

Abstract
Expression of the inducible form of nitric oxide synthase (iNOS) in brain may contribute to neurotoxicity in Alzheimer's disease (AD). Expression of iNOS can be induced in cerebellar granule cells (CGCs)in vivo as well as in vitro, allowing these cells to be used to study regulation of neuronal iNOS expression. We report here that microinjection of bacterial lipopolysaccharide and interferon gamma into rat cerebellum induced iNOS expression in CGCs and subsequent cell death assessed by staining for DNA fragmentation. Co-injection of three structurally distinct agonists of the peroxisome proliferator-activated receptor gamma (PPARγ), including the antidiabetic thiazolidinedione troglitazone, the nonsteroidal anti-inflammatory drug (NSAID) ibuprofen, and the prostanoid 15-deoxy-Δ12,14 prostaglandin J2, reduced both iNOS expression and cell death, whereas co-injection of the selective cyclo-oxygenase inhibitor NS-398 had no effect. These data demonstrate that PPARγ agonists can modulate inflammatory responses in brain. Because sustained medication with NSAIDs reduces the risk and delays the onset of AD, these results further suggest that NSAIDs provide therapeutic value by binding to PPARγ present in AD brain, thereby preventing iNOS expression and neuronal cell death.
Keywords: iNOS, PPARγ, cerebellar granule neurons, NSAIDs, Alzheimer's disease, apoptosis
Inflammatory activation of neuronal, as well as glial cells is believed to contribute to cell death and damage during neurological disease. In Alzheimer's disease (AD), inflammatory responses include transcription factor NFκB activation (Kuner et al., 1998; Kaltschmidt et al., 1999) and cytokine expression near plaques (Aisen and Davis, 1994). Accumulating data (Minc-Golomb et al., 1994; Sato et al., 1995; Heneka et al., 1998) indicates that neurons can express the inducible nitric oxide synthase (iNOS), whose production of NO can be neurotoxic (Dawson et al., 1994;Skaper et al., 1995). iNOS expression can cause neuronal apoptosisin vivo (Matsuoka et al., 1999; Quan et al., 1999) and induce apoptosis in macrophages (Sarih et al., 1993), astrocytes (Hu and Van, 1996), and differentiated PC12 cells (Heneka et al., 1998)in vitro. A role for iNOS in AD is suggested by findings that neurons within tangles (Vodovotz et al., 1996) and around Hirano bodies (Lee et al., 1999) express iNOS and that staining for nitrotyrosine is increased near lesion sites (Smith et al., 1997). Suppression of neuronal iNOS expression may therefore reduce neuronal damage in AD.
Recently it was shown that activation of the peroxisome proliferator-activated receptor gamma (PPARγ) reduces proinflammatory cytokine and iNOS expression in macrophages (Lemberger et al., 1996a;Colville-Nash et al., 1998; Ricote et al., 1998), microglial cells (Petrova et al., 1999), and monocytes (Jiang et al., 1998; Combs et al., 2000). PPARγ is a member of the nuclear hormone receptor superfamily implicated in adipocyte differentiation, insulin sensitivity, and inflammatory processes (Lemberger et al., 1996b;Vamecq and Latruffe, 1999). The anti-inflammatory actions of PPARγ are activated by structurally distinct ligands, including NSAIDs (Lehmann et al., 1997), antidiabetic thiazolidinediones (TZDs) (Thieringer et al., 2000), and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), a naturally occurring agonist (Forman et al., 1995; Kliewer et al., 1995). Activated PPARγ heterodimerizes with the retinoic acid receptor, binds to PPAR response elements, and induces gene transcription. The anti-inflammatory actions of PPARγ are not attributable to inhibition of cyclooxygenase (Colville-Nash et al., 1998; Heneka et al., 1999;Combs et al., 2000; Willson et al., 2000) but may be mediated by suppression of transcription factor activity (Colville-Nash et al., 1998; Ricote et al., 1999).
Recently, PPARγ agonists were shown to protect neuroblastoma cells against neurotoxic effects of conditioned media from monocytes stimulated with β-amyloid (Combs et al., 2000) or with lipopolysaccharide (LPS) and cytokines (Klegeris et al., 1999). Because NSAID treatment reduces the risks and delays the onset of AD (McGeer et al., 1996; Stewart et al., 1997), these results suggest that PPARγ activation by NSAIDs may mediate their therapeutic effects. Because neurons express iNOS in AD, it is possible that suppression of neuronal inflammation also contributes to the beneficial effects of NSAIDs. We recently demonstrated that iNOS expression in cerebellar granule cells (CGCs) induces cell death, which was blocked by selective iNOS inhibitors (Heneka et al., 1999) and by three structurally diverse PPARγ ligands (Fig. 1). In the present study we demonstrate that these ligands also downregulate neuronal iNOS expression and CGC death in vivo.
Fig. 1.

Structural relationship of PPARγ ligands used in this study.
MATERIALS AND METHODS
Materials. LPS (Salmonella typhirium), phenylmethylsulfonyl fluoride, aprotinin, leupeptin, and ibuprofren (IBU) were from Sigma (St. Louis, MO); 15-deoxy-Δ12,14 prostaglandin J2 (15d-PGJ2) was from Alexis (San Diego, CA); NS-398 was from Calbiochem (San Diego, CA); and troglitazone (Trog) was a gift of Parke-Davis (Ann Arbor, MI)
Animals. Male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 250–300 gm, were housed in groups of four under standard conditions at 22°C and a 12 hr light/dark cycle with ad libitum access to food and water.
Injection of immunostimulants. Rats were anesthetized with pentobarbital (50 mg/kg, i.p.; dissolved in 0.9% sodium chloride) and placed on a heating blanket. Body temperature was monitored by a rectal probe connected to the heating blanket and maintained at 37 ± 0.5°C for the time of surgery. Thereafter animals were placed in a stereotaxic frame. After exposure of the skull, a hole was drilled at the injection site, and 2 μl of a mixture containing recombinant rat interferon gamma (IFN-γ) (20 U; Life Technologies, Gaithersburg, MD), bacterial endotoxin LPS (10 μg; Salmonella typhirium; Sigma), and indicated anti-inflammatory agents (either none, 100 nmol of troglitazone, 100 nmol of ibuprofen, 10 nmol of 15d-PGJ2, or 10 nmol of NS-398) in PBS, pH 7.4, were injected over a period of 120 sec into cerebellum using a 5 μl Hamilton syringe, at anteroposterior (AP) −12.5, lateral (L) 0.0, and ventral (V) 5.0 mm relative to bregma (Paxinos et al., 1985). Controls received 2 μl of PBS. The needles were left in place for a further 5 min to prevent reflux up the needle tract. To maintain constant body temperature, animals were placed under a heating lamp until complete recovery from anesthesia. Twenty-four hours after intracerebellar injection, animals were killed by an overdose of pentobarbital and then perfused transcardially with 200 ml of heparinized sodium chloride (0.9%) and 200 ml of fixative containing 10% formaldehyde, 10% acetic acid, and 80% methanol. Brains were removed, immersed in fixative for 72 hr at room temperature, then paraffin-embedded. In some cases brains were removed without perfusion, and protein lysates were prepared for immunoblot detection. All experiments were performed in accordance with the declaration of Helsinski and the animal welfare guidelines and laws of the United States of America and were approved by the local ethical committee for animal experiments.
Processing of brain for immunohistochemistry.Immunohistochemistry was performed as previously described (Heneka et al., 2000). Serial coronal sections of the cerebellum were cut 8-μm-thick using a Leitz microtome and mounted on poly-l-lysine-coated slides. Slides were immersed in 10 mm citrate buffer, pH 6.0, and heated in a microwave oven, four cycles of 5 min each, to unmask antigen sites. Slides were cooled for 20 min at room temperature, then washed in PBS. Endogenous peroxidase activity was inhibited by rinsing slides in 0.1% hydrogen peroxide for 10 min. Nonspecific binding was blocked by 10% normal goat serum in PBS for 1 hr at room temperature. After washing in PBS, sections were incubated overnight at 4°C with primary antibodies: (1) mAb N32020 directed against iNOS (1:200 dilution; Transduction Laboratories, Lexington, KY); (2) mAb MCA 341 raised against rat brain NOS (bNOS) (1:500 dilution; Serotec, Raleigh, NC). Sections were washed extensively with PBS, then incubated with biotinylated anti-rabbit or anti-mouse IgG (1:200 dilution; Vector Laboratories, Burlingame, CA) for 30 min at room temperature. Immunohistochemical localization was performed using the avidin–biotin peroxidase complex method (ABC kit; Vector Laboratories) with 3,3′-diaminobenzidine as chromogen.
Quantification of immunohistochemistry. Quantitative analysis of iNOS- and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive cells was performed on brain sections from animals (three from each group). Antigens were detected in five sections having a defined distance relative to the level of cerebellar injection. The sections were the middle section corresponding to the level of injection, with the injection site discernible, and the other four sections were taken at a distance of 25 and 50 μm rostral and caudal of the injection site. Three different areas were defined and evaluated: caudal, and left and right from the injection side. The number of cells within the respective fields was determined using a counting grid.
Western blot analysis. Protein extracts were prepared by sonication of whole brain cerebellum (25 mg of wet weight tissue) in 10 volumes of 8 m urea. Aliquots were immediately mixed with SDS sample buffer, boiled, and either used immediately or frozen at −80°C. Twenty micrograms of protein were separated through 10% polyacrylamide SDS gels, and proteins were transferred by semidry blotting to polyvinylidene difluoride membranes. The membranes were blocked in Tris-buffered saline with 0.05% Tween 20 containing 0.5% BSA, washed, incubated with primary antibodies to iNOS (mAb, 1:1,000 dilution; Cayman, Ann Arbor, MI) or bNOS overnight at 4°C, washed extensively, incubated with peroxidase conjugated goat anti-mouse IgG, and then bands were visualized with enhanced chemiluminescence reagents (Pierce, Rockford, IL).
TUNEL staining. For TUNEL staining, slides were deparaffinized, washed three times with PBS, and preincubated with 0.1m sodium cacodylate (TDT) buffer for 5 min. Thereafter, the slides were exposed for 10 min to the reaction mixture (50 U of terminal transferase, 10 nm biotin-dUTP, and 25 mm cobalt chloride in TDT buffer). The reaction was stopped by incubating the slides for 10 min with 0.1 m sodium acetate. After blocking with 2% BSA, slides were incubated for 5 min with streptavidin—alkaline–phosphatase conjugate and developed with 0.41 mm nitroblue tetrazolium chloride and 0.38 mm 5-bromo-4-chloro-3-indolyl phosphate in 200 mm Tris-HCl, pH 9.5, containing 10 mmMgCl2. Technical controls were done in the absence of cobalt chloride.
Protein content determination. Protein concentration was determined spectrophotometrically in 96 well plates with Bradford reagent using bovine serum albumin as standard (Bradford, 1976).
Statistical analysis. Data are shown as mean ± SD of the number of positive cells per square millimeter. Differences between controls, immunostimulated, and treated animals were assessed by one-way ANOVA followed by a Tukey test (Systat, Evanston, IL).
RESULTS
Microinjection of a mixture of LPS and IFN-γ into the vermis of the rat cerebellum (Fig. 2) induced expression of iNOS, as assessed by immunoblot analysis for iNOS protein 24 hr after injection (Fig.3A). Injection of PBS served as control and failed to induce iNOS expression. Co-injection of three structurally distinct PPARγ ligands (Ibu, Trog, and 15d-PGJ2) reduced LPS/IFNγ-induced iNOS expression (Fig. 3A). Expression of bNOS, the constitutive NOS isoform expressed by CGCs, was neither affected by immunostimulation nor by co-injection with any of the PPARγ agonists (Fig. 3B).
Fig. 2.

Schematic of injection site into cerebellum. The diagram is of a coronal section showing the position of the microinjection cannula in the rat cerebellum. The injection cannula was placed stereotaxically into the cerebellum at AP −12.5, L 0.0, and V 5.0 mm relative to bregma (Paxinos et al., 1985). The scale given applies in both horizontal and vertical directions.
Fig. 3.

Suppression of cerebellar iNOS expression by PPARγ agonists. A mixture of LPS (10 μg) plus IFN-γ (20 U) dissolved in PBS was microinjected into rat cerebellum alone (none) or with ibuprofen (Ibu), troglitazone (Trog), or 15d-PGJ2(PGJ2). Injection of PBS only served as control (PBS). Levels of iNOS (A) and bNOS (B) were determined 24 hr after injections by Western blot analysis. The blots shown are representative of two independent experiments.
Immunocytochemical staining of rat cerebella 24 hr after injection of LPS/IFNγ revealed that the iNOS protein was primarily derived from CGCs (Fig. 4) and not from glial cells or macrophages. Consistent with the immunoblot results, iNOS-positive cells were not detectable after PBS injection (Fig.4A). Injection of LPS/IFNγ induced iNOS immunoreactivity almost exclusively in the granule cell layer of the cerebellum (Fig. 4B) and only occasionally in the molecular layer (data not shown). Immunostimulation in the presence of ibuprofen (Fig. 4C), troglitazone (Fig.4D), or 15d-PGJ2 (Fig.4E) significantly decreased iNOS-positive staining. Immunostimulation in the presence of the cyclooxygenase (COX)-2-selective inhibitor NS-398 did not reduce iNOS-positive staining (Fig. 4F). For quantitative assessment of iNOS-immunopositive cells, cerebellar sections with a defined distance rostral and caudal to the level of injection were evaluated. The number of iNOS-positive cells was maximal at the level of injection (Fig.5A). Ibuprofen was the most potent inhibitor and reduced iNOS-positive cell staining to background (PBS-injected) values at all distances from the injection site (p > 0.05 vs noninjected values; Fig.5B). Troglitazone reduced overall iNOS staining across all sections by ∼75% (from 70 ± 4 to 16 ± 1 positive cells per square millimeter per section), whereas 15d-PGJ2 was least effective and reduced overall iNOS staining by ∼50% (to 31 ± 5 positive cells per square millimeter per section). In all cases the number of bNOS-expressing cells was unaffected by immunostimulation or by treatment with PPARγ agonists (data not shown). The presence of NS-398 had no significant effect on iNOS-positive staining.
Fig. 4.

Immunhistocytochemical localization of iNOS expression and DNA strand breaks. PBS (A,G) or LPS plus IFN-γ in PBS (B–F, H–L) were injected into rat cerebellum, together with ibuprofen (C, I), troglitazone (D, J), 15d-PGJ2 (E, K), or NS-398 (F, L). After 24 hr the brains were removed and prepared for immunohistochemical detection of either iNOS (A–F) or DNA breaks by the TUNEL method (G–L). Scale bar, 50 μm.
Fig. 5.
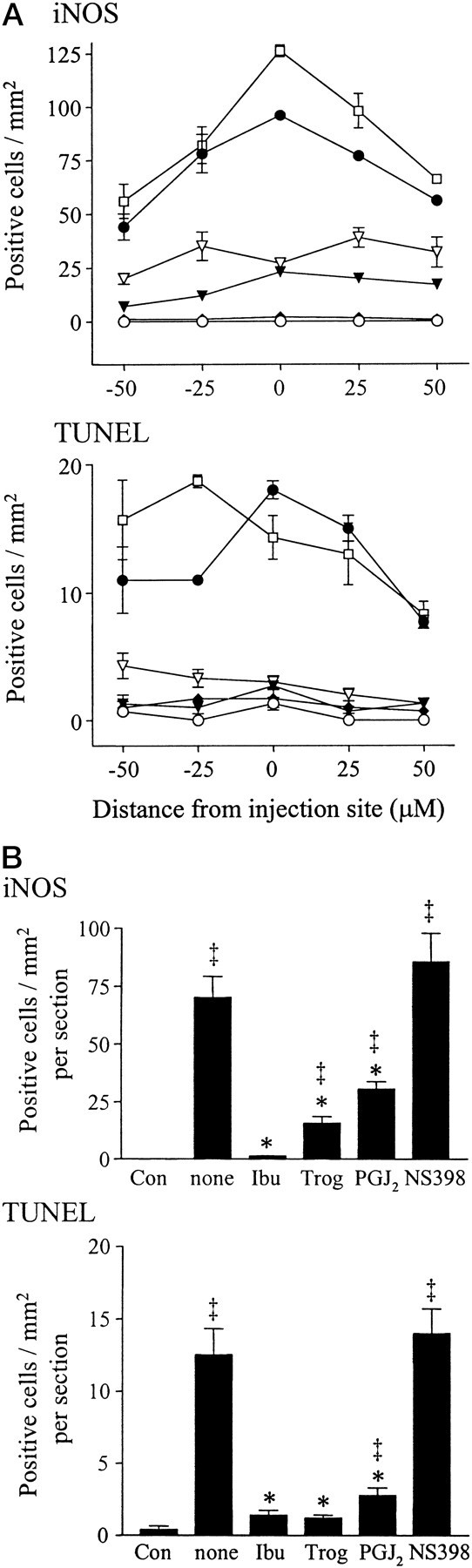
Quantitative analysis of iNOS expression and DNA strand breaks. A, Coronal cerebellar sections with a defined distance rostral and caudal to the level of injection were evaluated for iNOS-immunopositive or TUNEL-positive cells. Positive cells were counted using a counting grid and are given as positive cells per square millimeter. PBS injection (○) and immunostimulation with LPS plus IFN-γ (●) were compared to immunostimulation done in the presence of ibuprofen (♦), troglitazone (▾), 15d-PGJ2 ▿, or NS-398 (■). For all drugs except NS-398, significance was p < 0.01 versus immunostimulation alone (n = 3 for each group). B, The data in A were analyzed as the average number of positive staining cells per square millimeter per section. *p < 0.001 versus immunostimulation alone (none); ‡p < 0.01 versus control brain (no immunostimulation) (n = 15 for each group).
To evaluate the consequences of iNOS induction in CGCs, TUNEL staining was performed in adjacent cerebellar sections to provide an index of cell damage or death. Injection of PBS did not result in TUNEL labeling of CGCs (Fig. 4G). In contrast, induction of iNOS expression was paralleled by appearance of TUNEL-positive granule cells and appearance of chromatin condensation (Fig. 4H). As seen for iNOS staining, the number of TUNEL-positive cells was maximal at the level of injection (Fig. 5A) and markedly decreased in animals co-injected with either ibuprofen (Fig.4I), troglitazone (Fig. 4J), or 15d-PGJ2 (Fig. 4K). Across all sections examined, ibuprofen and troglitazone comparably reduced TUNEL-positive staining (to ∼10% of maximal values), whereas 15d-PGJ2 reduced TUNEL staining to ∼20% of maximal values. The degree of TUNEL-positive staining was not effected by immunostimulation in the presence of NS-398 (Fig.4L).
DISCUSSION
Induction of iNOS expression in CGCs both in vitro andin vivo has been previously described (Minc-Golomb et al., 1994; Sato et al., 1995; Heneka et al., 1999). We previously demonstrated that immunostimulation of CGCs in vitro results in transcription and expression of iNOS, subsequent release of NO, and induction of NO-dependent apoptotic cell death. Furthermore, we demonstrated that agonists of the PPARγ downregulate iNOS, thereby protecting CGCs from LPS and cytokine-induced cell death in vitro (Heneka et al., 1999). Because CGCs express the PPARγin vivo (Braissant et al., 1996), those results prompted us to test the consequences of iNOS induction in vivo and the possible modulation by agonists of PPARγ. Injection of bacterial LPS and IFN-γ into the vermis of the rat cerebellum induced iNOS expression in CGCs detected by immunoblot analysis and immunohistochemistry. In contrast, expression of bNOS, the constitutive isoform of NOS enzymes expressed by granule cells, was not affected by immunostimulation. Total iNOS protein and the number of iNOS-expressing granule cells were markedly reduced by three structurally distinct PPARγ agonists. These results suggest that PPARγ agonists provide similar anti-inflammatory actions in vivo as in vitro.
Our results demonstrate that increased CGC iNOS expression is accompanied by an increase in TUNEL-positive staining cells, suggesting that in vivo, as in vitro, iNOS-derived NO has neurotoxic consequences. This is supported by our findings that the decrease in iNOS expression caused by co-injection of PPARγ ligands with LPS and cytokines is accompanied by a parallel decrease in the appearance of TUNEL-positive staining cells. However, because PPARγ agonists can decrease expression of other cytokines, it is likely that other mechanisms contribute to their neuroprotective actions in vivo. Because the TUNEL method provides an index of DNA fragmentation and does not unequivocally distinguish between apoptotic versus necrotic pathways that can both lead to DNA damage, we cannot conclude if injection of LPS and cytokines causes induction of CGC apoptosis or necrosis. However, our previous findings that immunostimulation of primary CGC cultures resulted in iNOS expression, NO-dependent caspase-3 activation, DNA fragmentation, and CGC cell death suggests that at least a portion of the TUNEL-positive staining cells observed in vivo are undergoing apoptosis. Finally, although our data does not rule out that cells other than CGCs also show DNA damage, the fact that virtually all TUNEL staining is prevented by ibuprofen or troglitazone suggests that these drugs can act at multiple cell types.
Our in vivo data complements and extends recent in vitro studies describing anti-inflammatory actions of PPARγ agonists in human monocytes (Klegeris et al., 1999; Combs et al., 2000). In these studies, the neurotoxic effects of conditioned media from β-amyloid or LPS plus IFNγ-stimulated human THP-1 monocytes on human neuroblastoma cells were reduced when the THP-1 cells were stimulated in the presence of NSAIDs, troglitazone, or 15d-PGJ2, suggesting that PPARγ ligands could reduce neurotoxicity by blocking microglial inflammatory activation. Our observations that both iNOS expression and DNA fragmentation was induced in CGCs suggests that in this model of inflammation, the neuroprotective effects of PPARγ ligands are mediated neuronally, although effects on surrounding glial cells are not ruled out.
The exact mechanisms by which 15d-PGJ2, ibuprofen, and troglitazone reduce iNOS expression are not yet clear. With respect to pharmacological specificity and selectivity, although the above drugs are agonists of PPARγ, these agents may have additional actions in the brain. The identification of 15d-PGJ2 as an endogenous ligand of PPARγ (Kliewer et al., 1995) suggested that, at least in some cases, 15d-PGJ2 may be acting via PPARγ activation. This was clearly demonstrated by findings that 15d-PGJ2 reduced iNOS expression in RAW macrophages (which did not express PPARγ) if they were first transfected with a PPARγ expression plasmid (Ricote et al., 1998). However, the recent demonstration that 15d-PGJ2(and other cyclopentenones including PGA1) directly inhibit the activity of the IκB kinase IKKβ (Rossi et al., 2000), which is necessary to target IκB proteins for degradation, provides a mechanism by which 15d-PGJ2 can block NFκB activation in the absence of PPARγ. This may account for the ability of 15d-PGJ2 to block iNOS expression in microglial cells, despite lack of activation of PPAR gene transcription (Petrova et al., 1999).
The TZD troglitazone has also been shown to exert potent anti-inflammatory effects on cells. Troglitazone decreased tumor necrosis factor-α synthesis and expression in phorbol myristyl acetate-activated human monocytes (Jiang et al., 1998), and ciglitazone (a closely related TZD) blocked iNOS expression in rat astrocytes (Kitamura et al., 1999b). Activation of PPARγ by troglitazone reduced transcription from numerous promoter elements including NFκB, GAS, AP1 (Ricote et al., 1998), and NFAT (Yang et al., 2000), which can all contribute to activation of inflammatory gene expression. Although the TZDs were developed to be high-affinity, PPAR subtype-selective agonists, with binding affinities in the submicromolar range (Willson et al., 2000), and there are as yet no data to suggest binding to other proteins, troglitazone was reported to block iNOS expression in microglial cells without activating a PPAR-responsive reporter gene (Petrova et al., 1999). Thus, anti-inflammatory actions of troglitazone may, as the case for 15d-PGJ2, be mediated by mechanisms in addition to PPARγ activation.
The NSAIDs are known inhibitors of COXs, raising the possibility that the protective effects we observed after co-injection of ibuprofen were caused by inhibition of brain COX activity or expression. To address this possibility, we directly tested the effects of the selective COX-2 inhibitor NS-398 and found that this inhibitor neither reduced iNOS-positive nor TUNEL-positive staining. This finding is consistent with our previous in vitro studies in which we showed that concentrations of NSAIDs sufficient to inhibit COX and block prostaglandin synthesis were without effect on iNOS expression or CGC death (Heneka et al., 1999). Similarly, the neuroprotective effects of ibuprofen and indomethacin were not attributable to COX inhibition because their actions were not replicated by NS-398 (Klegeris et al., 1999; Combs et al., 2000), and neither 15d-PGJ2nor troglitazone have been shown to inhibit COX activity (Colville-Nash et al., 1998; Fujiwara et al., 1998). These findings suggest that the therapeutic actions of NSAIDs in AD are mediated via mechanisms other than COX inhibition, consistent with the fact that the therapeutic effects NSAIDs occur at concentrations greater than those that inhibit COX (Lehmann et al., 1997; Jiang et al., 1998) and that the COX inhibitor aspirin does not exert protective effects in AD (Stewart et al., 1997).
Observations of iNOS expression in tangle-bearing neurons (Vodovotz et al., 1996), in hippocampal neurons (Lee et al., 1999), and of increased nitrotyrosine-labeling of lesion sites in Alzheimer's disease (Smith et al., 1997) suggests that these results may have direct clinical implications. Stimulation of PPARγ by NSAIDs has been suggested to account for the beneficial effects observed in the treatment of rheumatoid arthritis at plasma drug concentrations substantially higher than required to inhibit cyclooxygenase (Breitner et al., 1995). The epidemiological observation that long-term treatment of patients suffering from rheumatoid arthritis with NSAIDs results in reduced risk and delayed onset of AD (McGeer et al., 1996; Stewart et al., 1997), and the finding that PPARγ is expressed in neurons (Braissant et al., 1996) and increased in AD (Kitamura et al., 1999a) suggests that PPARγ could play a pivotal role in the pathophysiology of neurodegenerative diseases. The beneficial effects of PPARγ agonists demonstrated in this in vivo study suggest that such compounds may have neuroprotective and anti-inflammatory properties. Because thiazolidinedione drugs act as PPARγ-agonists and currently are in clinical use as antidiabetic drugs, these compounds should be considered as candidates for clinical trials in AD and neuroinflammatory disease.
Footnotes
This work was supported by a grant to M.T.H. from the Deutsche Forschungsgemeinschaft (SFB 400-A8) and by National Institutes of Health Grant NS-31556 to D.L.F. We thank Anthony Sharp and Lucia Dumitrescu for technical assistance.
Correspondence should be addressed to Michael T. Heneka, Department of Neurology, University of Bonn, Sigmund-Freud-Strasse 25, 53105 Bonn, Germany. E-mail: m.heneka@uni-bonn.de.
REFERENCES
- 1.Aisen PS, Davis KL. Inflammatory mechanisms in Alzheimer's disease: implications for therapy. Am J Psychiatry. 1994;151:1105–1113. doi: 10.1176/ajp.151.8.1105. [DOI] [PubMed] [Google Scholar]
- 2.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 4.Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, Pericak VM, Saunders AM. Delayed onset of Alzheimer's disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging. 1995;16:523–530. doi: 10.1016/0197-4580(95)00049-k. [DOI] [PubMed] [Google Scholar]
- 5.Colville-Nash PR, Qureshi SS, Willis D, Willoughby DA. Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: correlation with induction of heme oxygenase 1. J Immunol. 1998;161:978–984. [PubMed] [Google Scholar]
- 6.Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson VL, Brahmbhatt HP, Mong JA, Dawson TM. Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology. 1994;33:1425–1430. doi: 10.1016/0028-3908(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 8.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 9.Fujiwara T, Ohsawa T, Takahashi S, Ikeda K, Okuno A, Ushiyama S, Matsuda K, Horikoshi H. Troglitazone, a new antidiabetic agent possessing radical scavenging ability, improved decreased skin blood flow in diabetic rats. Life Sci. 1998;63:2039–2047. doi: 10.1016/s0024-3205(98)00482-2. [DOI] [PubMed] [Google Scholar]
- 10.Heneka MT, Loschmann PA, Gleichmann M, Weller M, Schulz JB, Wullner U, Klockgether T. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem. 1998;71:88–94. doi: 10.1046/j.1471-4159.1998.71010088.x. [DOI] [PubMed] [Google Scholar]
- 11.Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wullner U, Klockgether T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol. 1999;100:156–168. doi: 10.1016/s0165-5728(99)00192-7. [DOI] [PubMed] [Google Scholar]
- 12.Heneka M, Klockgether T, Sharp A, Feinstein DL. The heat shock response inhibits NF-κB activation, nitric oxide synthase type 2 expression and macrophage/microglial activation in brain. J Cereb Blood Flow Metab. 2000;20:800–813. doi: 10.1097/00004647-200005000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Hu J, Van Eldik LJ. S100 beta induces apoptotic cell death in cultured astrocytes via a nitric oxide-dependent pathway. Biochim Biophys Acta. 1996;1313:239–245. doi: 10.1016/0167-4889(96)00095-x. [DOI] [PubMed] [Google Scholar]
- 14.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 15.Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:9409–9414. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitamura Y, Shimohama S, Koike H, Kakimura J, Matsuoka Y, Nomura Y, Gebicke HP, Taniguchi T. Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem Biophys Res Commun. 1999a;254:582–586. doi: 10.1006/bbrc.1998.9981. [DOI] [PubMed] [Google Scholar]
- 17.Kitamura Y, Kakimura J, Matsuoka Y, Nomura Y, Gebicke-Haerter PJ, Taniguchi T. Activators of peroxisome proliferator-activated receptor-gamma (PPARgamma) inhibit inducible nitric oxide synthase expression but increase heme oxygenase-1 expression in rat glial cells. Neurosci Lett. 1999b;262:129–132. doi: 10.1016/s0304-3940(99)00055-5. [DOI] [PubMed] [Google Scholar]
- 18.Klegeris A, Walker DG, McGeer PL. Toxicity of human THP-1 monocytic cells towards neuron-like cells is reduced by non-steroidal anti-inflammatory drugs (NSAIDs). Neuropharmacology. 1999;38:1017–1025. doi: 10.1016/s0028-3908(99)00014-3. [DOI] [PubMed] [Google Scholar]
- 19.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 20.Kuner P, Schubenel R, Hertel C. Beta-amyloid binds to p57NTR and activates NFkappaB in human neuroblastoma cells. J Neurosci Res. 1998;54:798–804. doi: 10.1002/(SICI)1097-4547(19981215)54:6<798::AID-JNR7>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 21.Lee SC, Zhao ML, Hirano A, Dickson DW. Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: association with Hirano bodies, neurofibrillary tangles, and senile plaques. J Neuropathol Exp Neurol. 1999;58:1163–1169. doi: 10.1097/00005072-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 23.Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996a;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 24.Lemberger T, Braissant O, Juge-Aubry C, Keller H, Saladin R, Staels B, Auwerx J, Burger AG, Meier CA, Wahli W. PPAR tissue distribution and interactions with other hormone-signaling pathways. Ann NY Acad Sci. 1996b;804:231–251. doi: 10.1111/j.1749-6632.1996.tb18619.x. [DOI] [PubMed] [Google Scholar]
- 25.Matsuoka Y, Kitamura Y, Takahashi H, Tooyama I, Kimura H, Gebicke-Haerter PJ, Nomura Y, Taniguchi T. Interferon-gamma plus lipopolysaccharide induction of delayed neuronal apoptosis in rat hippocampus. Neurochem Int. 1999;34:91–99. doi: 10.1016/s0197-0186(98)00053-9. [DOI] [PubMed] [Google Scholar]
- 26.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 27.Minc-Golomb D, Yadid G, Tsarfaty I, Resau JH, Schwartz JP. Expression of the inducible nitric oxide synthase by neurons following exposure to endotoxin and cytokine. J Neurochem. 1994;66:1504–1509. doi: 10.1046/j.1471-4159.1996.66041504.x. [DOI] [PubMed] [Google Scholar]
- 28.Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J Neurosci Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- 29.Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quan N, Mhlanga JD, Whiteside MB, McCoy AN, Kristensson K, Herkenham M. Chronic overexpression of proinflammatory cytokines and histopathology in the brains of rats infected with Trypanosoma brucei. J Comp Neurol. 1999;414:114–130. doi: 10.1002/(sici)1096-9861(19991108)414:1<114::aid-cne9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 31.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 32.Ricote M, Huang JT, Welch JS, Glass CK. The peroxisome proliferator-activated receptor(PPARgamma) as a regulator of monocyte/macrophage function. J Leukoc Biol. 1999;66:733–739. doi: 10.1002/jlb.66.5.733. [DOI] [PubMed] [Google Scholar]
- 33.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 34.Sarih M, Souvannavong V, Adam A. Nitric oxide synthase induces macrophage death by apoptosis. Biochem Biophys Res Commun. 1993;191:503–508. doi: 10.1006/bbrc.1993.1246. [DOI] [PubMed] [Google Scholar]
- 35.Sato I, Kim Y, Himi T, Murota S. Induction of calcium-independent nitric oxide synthase activity in cultured cerebellar granule neurons. Neurosci Lett. 1995;184:145–148. doi: 10.1016/0304-3940(94)11191-k. [DOI] [PubMed] [Google Scholar]
- 36.Skaper SD, Facci L, Leon A. Inflammatory mediator stimulation of astrocytes and meningeal fibroblasts induces neuronal degeneration via the nitridergic pathway. J Neurochem. 1995;64:266–276. doi: 10.1046/j.1471-4159.1995.64010266.x. [DOI] [PubMed] [Google Scholar]
- 37.Smith MA, Richey HP, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 39.Thieringer R, Fenyk-Melody JE, Le Grand CB, Shelton BA, Detmers PA, Somers EP, Carbin L, Moller DE, Wright SD, Berger J. Activation of peroxisome proliferator-activated receptor gamma does not inhibit IL-6 or TNF-alpha responses of macrophages to lipopolysaccharide in vitro or in vivo. J Immunol. 2000;164:1046–1054. doi: 10.4049/jimmunol.164.2.1046. [DOI] [PubMed] [Google Scholar]
- 40.Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999;354:141–148. doi: 10.1016/S0140-6736(98)10364-1. [DOI] [PubMed] [Google Scholar]
- 41.Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med. 1996;184:1425–1433. doi: 10.1084/jem.184.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPA Rs from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 43.Yang XY, Wang LH, Chen T, Hodge DR, Resau JH, DaSilva L, Farrar WL. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. Ppargamma Co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4544. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]