural era of endocytosis. Science. 1999;285:215–220. doi: 10.1126/science.285.5425.215. [DOI] [PubMed] [Google Scholar]
- 29.McMahon HT, Wigge P, Smith C. Clathrin interacts specifically with amphiphysin and is displaced by dynamin. FEBS Lett. 1997;413:319–322. doi: 10.1016/s0014-5793(97)00928-9. [DOI] [PubMed] [Google Scholar]
- 30.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud D, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;2:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 31.Mobley WC, Schenker A, Shooter EM. Characterization and isolation of proteolytically modified nerve growth factor. Biochemistry. 1976;15:5543–5552. doi: 10.1021/bi00670a019. [DOI] [PubMed] [Google Scholar]
- 32.Nathke IS, Heuser J, Lupas A, Stock J, Turck CW, Brodsky FM. Folding and trimerization of clathrin subunits at the triskelion hub. Cell. 1992;68:899–910. doi: 10.1016/0092-8674(92)90033-9. [DOI] [PubMed] [Google Scholar]
- 33.Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF–TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- 34.Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- 35.Schmid SL. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu Rev Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt A, Wolde M, Thiele C, Fest W, Kratzin H, Podtelejnikov AV, Witke W, Huttner WB, Soling HD. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature. 1999;401:133–141. doi: 10.1038/43613. [DOI] [PubMed] [Google Scholar]
- 37.Senger DL, Campenot RB. Rapid retrograde tyrosine phosphorylation of TrkA and other proteins in rat sympathetic neurons in compartmented cultures. J Cell Biol. 1997;138:411–421. doi: 10.1083/jcb.138.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silos-Santiago I, Greenlund LJ, Johnson EM, Jr, Snider WD. Molecular genetics of neuronal survival. Curr Opin Neurobiol. 1995;5:42–49. doi: 10.1016/0959-4388(95)80085-9. [DOI] [PubMed] [Google Scholar]
- 39.Slepnev VI, Ochoa GC, Butler MH, Grabs D, De Camilli P. Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science. 1998;281:821–824. doi: 10.1126/science.281.5378.821. [DOI] [PubMed] [Google Scholar]
- 40.Snider WD. Functions of the neurotrophins during nervous system development: what the knock-outs are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 41.Tebar F, Sorkina T, Sorkin A, Ericsson M, Kirchhausen T. Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J Biol Chem. 1996;271:28727–28730. doi: 10.1074/jbc.271.46.28727. [DOI] [PubMed] [Google Scholar]
- 42.Tsui-Pierchala BA, Ginty DD. Characterization of an NGF-P–TrkA retrograde signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J Neurosci. 1999;19:8207–8218. doi: 10.1523/JNEUROSCI.19-19-08207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Delft S, Schumacher C, Hage W, Verkleij AJ, van Bergen en Henegouwen PM. Association and colocalization of Eps15 with adaptor protein-2 and clathrin. J Cell Biol. 1997;136:811–821. doi: 10.1083/jcb.136.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson FL, Porcionatto MA, Bhattacharyya A, Stiles CD, Segal RA. TrkA glycosylation regulates receptor localization and activity. J Neurobiol. 1999;39:323–336. doi: 10.1002/(sici)1097-4695(199905)39:2<323::aid-neu15>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 45.Wilde A, Brodsky FM. In vivo phosphorylation of adaptors regulates their interaction with clathrin. J Cell Biol. 1996;135:635–645. doi: 10.1083/jcb.135.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- 47.Yuen EC, Mobley WC. Early BDNF, NT-3, and NT-4 signaling events. Exp Neurol. 1999;159:297–308. doi: 10.1006/exnr.1999.7148. [DOI] [PubMed] [Google Scholar]
- 48.Yuen EC, Howe CL, Li Y, Holtzman DM, Mobley WC. Nerve growth factor and the neurotrophic factor hypothesis. Brain Dev. 1996;18:362–368. doi: 10.1016/0387-7604(96)00051-4. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, Valletta JS, Grimes ML, Mobley WC. Multiple levels for regulation of TrkA in PC12 cells by nerve growth factor. J Neurochem. 1995;65:1146–1156. doi: 10.1046/j.1471-4159.1995.65031146.x. [DOI] [PubMed] [Google Scholar]