

Abstract
Matrix metalloproteinases (MMPs) belong to a class of extracellular proteinases responsible for maintaining and remodeling the extracellular matrix. In addition to multiple functions in normal physiology, abnormal MMP expression and activity may also participate in the pathophysiology of cerebral disease. Here, we show that MMP-9 (gelatinase B; EC.3.4.24.35) contributes to the pathophysiology of traumatic brain injury. After controlled cortical impact in mice, MMP-9 was increased in traumatized brain. Total MMP-9 levels at 24 hr were significantly increased as measured by a substrate cleavage assay. Zymograms showed that MMP-9 was elevated as early as 3 hr after traumatic brain injury, reaching a maximum at ∼24 hr. Increased MMP-9 levels persisted for up to 1 week. Western blot analysis indicated increased profiles of MMP-9 expression that corresponded with the zymographic data. Knock-out mice deficient in MMP-9 gene expression were compared with wild-type littermates in terms of morphological and motor outcomes after trauma. Motor outcomes were measured at 1, 2, and 7 d after traumatic brain injury by the use of a rotarod device. MMP-9 knock-out mice had less motor deficits than wild-type mice. At 7 d, traumatic brain lesion volumes on Nissl-stained histological sections were significantly smaller in MMP-9 knock-out mice. These data demonstrate that MMP-9 contributes to the pathophysiology of traumatic brain injury and suggest that interruption of the MMP proteolytic cascade may be a possible therapeutic approach for preventing the secondary progression of damage after brain trauma.
Keywords: brain trauma, controlled cortical impact, extracellular matrix, proteolysis, neurodegeneration, mouse
Matrix metalloproteinases (MMPs) form a family of zinc-dependent endopeptidases that are collectively able to degrade or modify essentially all components of the extracellular matrix (Nagase and Woessner, 1999). Target substrates include collagens, gelatin, fibronectin, laminin, elastin, and proteoglycans. Because MMPs can rapidly degrade critical protein components in the extracellular matrix, their biological activity is strictly regulated (Fini et al., 1998; Westermarck and Kahari, 1999). Primary modes of regulation occur via gene transcription, proenzyme activation, and dynamic inhibition by tissue inhibitors of metalloproteinases. MMPs participate in a wide array of important physiological processes including embryological remodeling, wound healing, angiogenesis, bone remodeling, ovulation, and implantation. Considering their central involvement in normal biology, it is not surprising that overactivity of MMPs is also involved in diverse disease processes (Lukashev and Werb, 1998). In the CNS, MMPs have been implicated in degenerative disorders such as multiple sclerosis and Alzheimer's disease (Yong et al., 1998). More recently, evidence is accumulating to show that MMPs can be involved in acute brain injury. MMP activity is upregulated after cerebral ischemia (Rosenberg et al., 1996; Romanic et al., 1998; Gasche et al., 1999; Heo et al., 1999) and edema (Morita-Fijimura et al., 1999).
In this study, we investigated the hypothesis that MMP-9 (gelatinase B; EC.3.4.24.35) may be involved in the pathophysiological cascade of neuronal damage after traumatic brain injury. This hypothesis was tested by performing experiments using a standard controlled cortical impact model of traumatic brain injury in mice. First, we documented via enzyme activity assays, zymography, and Western blots that MMP-9 was upregulated after trauma. Then, we demonstrated that behavioral motor deficits and cerebral lesion volumes were significantly reduced in mutant mice with a targeted knock-out of the MMP-9 gene.
MATERIALS AND METHODS
Controlled cortical impact model of traumatic brain injury. All experiments were performed following an institutionally approved protocol in accordance with the NIH Guide for the Care and Use of Laboratory Animals. For initial MMP activity measurements, normal C57/B6 and CD1 mice were used. For all other experiments, the MMP-9 knock-out strain and their corresponding wild-type littermates (Vu et al., 1998) were used. The standard controlled cortical impact model of traumatic brain injury (Dixon et al., 1991; Smith et al., 1995; Whalen et al., 1999) was performed as follows. Briefly, mice were anesthetized with 1–1.5% halothane in an air/oxygen mixture via a face mask. An electronic thermostat-controlled warming blanket was used to maintain the core temperature at 37.5°C. Mice were placed into the head holder within the device, and a 5 mm craniotomy was performed over the right cerebral hemisphere between the bregma and lambda by the use of a trephine drill, taking care to keep the dura intact. A 3 mm flat-tipped impactor was placed on the dural surface at a 20° angle, and injury was induced by the use of 4 m/sec impact velocity, 1 mm impact depth, and 150 msec impact dwell time. After traumatic brain injury, the bone flap was replaced over the craniotomy, and mice were allowed to recover back in their cages. Because the duration of the entire cortical impact procedure is ∼10 min, it was believed that it was unnecessary to prolong anesthesia/surgical time to insert catheters for measuring blood pressure, pH, and gases. However, measurements from previous series demonstrated that all physiological parameters remained within the normal range for this model in our laboratory.
MMP activity assay. At 24 hr after traumatic brain injury, mice were killed with a lethal overdose of sodium pentobarbital intraperitoneally and transcardially perfused with cold saline (4°C). MMP-9 activity present in supernatants from brain homogenates was measured by the use of a colorimetric antibody-specific substrate cleavage method (Amersham Pharmacia Biotech, Piscataway, NJ). Briefly, MMP-9 was captured in microtiter wells coated with anti-MMP-9 antibody. Total MMP-9 activity is measured by converting MMP-9 proforms into active forms by the use ofp-aminophenylmercuric acetate. Note that this method does not distinguish between latent zymogen versus cleaved active forms of the enzyme; therefore this assay measures total MMP-9 that can be activated. MMP-9 enzyme activity was then assessed by cleavage of a chromogenic peptide substrate (S-2444) and quantified at 405 nm with a spectrophotometric plate reader (Bio-Tek Instruments). Traumatized brains (n = 5) were compared with sham-operated controls (n = 3).
MMP zymography. At 1 hr, 3 hr, 6 hr, 1 d, 3 d, 5 d, and 7 d after trauma, mice were killed with an overdose of sodium pentobarbital (100 mg/kg, i.p.) and transcardially perfused with chilled (4°C) PBS, pH 7.4. The brains were rapidly removed, and damaged brain tissue within the traumatized hemisphere (∼100–150 mg) was homogenized in lysis buffer (50 mmTris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, and 0.1% deoxycholic acid) including protease inhibitors (2 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 mm PMSF) for 30 min on ice. The homogenate was then centrifuged at 14,000 × g for 10 min at 4°C. Protein concentrations in the supernatants were measured with the Bradford assay (Bio-Rad, Hercules, CA). Three brains were used per lane. Approximately 10 μg of each sample was loaded per lane into the wells of precast gels (10% polyacrylamide minigels containing 0.1% gelatin; Novex) with SDS sample buffer (2×; Novex). Electrophoresis was performed with a Tris-glycine running buffer at 125 V constant voltage for 1.5–2 hr. The gel was removed and incubated for 1 hr at room temperature in 100 ml of 2.7% Triton X-100 on a rotary shaker. Each gel was incubated with 100 ml of development buffer (50 mm Tris base, 40 mm HCl, 200 mmNaCl, 5 mm CaCl2, and 0.2% Brij 35; Novex) at 37°C for 14–18 hr on a rotary shaker. Staining was performed with 100 ml of 0.5% Coomassie blue G-250 in 30% methanol and 10% acetic acid for 1 hr, and gels were then destained with three changes of solutions. Gelatinolytic activity was demonstrated as clear zones or bands at the appropriate molecular weights. Mouse MMP-9 and human MMP-2 (from Chemicon, Temecula, CA) were used as standards.
Western blots. At 1 hr, 3 hr, 6 hr, 1 d, 3 d, 5 d, and 7 d after trauma, mice were killed with an overdose of sodium pentobarbital (100 mg/kg, i.p.) and transcardially perfused with chilled (4°C) PBS, pH 7.4. The brains were rapidly removed, and supernatants from homogenates were prepared as described above for Western blot analysis using a monoclonal antibody targeted toward the catalytic subunit of mouse MMP-9 (donated by Robert Senior, Washington University, St. Louis, MO). Three brains were used per lane. Approximately 10 μg of each sample per lane was loaded onto 4–20% Tris-glycine gels with equal volumes of SDS sample buffer (Novex) and then transferred to polyvinylidene difluoride membranes (Novex). Membranes were blocked for 1 hr at room temperature in PBS and 0.1% Tween 20 with 10% low-fat dried milk and then incubated with the primary anti-MMP-9 antibody for 1 hr at room temperature. After the primary incubation, the membranes were washed and then incubated again with horseradish peroxidase-linked anti-rabbit IgG and developed by the use of enhanced chemiluminescence (ECL; Amersham). Mouse MMP-9 (from Chemicon) was used as a standard.
Rotarod motor performance tests. To assess behavioral deficits after traumatic brain injury, the standard rotarod test (Hamm et al., 1994) was performed at 1, 2, and 7 d after controlled cortical impact. All mice were initially trained on the rotarod for 2 d before the day of trauma. One hour before traumatic brain injury, mice were assessed on the rotarod to obtain preinjury baselines. Thereafter, all rotarod scores were expressed as a percentage of preinjury baselines to reduce interanimal variability. Scores were measured as the latency or time successfully spent running on the rotating rod (35 rpm); 10–13 animals were measured for each time point. A repeated measures ANOVA was used for multiple comparisons over time between wild-type littermates and MMP-9 knock-out mice. Comparisons at each time point was performed by the use of Fisher's PLSD test.
Traumatic lesion volume quantitation. After traumatic brain injury, mice were allowed to survive for 7 d before being killed with a lethal overdose of sodium pentobarbital intraperitoneally. All mice were transcardially perfused with chilled (4°C) PBS, pH 7.4, followed by 4% paraformaldehyde in 0.1 m PBS. The brains were rapidly removed and fixed in fresh paraformaldehyde solution overnight at 4°C and then stored in 30% sucrose and 0.1m PBS. Brains were sectioned by the use of a freezing microtome into 25-μm-thick coronal slices. Every 20th section was mounted onto a glass slide and stained with 0.1% cresyl violet. Histological lesion areas were quantified with a standard computer-assisted image analysis program, and lesion areas were then integrated to obtain total lesion volumes in cubic millimeters. Differences between wild-type littermates (n = 11) and MMP-9 knock-out mice (n = 9) were assessed with two-tailed t tests.
RESULTS
MMP-9 is upregulated after traumatic brain injury
Traumatic brain injury induced via controlled cortical impact resulted in significant elevations of MMP-9 protein levels. At 24 hr after trauma, total enzyme measured by colorimetric detection of substrate cleavage showed that MMP-9 levels were increased by almost threefold compared with that in the contralateral cortex (Fig.1). Interestingly, MMP-9 was slightly elevated in contralateral brain as well, suggesting that after trauma, perturbations in cerebral status was not restricted to the impacted hemisphere alone and subtle mechanical- and biochemical-induced alterations may propagate across to the opposite hemisphere (Fig. 1). In sham-operated brains, the craniotomy procedure alone (without impact trauma) also appeared to increase MMP-9 protein levels slightly, although this did not reach statistical significance (Fig. 1).
Fig. 1.

MMP-9 enzyme activity (mean ± SEM) in C57/B6 mouse brain at 24 hr after trauma or sham operation. In sham-operated controls (n = 3), no constitutive MMP-9 activity was detected in the contralateral hemisphere. In the ipsilateral side, the craniotomy procedure alone appeared to induce a slight elevation in enzyme activity. However, this increase was not statistically significant (p = 0.16). In mice subjected to brain trauma (n = 5), MMP-9 activity was significantly higher in the traumatized hemisphere (*p < 0.05). Interestingly, however, MMP-9 enzyme activity was also significantly elevated in the contralateral side compared with that in the shams (p < 0.05).CCI, Controlled cortical impact.
Alterations in MMP-9 was assessed over the course of 1 week after trauma by the use of zymography. MMP-9 levels in traumatized brain appeared to be elevated beginning as early as 3 hr after trauma and remained elevated for the entire 1 week period of analysis (Fig.2A). MMP-9 was primarily expressed as the higher molecular weight zymogen (105 kDa). No clear evidence of the lower molecular weight cleaved (activated) form of MMP-9 was observed. MMP-9 was not detected in normal brains from unoperated mice, but sham-operated control brains subjected to craniotomy alone appeared to show slight elevations in MMP-9 levels (Fig. 2A). Faint bands of zymographic digestion were also observed at a level close to that of the human MMP-2 standard (72 kDa), suggesting a possible presence of MMP-2 (gelatinase A) activity.
Fig. 2.

A, Representative zymograms of brain homogenates from wild-type CD1 mice. No constitutive MMP-9 activity was detected in normal brains from unoperated mice. Slight induction of MMP-9 was detected in sham-operated controls at 24 hr after craniotomy alone without brain trauma. In traumatized brain, increased activity was present by 3 hr after cortical impact. Elevated MMP-9 levels persisted until the last time point measured at 7 d after trauma. Primarily MMP-9 zymogen (105 kDa) was detected, and only faint bands were present for the cleaved active forms (97 kDa). Controls included normal brains from unoperated mice, brains from sham-operated mice subjected to craniotomy without cortical impact, the mouse MMP-9 standard (Chemicon), and the human MMP-2 standard (Chemicon). Samples derived from three brains were used perlane. Slight induction of gelatinolytic activity was also observed close to the human MMP-2 standard, suggesting a possible induction of MMP-2 after trauma. Note that (1) mouse MMP-9 has a higher molecular weight (105 kDa) than human MMP-9 (92 kDa) and (2) human and mouse MMP-2 has a molecular weight of 72 kDa but is typically detected at 65 kDa when run with a nonreducing buffer.B, Western blots showing that protein expression of MMP-9 is increased after brain trauma in CD1 mice. MMP-9 was not detectable in either normal brains from unoperated mice or sham-operated controls. In all cases, only MMP-9 zymogen was detected. Samples derived from three brains were used for eachlane.
Western blot analysis showed that MMP-9 protein in the brain was increased beginning as early as 3 hr after trauma, with a maximum occurring at ∼24 hr. However, MMP-9 levels remained clearly elevated for the entire 1 week experimental period (Fig. 2B). Once again, only the 105 kDa zymogen was seen, although the antibody recognizes both the zymogen and the cleaved forms of MMP-9 (R. Senior, personal communication).
MMP-9-deficient knock-out mice are resistant to traumatic brain injury
The motor response to traumatic brain injury in MMP-9 knock-out mice was compared with that in wild-type littermates. Initial pretrauma performance on the rotarod device seemed to suggest that the MMP-9 knock-outs had somewhat lower motor endurance compared with that of the wild-type littermates, although the difference did not reach statistical significance (latencies were 63 ± 12 sec for knock-outs and 87 ± 14 sec for wildtypes; p = 0.21; Fig. 3A). However, to normalize for this potential difference in pretrauma performance, all post-trauma latencies were also calculated as a percentage of preinjury baselines. After traumatic brain injury induced by controlled cortical impact, rotarod performance was significantly reduced in all mice. However, over 1, 2, and 7 d after injury, a repeated measures ANOVA demonstrated that knock-out mice had significantly fewer deficits compared with that of wild-type mice (p < 0.05; Fig. 3).
Fig. 3.
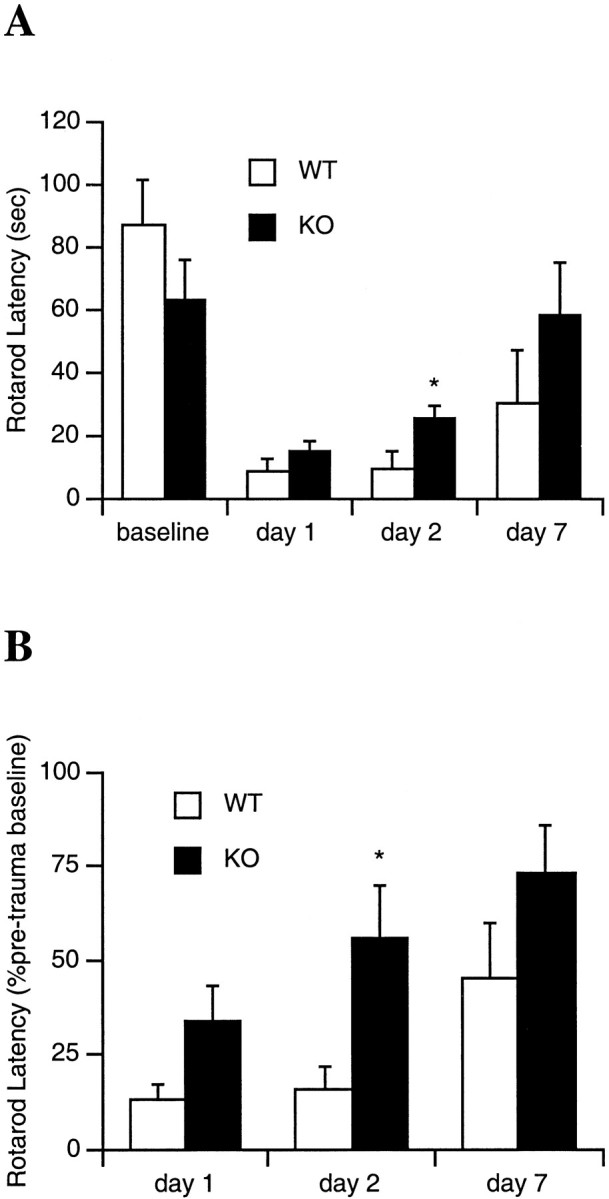
Motor performance (mean ± SEM) on a rotarod device after brain trauma expressed either as the absolute time in seconds (A) or a percent of pretrauma latency (B) on the rotating rod. After brain injury, latencies were significantly reduced in all mice. A repeated measures ANOVA showed that overall rotarod performance was significantly improved in knock-outs (KO) versus wild-type littermates (WT). However, single time point comparisons showed significantly improved performance in the MMP-9 knock-out mice versus wild-type littermates only at 2 d after trauma (*p < 0.05). Although the average rotarod latency was also improved in knock-outs at 1 and 7 d, the difference did not reach statistical significance (p = 0.09 at day 1, p = 0.18 at day 7 for percentage comparisons). This may be caused by the somewhat variable rotarod response after trauma and the need for larger numbers of mice to increase the statistical power (n = 10–13 per time point).
At 7 d after injury, traumatized brains showed well demarcated lesions with cavitations comprising the cortex as well as underlying hippocampal structures (Fig.4A). Parenchyma immediately surrounding the necrotic cavitation also showed decreased cresyl violet staining. Although hippocampal structures were disrupted, neuronal loss could also be discerned in CA1 and CA3 regions that remained. Overall traumatic lesion volumes were significantly smaller in the MMP-9 knock-outs compared with wild-type littermates (7.44 ± 0.5 mm3 in wildtypes vs 5.67 ± 0.6 mm3 in knock-outs; p< 0.05; Fig. 4B).
Fig. 4.

A, Representative Nissl-stained brain sections showing that MMP-9 knock-outs (KO) suffer less brain injury compared with that in wild-type littermates (WT) at 7 d after trauma. B,Lesion volumes at 7 d after traumatic brain injury (in cubic millimeters; mean ± SEM). MMP-9 KO(n = 9) had significantly smaller lesion volumes compared with that of WT (n = 11; *p < 0.05).
DISCUSSION
In addition to primary damage induced by the initial mechanical impact, secondary mechanisms of cell death can significantly contribute to traumatic brain injury (McIntosh et al., 1998a,b). The progression of nervous tissue damage after trauma involves a multifactorial cascade of secondary pathophysiology including excitotoxicity (Hayes et al., 1988; Faden et al., 1989; Palmer et al., 1993), oxidative stress (Kontos and Povlishock, 1986), inflammation (Arvin et al., 1996), and abnormal apoptosis (Rink et al., 1995; Colicos and Dash, 1996;Pravdenkova et al., 1996; Clark et al., 1997; Crowe et al., 1997; Liu et al., 1997; Yakovlev et al., 1997; Conti et al., 1998; Fox et al., 1998; Kaya et al., 1999; Springer et al., 1999). However, in this report, we demonstrate that upregulation of MMP-9 may participate in the pathophysiology of traumatic brain injury. In normal mouse brain, cortical impact injury induced a significant upregulation of MMP-9 levels as measured by a variety of methods including colorimetric substrate cleavage assay, zymography, and Western blot analysis. The role for MMP-9 was further explored by the use of knock-out mice deficient in MMP-9 gene expression. These studies suggested that MMP-9 may play a deleterious role in traumatic brain injury because knock-out mice had significantly smaller lesions compared with that in wild-type littermates. Importantly, motor deficits measured by the use of a rotarod device also showed that MMP-9 knock-out mice had less behavioral injury compared with that in the wild-type mice.
As expected, no significant MMP-9 was detectable in normal brain, consistent with the understanding that MMP-9 is not constitutively expressed in mammalian brain (Rosenberg et al., 1996; Gasche et al., 1999; Heo et al., 1999). However, after traumatic brain injury, MMP-9 levels were observed to increase as early as 3 hr after injury, reaching an apparent maximum at ∼24 hr and remaining elevated for up to 1 week. It will be important to document carefully the temporal profile of MMP-9 levels over longer periods of time in future studies to understand fully its contribution to long-term pathophysiology. Long-term changes have been described after traumatic brain injury (Dixon et al., 1999) so chronic MMP activity may be involved. Although the results of our study make a mechanistic connection between increased levels of MMP-9 protein and brain tissue damage, we have not directly measured proteolytic activity. Therefore, we cannot say whether tissue damage is caused by proteolysis by MMP-9 or whether some undocumented activity of MMP-9 is involved. The MMP-9 protein detected by the various methods used in this study was the 105 kDa proenzyme form; none of the truncated forms of the enzyme associated with proteolytic activity in vitro was detectable. This in vivo finding is often reported (Matsubara et al., 1991; Fini et al., 1996; Mohan et al., 2000) and is not inconsistent with functionality of MMP-9 as a protease. A number of studies suggest that enzymatic activation of MMP-9 proenzyme occurs by binding at the cell surface and that activated enzyme is then rapidly degraded (Yu and Stamenkovic, 1999). Therefore, little active MMP-9 may be present in tissue at any given time.
Our zymographic data showed that after traumatic brain injury, the primary gelatinase elevated was MMP-9. Faint zones of activity were observed at a level close to that of the human MMP-2 standard (72 kDa), suggesting that small elevations in MMP-2 may also occur in our model. This is in contrast to studies of cerebral ischemia in which large elevations in MMP-2 activity were observed (Rosenberg et al., 1996;Romanic et al., 1998; Gasche et al., 1999; Heo et al., 1999). Although traumatic brain injury shares many common pathways with cerebral ischemia, there are critical differences in pathophysiology (McIntosh et al., 1998a,b), and the present data are consistent with this idea. On the other hand, focal regions of tissue ischemia can accompany traumatic brain injury (Bryan et al., 1995; Dietrich et al., 1998) so common upstream triggers of MMP upregulation may also be involved.
Although the present data suggest that MMP-9 contributes to brain damage after trauma, the precise mechanisms involved remain to be defined. In cerebral ischemia, an emphasis has been placed on the fact that MMP-9 can digest matrix proteins present in the vascular basal lamina including collagen, fibronectin, and laminin. Damage to vascular integrity would then lead to disrupted blood–brain barrier function and increased vasogenic edema (Rosenberg et al., 1996; Gasche et al., 1999; Heo et al., 1999). Because edema can play a critical role in traumatic brain injury, this specific pathway may be involved here as well. However, it is important to recognize that matrix proteins such as laminin are also widely disseminated throughout the brain parenchyma, and loss of parenchymal laminin may affect cell–matrix interactions and cell survival (Hagg et al., 1989; Murtomaki et al., 1995; Chen and Strickland, 1997; Tsirka et al., 1997). Therefore, it remains conceivable that MMP-mediated matrix protein degradation may disrupt cell–matrix interactions beyond the vascular compartment as well and further contribute to neuronal cell death. It must be acknowledged, however, that laminin can be degraded by other enzymes [e.g., plasmin (Tsirka et al., 1997)]. Cascades of extracellular proteolysis after traumatic brain injury may involve other proteases besides MMPs.
There are a few caveats associated with the present study. First, we measured traumatic lesion volumes at a relatively early time point (1 week) after cortical impact. We cannot exclude the possibility that long-term effects may occur that overwhelm the acute protective effects of MMP-9 gene knock-out. It has been shown that degenerative and/or regenerative processes may extend for periods up to a year after traumatic brain injury (Dixon et al., 1999). During these prolonged periods, MMPs may play critical roles in cerebral reorganization. The possibility of biphasic responses to traumatic brain injury has been demonstrated recently in the tumor necrosis factor- (TNF-) and TNF receptor-deficient mutant mice, in which absence of the gene appeared to protect the brain during acute time frames but retard functional recovery during prolonged periods after injury (Scherbel et al., 1999; Sullivan et al., 1999). Second, we only used a primarily motor behavioral task (i.e., the rotarod assay). It is known that cognitive and/or memory deficits are an important part of the controlled cortical impact model of traumatic brain injury (Dixon et al., 1999). Indeed, we find that lesion volumes tend to include extensive destruction of hippocampal structures, especially in the wild-type mice. It will be important to determine whether cognitive deficits are also improved by MMP-9 gene deletion; these studies are being pursued in our laboratory presently. Third, we do not have data on the cell types that serve as sources for MMP-9. It is known that macrophages and neutrophils can be an important source for MMPs and other deleterious proteases (Pagenstecher et al., 1998), and inflammatory macrophage and neutrophil infiltration is clearly an important event after trauma (Carlos et al., 1997; Bethea et al., 1998;Chatzipantelli et al., 2000). However, it has been shown that resident brain cells (neurons, astrocytes, and oligodendrocytes) can also produce MMPs (Gottschall and Deb, 1996; Zuo et al., 1998; Oh et al., 1999). It will be important to define carefully the cellular sources of deleterious MMP activity after traumatic brain injury. Finally, the MMP family comprises many other members besides MMP-9. MMPs (along with plasminogen activators) form a protease cascade that bears some similarity to the caspase cascade in terms of amplifying cell death (Cuzner and Opdenakker, 1999). Much like the caspase cascade in which caspase-3 may be a possible final executioner, it has also been proposed that MMP-9 may be a terminal member of the MMP cascade (Cuzner and Opdenakker, 1999). Nevertheless, it will be critical to assess the roles for more upstream MMPs by the use of other specific antibodies and knock-out mouse models.
In conclusion, the present study demonstrated that MMP-9 activity was increased in a mouse model of traumatic brain injury, and knock-out mice deficient in MMP-9 gene expression showed reduced morphological damage and motor deficits compared with that in wild-type mice. These data demonstrate that MMP-9 contributes to the pathophysiology of traumatic brain injury. Interrupting the MMP cascade may be a relevant therapeutic approach for preventing the devastating progression of secondary injury.
Footnotes
This work was funded in part by National Institutes of Health Grants R01-NS37074, R01-NS38731, R01-NS40529, and R01-EY12651. M.E.F. is a Stein Research to Prevent Blindness Professor. We thank Drs. Robert Senior and J. Michael Shipley for kindly donating the anti-mouse MMP-9 antibody and for permission to use the MMP-9 knock-out mouse in these studies. Drs. Michael Schwartzchild and Jiang-Fan Chen provided critical advice on the rotarod motor test, and we are grateful to Dr. Michael Whalen for critical discussions on the pathophysiology of traumatic brain injury.
Correspondence should be addressed to Dr. Eng H. Lo, Neuroprotection Research Laboratory, Departments of Neurology and Radiology, Harvard Medical School, Massachusetts General Hospital East 149-2322, Charlestown, MA 02129. E-mail: Lo@helix.mgh.harvard.edu.
REFERENCES
- 1.Arvin B, Neville LF, Barone FC, Feuerstein GZ. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996;20:445–452. doi: 10.1016/0149-7634(95)00026-7. [DOI] [PubMed] [Google Scholar]
- 2.Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor kappa B activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryan RM, Cherian L, Robertson C. Regional cerebral blood flow after controlled cortical impact. Anesth Analg. 1995;80:687–695. doi: 10.1097/00000539-199504000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Carlos TM, Clark RSB, Higgins DF, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61:279–285. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- 5.Chatzipantelli K, Alonso OF, Kraydieh S, Dietrich WD. Importance of posttraumatic hypothermia and hyperthermia on the inflammatory response after fluid percussion brain injury: biochemical and immunocytochemical studies. J Cereb Blood Flow Metab. 2000;20:531–542. doi: 10.1097/00004647-200003000-00012. [DOI] [PubMed] [Google Scholar]
- 6.Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 7.Clark RSB, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci. 1997;17:9172–9182. doi: 10.1523/JNEUROSCI.17-23-09172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colicos MA, Dash PK. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 1996;739:120–131. doi: 10.1016/s0006-8993(96)00824-4. [DOI] [PubMed] [Google Scholar]
- 9.Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 11.Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteinases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999;94:1–14. doi: 10.1016/s0165-5728(98)00241-0. [DOI] [PubMed] [Google Scholar]
- 12.Dietrich WD, Alonso O, Busto R, Prado R, Zhao W, Dewanjee MK, Ginsberg MD. Posttraumatic cerebral ischemia after fluid percussion brain injury: an autoradiographic and histopathological study in rats. Neurosurgery. 1998;43:585–593. doi: 10.1097/00006123-199809000-00105. [DOI] [PubMed] [Google Scholar]
- 13.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 14.Dixon CE, Kochanek PM, Yan HQ, Schiding JK, Griffith RG, Baum E, Marion DW, DeKosky ST. One year study of spatial memory performance, brain morphology, and cholinergic markers after controlled cortical impact injury. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- 15.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 16.Fini ME, Parks WC, Rinehart WB, Girard MT, Matsubara M, Cook JR, West-Mays JA, Sadow PM, Burgeson RE, Jeffrey JJ, Raizman MB, Krueger RR, Zieske JD. Role of matrix metalloproteinases in failure to re-epithelialize after corneal injury. Am J Pathol. 1996;149:1287–1302. [PMC free article] [PubMed] [Google Scholar]
- 17.Fini ME, Cook JR, Mohan R, Brinckerhoff CE. Regulation of matrix metalloproteinase gene expression. In: Parks WC, Mecham RP, editors. Matrix metalloproteinases. Academic; New York: 1998. pp. 299–356. [Google Scholar]
- 18.Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- 19.Gasche Y, Fujimura Y, Copin J, Kawase M, Masengale J, Chan PH. Early appearance of activated MMP-9 after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Gottschall PE, Deb S. Regulation of matrix metalloproteinase expression in astrocytes, microglia and neurons. Neuroimmunomodulation. 1996;3:69–75. doi: 10.1159/000097229. [DOI] [PubMed] [Google Scholar]
- 21.Hagg T, Muir D, Engvall E, Varon S, Manthorpe M. Laminin antigens in rat CNS: distribution and changes upon brain injury. Neuron. 1989;3:721–732. doi: 10.1016/0896-6273(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 22.Hamm RJ, Pike BR, O'Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits after traumatic brain injury. J Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- 23.Hayes RL, Jenkins LW, Lyeth BG, Balster RL, Robinson SE, Clifton GL, Stubbins JF, Young HF. Pretreatment with phencyclidine, an NMDA antagonist attenuates long term behavioral deficits in the rat produced by traumatic brain injury. J Neurotrauma. 1988;5:259–274. doi: 10.1089/neu.1988.5.259. [DOI] [PubMed] [Google Scholar]
- 24.Heo JH, Lucero J, Abumiya T, Koizol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Kaya SS, Mahmood A, Li Y, Yavuz E, Goskel M, Chopp M. Apoptosis and expression of p53 proteins and cyclin D1 after cortical impact in rat brain. Brain Res. 1999;818:23–33. doi: 10.1016/s0006-8993(98)01204-9. [DOI] [PubMed] [Google Scholar]
- 26.Kontos HA, Povlishock JT. Oxygen radicals in brain injury. Cent Nerv Syst Trauma. 1986;3:257–263. doi: 10.1089/cns.1986.3.257. [DOI] [PubMed] [Google Scholar]
- 27.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukashev ME, Werb Z. ECM signalling: orchestrating cell behavior and misbehavior. Trends Cell Biol. 1998;8:437–441. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- 29.Matsubara M, Girard MT, Kublin CL, Cintron C, Fini ME. Differential roles for two gelatinolytic enzymes of the matrix metalloproteinase family in the remodelling cornea. Dev Biol. 1991;147:425–432. doi: 10.1016/0012-1606(91)90300-r. [DOI] [PubMed] [Google Scholar]
- 30.McIntosh TK, Juhler M, Wieloch T. Novel pharmacologic strategies in the treatment of experimental traumatic brain injury. J Neurotrauma. 1998a;15:731–769. doi: 10.1089/neu.1998.15.731. [DOI] [PubMed] [Google Scholar]
- 31.McIntosh TK, Saatman RE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. Molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol. 1998b;24:251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- 32.Mohan R, Sivak J, Ashton P, Russo LA, Pham BT, Raizman MB, Fini ME. Curcuminoids inhibit the angiogenic response stimulated by FGF-2 including expression of matrix metalloproteinase gelatinase B. J Biol Chem. 2000;275:10405–10412. doi: 10.1074/jbc.275.14.10405. [DOI] [PubMed] [Google Scholar]
- 33.Morita-Fijimura Y, Fujimura M, Gasche Y, Copin J, Chan PH. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury induced brain trauma. J Cereb Blood Flow Metab. 1999;20:130–138. doi: 10.1097/00004647-200001000-00017. [DOI] [PubMed] [Google Scholar]
- 34.Murtomaki S, Trekner E, Wright J, Saksela O, Liesi P. Increased proteolytic activity of the granule neurons may contribute to neuronal death in the weaver mouse cerebellum. Dev Biol. 1995;168:635–648. doi: 10.1006/dbio.1995.1108. [DOI] [PubMed] [Google Scholar]
- 35.Nagase H, Woessner JF. Matrix metalloproteinases: a minireview. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 36.Oh LYS, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pagenstecher A, Stalder AK, Kincaid CL, Shapiro SD, Campbell IL. Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in mouse CNS in normal and inflammatory states. Am J Pathol. 1998;152:729–741. [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, DeKosky ST. Traumatic brain injury induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- 39.Pravdenkova SV, Basnakian AG, James JS, Andersen BJ. DNA fragmentation and nuclear endonuclease activity in rat brain after severe closed head injury. Brain Res. 1996;729:151–155. [PubMed] [Google Scholar]
- 40.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 41.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 42.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 43.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor deficient mice to experimental brain injury. Proc Natl Acad Sci USA. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith DH, Soares HD, Pierce JS, Perlman K, Saatman K, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histological effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- 45.Springer JE, Azhill RD, Knapp PE. Activation of caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, St Clair DK, Mattson MP, Scheff SW. Execerbation of damage and altered NFkB activation in mice lacking TNF receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vu TH, Shipley JM, Bergers G, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9 is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westermarck J, Kahari W. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 50.Whalen MJ, Clarck RSB, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham SH, Virag L, Hasko G, Stachlewitz R, Szabo C, Kochanek PM. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly-ADP-ribose polymerase. J Cereb Blood Flow Metab. 1999;19:835–842. doi: 10.1097/00004647-199908000-00002. [DOI] [PubMed] [Google Scholar]
- 51.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and disease of the CNS. Trends Neurosci. 1998;21:75–80. doi: 10.1016/s0166-2236(97)01169-7. [DOI] [PubMed] [Google Scholar]
- 53.Yu Q, Stamenkovic I. Localization of metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuo J, Ferguson TA, Hernandez YJ, Stetler-Stevenson WG, Muir D. Neuronal matrix metalloproteinase 2 degrades and inactivates a neurite-inhibiting chondroitin sulfate proteoglycan. J Neurosci. 1998;18:5203–5211. doi: 10.1523/JNEUROSCI.18-14-05203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]