

Abstract
Trains of action potentials cause Ca2+-dependent facilitation and inactivation of presynaptic P/Q-type Ca2+ channels that can alter synaptic efficacy. A potential mechanism for these effects involves calmodulin, which associates in a Ca2+-dependent manner with the pore-forming α1A subunit. Here, we report that Ca2+ and calmodulin dramatically enhance inactivation and facilitation of P/Q-type Ca2+channels containing the auxiliary β2a subunit compared with their relatively small effects on channels with β1b. Tetanic stimulation causes an initial enhancement followed by a gradual decline in P/Q-type Ca2+ currents over time. Recovery of Ca2+ currents from facilitation and inactivation is relatively slow (30 sec to 1 min). These effects are strongly inhibited by high intracellular BAPTA, replacement of extracellular Ca2+ with Ba2+, and a calmodulin inhibitor peptide. The Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels observed here are consistent with the behavior of presynaptic Ca2+ channels in neurons, revealing how dual feedback regulation of P/Q-type channels by Ca2+ and calmodulin could contribute to activity-dependent synaptic plasticity.
Keywords: calcium channel, calmodulin, synaptic plasticity, inactivation, facilitation, action potential
Ca2+entry through presynaptic voltage-gated Ca2+ channels links membrane depolarization and exocytosis of synaptic vesicles in the nerve terminal. The amount of neurotransmitter released is steeply dependent on presynaptic Ca2+ concentrations (Dodge and Rahamimoff, 1967; Mintz et al., 1995) such that increases or decreases in Ca2+ influx can powerfully alter neurotransmission. At many central and peripheral synapses, transmission is mediated by N- and P/Q-type Ca2+ channels (Dunlap et al., 1995). These channels are inhibited by G-protein βγ subunits (Herlitze et al., 1996; Ikeda, 1996), and relief of this inhibition can produce short-term synaptic facilitation (Brody and Yue, 2000). In addition, P/Q-type channels are subject to feedback regulation by Ca2+. We have shown that Ca2+ influx through P/Q-type channels enhances inactivation, increases recovery from inactivation, and causes a long-lasting facilitation of the Ca2+current, effects that require direct association of Ca2+/calmodulin with the pore-forming α1A subunit (Lee et al., 1999).
High-frequency activation of presynaptic axons at a brainstem auditory synapse accelerates inactivation of presynaptic P/Q-type Ca2+ channels leading to post-tetanic depression of EPSPs, an effect that is enhanced by extracellular and intracellular Ca2+ (Forsythe et al., 1998). Both tetanic and paired-pulse stimulation also cause a transient facilitation of P/Q-type Ca2+ currents that depends on incoming Ca2+ and is reduced by intracellular dialysis with BAPTA (Borst and Sakmann, 1998;Cuttle et al., 1998). The modulation of recombinant P/Q-type channels by Ca2+/calmodulin (Lee et al., 1999) is consistent with, but smaller than, these effects of Ca2+ on inactivation and facilitation of native presynaptic Ca2+ channels.
Brain presynaptic Ca2+ channels formed from α1A subunits are often characterized by little voltage-dependent inactivation (Mintz et al., 1992; Usowicz et al., 1992). Slowly inactivating P/Q-type channels can be reproduced in heterologous systems by expression of a recently recognized splice variant of α1A, α1A-b(Bourinet et al., 1999), or by coexpression of α1A with the auxiliary Ca2+ channel β subunit β2a (Stea et al., 1994; De Waard and Campbell, 1995). Channels comprised of α1A, β1b, and α2δ subunits exhibit strong voltage-dependent inactivation that might occlude Ca2+-dependent modulation (Lee et al., 1999). Therefore, Ca2+-dependent inactivation and facilitation may be more prominent in P/Q-type Ca2+ channels containing β2a subunits, which are likely physiological partners of α1A in many regions of the brain (Stea et al., 1994; Tanaka et al., 1995). Here we demonstrate that substitution of β1b with β2a unmasks a surprisingly large Ca2+-dependent facilitation and inactivation of P/Q-type Ca2+ channels in response to step depolarizations and high-frequency activation with characteristics similar to those observed for presynaptic Ca2+ channels in the brain. Both effects are mediated in part by Ca2+ and calmodulin but differ in their kinetics and sensitivity to Ca2+. These results reveal a complex feedback regulation of P/Q-type channels by Ca2+ that may contribute to both the enhancement and depression of synaptic transmission.
MATERIALS AND METHODS
cDNA expression constructs. Mammalian expression constructs of rat Ca2+ channel subunits were α1A (rbA), β1b, and β2a that were subcloned in pMT2XS and α2δ that was subcloned in pZEM228 (Stea et al., 1994). Deletion of the α1Acalmodulin-binding domain (CBD; amino acids 1960–2000) was accomplished by amplifying by PCR an EcoRV/PmlI fragment that incorporated the deletion and subcloning into the corresponding sites of rbA in a pBluescript SK+ shuttle vector. From this construct, aSgraI/MluI fragment containing the deletion was subcloned into rba/pMT2XS. The adenylate cyclase I (ACI) expression construct was generated by amplifying amino acids 481–575 of adenylyl cyclase type I by PCR and subcloning into pCEP4 (Invitrogen, San Diego, CA). The same strategy was used for the ACI(F-R) construct used in control experiments except that the PCR template was adenylyl cyclase type I containing a single phenylalanine to arginine mutation that disrupts calmodulin binding (Wu et al., 1993). Both mutant and wild-type ACI cDNAs were provided by Dr. Daniel Storm (University of Washington, Seattle, WA).
Cell culture and transfection. tsA-201 cells were maintained in DMEM/Ham's F12 (1:1) supplemented with 10% fetal bovine serum (Life Technologies, Rockville, MD) at 37°C under 10% CO2. Cells plated in 35 mm tissue culture dishes were grown to ∼70% confluency and transfected by the calcium phosphate method with a total of 5 μg of DNA including a 1:1 molar ratio of Ca2+ channel subunits and 0.3 μg of a CD8 expression plasmid for identification of transfected cells. ACI peptide constructs were expressed at a 10:1 molar ratio with Ca2+ channel subunits.
Electrophysiological recordings. At least 48 hr after transfection, tsA-201 cells were incubated with CD8 antibody-coated microspheres (Dynal, Oslo, Norway) to allow visual identification of transfected cells. Ca2+ currents were recorded in the whole-cell configuration of the patch-clamp technique using a List EPC-7 patch-clamp amplifier and were filtered at 5 kHz. Voltage protocols were applied, and data were acquired using Fastlab software (Indec Systems). Leak and capacitive transients were subtracted using a P/-4 protocol. Extracellular recording solutions contained 150 mm Tris, 1 mmMgCl2, and 10 mmCaCl2 or BaCl2. Intracellular solutions consisted of 120 mmN-methyl-d-glucamine, 60 mm HEPES, 1 mmMgCl2, 2 mm Mg-ATP, and 0.5 or 10 mm EGTA. GDPβS (1 mm) and GTPγS (0.5 mm) (Sigma, St. Louis, MO) were included in some intracellular solutions. The pH of intracellular and extracellular recording solutions was adjusted to 7.3 with methanesulfonic acid. Because extracellular Ba2+ and intracellular BAPTA caused shifts in the voltage dependence of activation of 10 mV negative and positive, respectively, voltage protocols were adjusted to compensate for this difference as noted. All averaged data represent the mean ± SEM.
RESULTS
Ca2+-dependent inactivation of P/Q-type Ca2+ channels during step depolarizations
P/Q-type Ca2+ channels (α1A, β1b, and α2δ) transfected into tsA-201 cells inactivate faster and more completely when Ca2+ rather than Ba2+ is the permeant ion and when Ca2+ accumulates intracellularly with reduced concentrations of Ca2+ chelators in intracellular recording solutions (Lee et al., 1999). However, inactivation is not caused solely by incoming Ca2+ ions because P/Q-type Ca2+ currents inactivate rapidly at positive voltages even in the presence of high intracellular EGTA (Fig.1A). Because Ca2+-dependent inactivation of P/Q-type Ca2+ channels may be occluded by a voltage-dependent mechanism of inactivation, we tested whether Ca2+-dependent inactivation was more significant in P/Q-type channels containing the β2a subunit, which exhibit relatively little voltage-dependent inactivation (Stea et al., 1994). Unlike channels containing β1b subunits, Ca2+ currents (ICa) through P/Q-type channels containing β2a subunits inactivate slowly with high intracellular EGTA or BAPTA and when extracellular Ca2+ is replaced by Ba2+ (Fig. 1A). However, when intracellular EGTA is reduced to 0.5 mm,ICa inactivates almost completely during a 1 sec step depolarization (Fig. 1A). To estimate the magnitude of Ca2+-dependent inactivation, residual current at the end of the test pulse (Ires) was compared with the peak current (Ipk) (Fig.1B). With 0.5 mm intracellular EGTA, inactivation of ICa was approximately three times more complete (Ires/Ipk= 0.24 ± 0.02; n = 12) than that with 10 mm EGTA (Ires/Ipk= 0.66 ± 0.08; n = 9) or 10 mm BAPTA (Ires/Ipk= 0.64 ± 0.02; n = 10) or when Ba2+ was used as the permeant ion (Ires/Ipk= 0.66 ± 0.04; n = 10). As shown for P/Q-type channels containing β1b (Lee et al., 1999), calmodulin is important for Ca2+-dependent inactivation, because it is greatly diminished by deletion of the α1A CBD (ΔCBD;Ires/Ipk= 0.45 ± 0.03; n = 8) and by overexpression of a peptide from type I adenylyl cyclase that competes with Ca2+ channels for binding to calmodulin (Wu et al., 1993) (Ires/Ipk= 0.40 ± 0.04 [ACI; n = 12] vs 0.16 ± 0.04 [ACI(F-R), control peptide; n = 9]) (Fig. 1). The prominent Ca2+- and calmodulin-dependent inactivation revealed by the β2a subunit suggests that, under physiological conditions, the extent to which P/Q-type Ca2+ channels are regulated by Ca2+ depends critically on subunit composition. The robust effects of Ca2+and calmodulin, combined with the limited voltage-dependent inactivation conferred by β2a, facilitate detailed analysis of Ca2+-dependent modulation of P/Q-type Ca2+ channels. Therefore, further studies were restricted to channels containing the β2a subunit.
Fig. 1.

Ca2+- and calmodulin-dependent inactivation of P/Q-type Ca2+ channels in tsA-201 cells. A, Normalized Ca2+ currents recorded from cells cotransfected with α1A, β1b or β2a (as indicated), and α2δ subunits are shown. Left, The extracellular solution contained 10 mmCa2+, and the intracellular solution contained EGTA (0.5 or 10 mm) or BAPTA (10 mm) as indicated. Right, The intracellular solution contained 0.5 mm EGTA. Current traces are shown from channels containing α1A subunits recorded with 10 mm extracellular Ca2+or Ba2+ (top), from channels containing α1A or the α1A ΔCBD deletion mutant (middle), or from channels containing α1A with overexpression of either a calmodulin-binding inhibitor peptide (ACI) or an inactive mutant form of the peptide [ACI(F-R)] (bottom). Currents were elicited by a 1 sec step depolarization to +10 mV from a holding potential of −80 mV, except when BAPTA or Ba2+ was used and depolarization was stepped to +20 and 0 mV, respectively, to compensate for shifts in the voltage dependence of activation. B, Inactivation expressed as the ratio of residual current at the end of the 1 sec depolarization (Ires) to the peak current (Ipk) is shown.Numbers in parentheses indicate the number of cells recorded in each condition.
To confirm further the Ca2+ dependence of P/Q-type channel inactivation, we tested the effects of a conditioning prepulse (1 sec) to various voltages on Ca2+ currents elicited by a test pulse to +20 mV. If P/Q-type channel inactivation depends on previous Ca2+ entry, then inactivation of the test pulse current should be greatest at the prepulse voltage eliciting the peak inward Ca2+ current. As shown in Figure 2, A and B,ICa inactivation increased concomitantly with the amplitude of the prepulse-induced current and declined as the prepulse voltage approached the reversal potential for Ca2+. Unlike the proposed mechanism of preferential closed-state inactivation (Patil et al., 1998), inactivation measured here depended on Ca2+ entry and intracellular accumulation during the prepulse because no inactivation of the test current was observed when BAPTA was included in the recording pipette (Fig.2B).
Fig. 2.

Voltage dependence of inactivation and recovery from inactivation of P/Q-type Ca2+ currents.A, The voltage protocol for measuring inactivation caused by a 1 sec conditioning prepulse is shown above representative Ca2+ currents evoked by a 10 msec test pulse to +20 mV after prepulses to the indicated voltages. B, The relationship between the inactivating prepulse voltage and peak Ca2+ currents is shown. Peak Ca2+currents elicited by the +20 mV test pulse after the conditioning prepulse were recorded with intracellular solutions containing 0.5 mm EGTA (closed circles;n = 6) or 10 mm BAPTA (open circles; n = 9) and were normalized to Ca2+ currents evoked after a −30 mV prepulse.Open inverted triangles represent the current–voltage relationship for Ca2+ currents (0.5 mmintracellular EGTA) elicited by test pulses to the indicated voltages without a prepulse and normalized to the current amplitude of the +10 mV test pulse (n = 8). C, Recovery from inactivation induced by a 2 sec conditioning pulse to +10 mV (+20 mV for BAPTA) was monitored using +10 mV test pulses (+20 mV for BAPTA) (6 msec) at 0.2 Hz. Test currents were normalized to the noninactivated Ca2+ current evoked before the inactivating prepulse. The voltage protocol is shown above the sample current records. The current during the 5 sec interval between test pulses was not recorded. D, Fractional recovery from inactivation obtained by plotting Ca2+ current amplitudes normalized to the noninactivated test current against time after the inactivating prepulse is shown. Intracellular recording solutions contained either 0.5 mm EGTA (n = 6) or 10 mm BAPTA (n = 5).
Similar to presynaptic Ca2+ channels in rat brainstem (Forsythe et al., 1998), Ca2+ currents through P/Q-type Ca2+ channels expressed in tsA-201 cells recover slowly from inactivation. The time course of recovery from inactivation induced by a 2 sec prepulse to +10 mV was described by a single exponential (τ = 22.9 ± 3.5 sec; n= 6), and full recovery took >1.5 min (Fig. 2C,D). The time constant reflects primarily recovery from Ca2+-dependent inactivation because the 2 sec prepulse caused relatively little inactivation in cells recorded with intracellular BAPTA. The slow recovery ofICa inactivation would lead to cumulative inactivation of P/Q-type Ca2+channels during repetitive stimulation, a proposed mechanism for post-tetanic depression at some synapses (Branchaw et al., 1997;Forsythe et al., 1998).
Ca2+-dependent inactivation and facilitation during repetitive stimulation
To determine the significance of Ca2+-dependent modulation of P/Q-type channels during physiological stimuli, Ca2+ currents were elicited by 100 Hz trains of 5 msec test pulses. With 0.5 mm intracellular EGTA, the amplitude of ICa increased ∼30% (facilitation) during the first five depolarizations and then inactivated below the initial current level over the next 800 msec (Fig. 3A). Both the facilitation and inactivation of ICarequired Ca2+ and calmodulin because neither effect was observed for Ba2+currents or Ca2+ currents with intracellular BAPTA, and both were inhibited by overexpression of the calmodulin inhibitor peptide (Fig. 3). Surprisingly, raising intracellular EGTA to 10 mm blocked inactivation but not facilitation of ICa during repetitive stimulation, suggesting a difference in the Ca2+ sensitivity of the two processes (Fig. 3A). Because facilitation was blocked by 10 mm BAPTA, but not by EGTA that is a slower Ca2+ buffer, facilitation may result from rapid local increases in Ca2+ that are ineffectively buffered by 10 mm EGTA. By contrast, inactivation may require longer-lasting and/or more global increases in Ca2+ that are readily prevented by both EGTA and BAPTA.
Fig. 3.
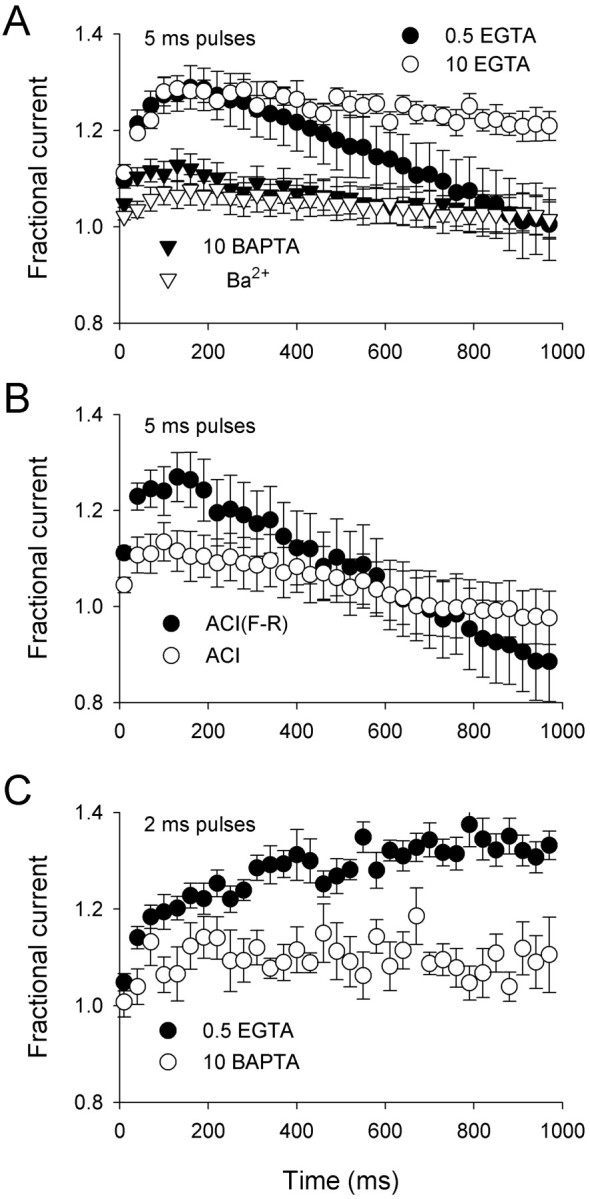
Ca2+- and calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+channels in response to repetitive depolarizations. A, Effects of intracellular Ca2+ chelators (0.5 mm EGTA; n = 7; 10 mm EGTA;n = 6; 10 mm BAPTA;n = 11) and extracellular Ba2+(n = 8) on currents elicited by 100 Hz trains of 5 msec pulses to +10 mV (+20 mV for BAPTA; 0 mV for Ba2+). Points represent averaged peak currents normalized to the peak current elicited by the first pulse of the train. Every third point of the train is plotted. B, Effect of a calmodulin inhibitor (ACI; n = 8) and control peptide [ACI(F-R); n = 7] on fractional current measured as described in A with 0.5 mm intracellular EGTA and 10 mm extracellular Ca2+. C, The same voltage protocol described in A except that the duration of test pulses during the train was reduced to 2 msec. Recordings were with 0.5 mm intracellular EGTA (n = 9) or 10 mm BAPTA (n = 8).
This explanation is strengthened by the strong facilitation ofICa without inactivation during a train of 2 msec depolarizations, in which the cumulative Ca2+ influx would be considerably less than that during repetitive 5 msec test pulses (Fig. 3C). Facilitation of ICa accumulated and was maintained during the train with 0.5 mmintracellular EGTA but not BAPTA. Because action potentials typically do not exceed 2 msec in duration, facilitation ofICa through P/Q-type Ca2+ channels would predominate during short, high-frequency bursts of action potentials, whereas inactivation of ICa would develop during prolonged trains causing a progressive accumulation of intracellular Ca2+. This could explain why tetanic stimulation causes an initial facilitation followed by inactivation of presynaptic P/Q-type channels in neurons (Forsythe et al., 1998), whereas short paired pulses cause only facilitation ofICa (Borst and Sakmann, 1998; Cuttle et al., 1998).
Ca2+-dependent facilitation in double-pulse protocols
The potentially large impact of P/Q-type Ca2+ channel facilitation on synaptic function motivated further analysis of this process using double-pulse protocols. Facilitation induced by a 50 msec prepulse to a variable voltage was measured by comparing ICaelicited by a test pulse before and after the conditioning prepulse (Fig. 4A). If facilitation depended on Ca2+ influx during the prepulse, then facilitation should be greatest at prepulse voltages eliciting the largest inward Ca2+current. With 0.5 mm intracellular EGTA, facilitation of ICa increased with prepulse voltages positive to −20 mV, reached a maximum of more than twofold near +20 mV, and declined at more positive prepulse voltages (Fig. 4B). The peak of this biphasic voltage dependence of facilitation correlated with the peak of theI–V relationship for ICaduring the prepulse (see Fig. 2B), underscoring the importance of prepulse-induced Ca2+ influx for the enhancement of the test current. Furthermore,ICa facilitation during the test pulse was significantly but incompletely reduced when extracellular Ba2+ was substituted for Ca2+ and when 10 mmBAPTA was included in the intracellular solution. A proportion of the residual facilitation seen with intracellular BAPTA may result from insufficient Ca2+ buffering because its voltage dependence corresponded to current activation during the prepulse (Fig. 4B). However, the small voltage-dependent facilitation of Ba2+currents (Fig. 4B) suggests that facilitation of P/Q-type Ca2+ channels may be initiated by a Ca2+-independent mechanism that is greatly enhanced by Ca2+ influx and calmodulin binding to the channel. This Ca2+-independent facilitation, revealed during double-pulse protocols using long (50 msec) prepulses, may be relatively insignificant under physiological conditions, in which Ca2+ is the charge carrier and action potentials typically do not surpass 2 msec in duration (see Fig.3).
Fig. 4.

Prepulse voltage dependence of P/Q-type Ca2+ channel facilitation. A, The voltage protocol shown above representative Ca2+currents recorded with 0.5 mm intracellular EGTA. Shown are currents elicited by a test pulse to 0 mV before (P1) and 5 msec after (P2) a 50 msec conditioning prepulse to the voltages (in millivolts) indicated below each current trace. B, Effects of intracellular Ca2+ chelators (0.5 mmEGTA; n = 8; 10 mm BAPTA;n = 6) and extracellular Ba2+(n = 6) on facilitation as a function of prepulse voltage. The facilitation ratio was obtained by normalizing the peak current from P2 to that from P1.
Presynaptic calcium currents are reduced by activation of G-proteins, and G-protein-modulated currents can be facilitated by double-pulse protocols (Dolphin, 1996). Such facilitation superficially resembles the Ca2+-dependent facilitation observed here. However, several lines of evidence argue against the involvement of G-proteins in the facilitation ofICa in our experiments. First, activation of G-proteins with intracellular GTPγS is necessary for significant G-protein-dependent facilitation of P/Q-type Ca2+ channels (Herlitze et al., 1996). Prepulses to +10 mV produce near maximal Ca2+-dependent facilitation ofICa (Fig. 4B), but GTPγS is not required for, and does not enhance, facilitation by this prepulse voltage (Fig. 5A,top, middle). Second, inhibiting basal G-protein activity by intracellular dialysis with GDPβS does not reduce facilitation of ICa (Fig.5A, bottom). Third, a strong depolarizing prepulse to +100 mV causes robust G-protein-dependent facilitation when intracellular GTPγS is included in the pipette (Fig. 5B,middle) but little facilitation ofICa when GTPγS is absent (Fig.5B, top) or when G-proteins are inhibited by GDPβS (Fig. 5B, bottom). Taken together, these results are inconsistent with an essential role for G-proteins in Ca2+-dependent facilitation of P/Q-type channels. Although the G-protein- and Ca2+-dependent forms of facilitation are clearly separated by prepulses to +100 and +10 mV, respectively, both would contribute to facilitation at the peak of the neuronal action potential (approximately +40 mV) under physiological conditions.
Fig. 5.
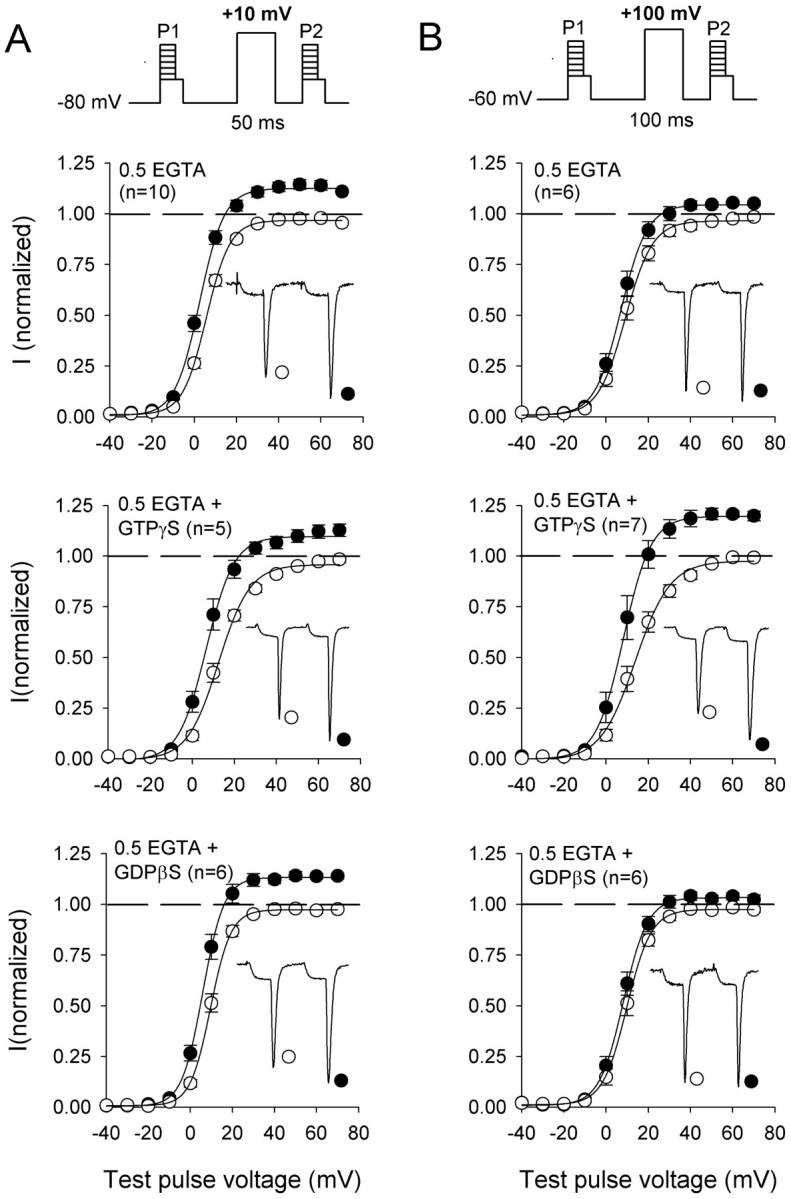
Effect of guanine nucleotides on prepulse facilitation of P/Q-type Ca2+ channels.A, Voltage dependence of activation of Ca2+ currents before (open circles;P1) and after (closedcircles; P2) a depolarizing prepulse from −80 to +10 mV with intracellular solutions containing 0.5 mm EGTA (top), 0.5 mm EGTA and 0.5 mm GTPγS (middle), or 0.5 mm EGTA and 1 mm GDPβS (bottom). Tail currents were measured by holding at −40 mV for 5 msec after test pulses (P1, P2) to variable voltages. Peak tail currents were normalized to the largest tail current measured during the nonfacilitated prepulses (P1) and plotted against the test pulse voltage.Insets, Representative currents elicited by test pulses to +40 mV before and after the prepulse. B, Voltage dependence of activation measured as described in Aexcept that P1, a +100 mV conditioning pulse, andP2 were stepped from a holding potential of −60 mV. Intracellular solutions contained 0.5 mm EGTA (top), 0.5 mm EGTA and 0.5 mmGTPγS (middle), or 0.5 mm EGTA and 1 mm GDPβS (bottom). Symbolsand insets are as described in A.
Onset and decay of Ca2-dependent facilitation
The effect of Ca2+ on enhancingICa facilitation (Fig.4B) may be caused by a Ca2+-dependent acceleration of the onset of facilitation and/or a slowing of its decay. To characterize the mechanism by which Ca2+ entry and intracellular accumulation promote facilitation of P/Q-type Ca2+ channels, the rates of onset and decay of ICa facilitation were determined with various degrees of intracellular Ca2+ buffering with or without the calmodulin inhibitor peptide.
In a double-pulse protocol, the onset of facilitation was determined by plotting facilitation of ICa as a function of prepulse duration (Fig.6A). With 0.5 mm intracellular EGTA, facilitation increased with prepulse duration according to a single-exponential time course (τ = 13.3 ± 1.3 msec; n = 8), withICa inactivation becoming evident with prepulses longer than 60 msec (Fig. 6B). With 10 mm intracellular BAPTA, facilitation ofICa was reduced, but its time course was similar to that in 0.5 mm EGTA (τ = 16 ± 1.3 msec; n = 7). At various prepulse voltages, the time constants for the onset ofICa facilitation were not significantly different (p > 0.05) with either Ca2+ buffer (Fig. 6C). These results are consistent with a role for Ca2+ in controlling the amount but not the rate of onset of ICa facilitation.
Fig. 6.

Onset of facilitation of P/Q-type Ca2+ channels. A, The voltage protocol (top) and representative currenttraces (bottom) recorded with 10 mm extracellular Ca2+ and 0.5 mm EGTA or 10 mm BAPTA in the intracellular recording solution. Currents were elicited by test pulses to 0 mV (+10 mV for BAPTA) before (P1) and 5 msec after (P2) conditioning prepulses (+10 mV for 0.5 mm EGTA; +20 mV for 10 mm BAPTA) of the indicated durations. B, Effect of intracellular Ca2+ chelators (0.5 mm EGTA;n = 8; 10 mm BAPTA;n = 7) on facilitation as a function of prepulse duration. Facilitation was obtained by normalizing the peak current from P2 to that from P1. Single-exponential fits of the data are shown; only the first seven points of the data obtained with 0.5 mm EGTA were included in the fit because of the onset of inactivation after longer prepulses.C, Effect of prepulse voltage on time constants for the onset of facilitation. Mean time constants were obtained from single-exponential fits to the data in B. Shown are results obtained with intracellular solutions containing 0.5 mm EGTA or 10 mm BAPTA at the indicated prepulse voltages. The number of cells recorded for each condition is indicated in parentheses.
Although Ca2+ entry during the prepulse did not affect the rate of onset ofICa facilitation, it did significantly slow its decay. As the interval between the 50 msec conditioning prepulse and the second test pulse was increased, facilitation ofICa decreased with a single-exponential time course (τ = 112.7 ± 17 msec;n = 9) with 0.5 mm intracellular EGTA. The time course was sensitive to Ca2+ influx during the prepulse because the decay of facilitation was almost twice as slow (τ = 221.5 ± 48.4 msec; n = 6) with a longer 200 msec prepulse and twice as fast (τ = 43.5 ± 5.6 msec;n = 6) with a 5 msec prepulse (Fig.7A,B). These data suggest that the persistence of facilitation is caused by the intracellular accumulation of Ca2+ ions that had entered during the prepulse.
Fig. 7.

Effects of Ca2+ and calmodulin on decay of facilitation of P/Q-type Ca2+channels. A, The voltage protocol (top) for measuring decay of facilitation and representative Ca2+ currents (bottom) elicited by test pulses to 0 mV (+10 mV for 10 mm BAPTA) before (P1) and after (P2) a conditioning prepulse to +10 mV (+20 mV for BAPTA) for 5 or 200 msec. Superimposed are currents in which P2 was given 0.1 msec (double asterisks) or 300 msec (single asterisk) after the conditioning prepulse. Intracellular recording solutions contained either 0.5 mm EGTA (left traces) or 10 mm BAPTA (right traces). B, Effect of prepulse duration and Ca2+ buffering on decay of facilitation. The facilitation ratio was obtained by normalizing the peak current fromP2 to that from P1 and was plotted against the interval between the conditioning prepulse andP2. Shown are results obtained with 5, 50, and 200 msec conditioning prepulses with 0.5 mm EGTA (left) or 10 mm BAPTA (right) in the recording pipette. C, Data obtained as described in B with a 50 msec conditioning prepulse and intracellular solutions containing 0.5 mm EGTA (n = 9), 10 mm BAPTA (n = 10), or 10 mm EGTA (n = 7) and with overexpression of the calmodulin inhibitor peptide (ACI; n = 5) with a 0.5 mm EGTA intracellular solution. D, Time constants (τ;msec) for the decay of facilitation obtained from single-exponential fits of the data in C. Residual facilitation is the percentage of the maximum facilitated current remaining 400 msec after the conditioning prepulse.
Consistent with this idea, the decay of facilitation depended on intracellular Ca2+ buffering. After a 50 msec prepulse, the decay of ICafacilitation with 10 mm intracellular BAPTA was significantly faster (τ = 47.6 ± 3.6 msec;n = 10) than that with 0.5 mmEGTA, and the time course changed little with prepulse duration (τ200msec = 43.4 ± 3.9 msec; n = 7; τ5msec = 46.7 ± 7.4 msec; n = 5) (Fig. 7A,B). The Ca2+-dependent slowing of the decay of facilitation was also diminished by overexpression of the calmodulin inhibitor peptide (τ = 67.7 ± 9.2 msec; n = 6) (Fig. 7C) and by substitution of extracellular Ca2+ with Ba2+ (data not shown). Interestingly, 10 mm intracellular EGTA, which supported facilitation equal to that in 0.5 mm EGTA (see Fig. 7C, τ = 0 intercept), also greatly accelerated its decay (τ = 55.3 ± 2.7 msec; n = 7).
In addition to slowing the decay ofICa facilitation, Ca2+ influx and intracellular accumulation prevented complete recovery of ICa to initial levels. With 0.5 mm EGTA,ICa was still potentiated by 26.1 ± 8.3% (n = 9) 400 msec after a 50 msec conditioning prepulse. In contrast, little facilitation ofICa remained at this time point with intracellular BAPTA (4.6 ± 1.6%; n = 9) or 10 mm EGTA (0.5 ± 1.2%; n = 7) or with overexpression of the calmodulin inhibitor peptide (5.5 ± 2.2% msec; n = 6) (Fig. 7C,D). Complete decay of ICa to initial levels was also observed when the prepulse was limited to 5 msec (Fig.7B). Thus, Ca2+ entry during a conditioning prepulse both enhances and prolongs facilitation ofICa for >0.5 sec after a depolarizing stimulus. ICa remained facilitated for at least 10 sec but had decayed completely by 30 sec, the interval between trials (data not shown). This prolonged facilitation would greatly enhance Ca2+influx in presynaptic nerve terminals over time and may have important implications for relatively long-lasting changes in synaptic strength.
DISCUSSION
We have shown that incoming Ca2+triggers both a negative and positive feedback regulation of subsequent Ca2+ entry through P/Q-type Ca2+ channels. Repetitive stimuli and step depolarizations enhance facilitation and accelerate inactivation of P/Q-type Ca2+ currents. Both effects are suppressed by intracellular dialysis with BAPTA, extracellular Ba2+ in place of Ca2+, and overexpression of a calmodulin-binding inhibitor peptide. These modulatory effects of Ca2+ and calmodulin are distinct from previously described mechanisms of facilitation and inactivation of P/Q-type Ca2+ channels (Brody et al., 1997; Patil et al., 1998) and are consistent with the behavior of presynaptic P/Q-type Ca2+ channels in nerve terminals (Borst and Sakmann, 1998; Cuttle et al., 1998; Forsythe et al., 1998).
Auxiliary β subunits and Ca2+-dependent modulation of P/Q-type Ca2+ channels
Differences in the properties of P/Q-type Ca2+ channels in the nervous system arise in part from the association of α1A with distinct β subunits. Unlike β1, β3, and β4 subunits, which accelerate inactivation of P/Q-type Ca2+ channels, β2asignificantly slows inactivation kinetics and causes a large depolarizing shift in the voltage dependence of steady-state inactivation (Stea et al., 1994; De Waard and Campbell, 1995). However, in P/Q-type Ca2+ channels containing β2a, the relatively limited voltage-dependent inactivation unmasks powerful negative and positive feedback regulation by Ca2+. Ca2+-dependent modulation of strongly inactivating P/Q-type Ca2+ channels, such as those containing β1b (Lee et al., 1999), is modest compared with that of Ca2+ channels with β2a. Because α1Ais widely distributed throughout the nervous system (Stea et al., 1994;Westenbroek et al., 1995), the significance of Ca2+-dependent inactivation and facilitation of P/Q-type Ca2+ channels in neurons would depend critically on coexpression with a specific β subunit. The prominence of Ca2+-dependent regulation of P/Q-type channels may also depend on the splice variant of α1A expressed. Voltage-dependent inactivation of the α1A-b variant coexpressed with either β1b or β2ais minimal (Bourinet et al., 1999). Based on our findings, this should reveal a marked Ca2+-dependent modulation of α1A-b channels, regardless of the β subunit expressed. Thus, the functional diversity of P/Q-type Ca2+ channels is potentially quite complex, and various combinations of α1A and β subunits will yield channels distinguished by their biophysical properties as well as their regulation by Ca2+.
Mechanism of Ca2+-dependent inactivation and facilitation of P/Q-type Ca2+ channels
Ca2+ entry through P/Q-type Ca2+ channels promotes the binding of calmodulin to an atypical α1A CBD in the cytoplasmic C-terminal domain of the α1Asubunit (Lee et al., 1999). This interaction underlies Ca2+-dependent inactivation and facilitation of P/Q-type channels with β1b, because these effects are reversed by deletion of the α1A CBD. Our studies of P/Q-type channels with β2a reveal important new insights into the role of Ca2+ and calmodulin in the feedback regulation by Ca2+. First, deletion of the α1A CBD strongly reduces but does not eliminate Ca2+-dependent inactivation and facilitation (Fig. 1; data not shown). Also, the ACI peptide, which should eliminate the influence of Ca2+/calmodulin, does not completely abolish the effects of Ca2+ on inactivation or facilitation (Figs. 1, 3). The incomplete blockade of Ca2+-dependent inactivation and facilitation by these two manipulations suggests additional modulation by Ca2+, independent of calmodulin. Such calmodulin-independent modulation might be caused by Ca2+ ions binding to an EF-hand motif in the C-terminal domain of α1A (De Leon et al., 1995) or to other, as yet unrecognized, Ca2+-binding sites. In addition, Ca2+ entry through P/Q-type channels could activate second messenger-regulated kinases, such as protein kinase C, that potentiate ICa through P/Q-type channels (Stea et al., 1995; Bourinet et al., 1999) and could produce effects additive to those of calmodulin on facilitation.
A second surprising feature of P/Q-type Ca2+ channel modulation by Ca2+ was revealed in the block of inactivation but not facilitation by 10 mm intracellular EGTA (Figs. 1, 3), which suggests a difference in the Ca2+ dependence of the two processes. An intriguing explanation for the distinct effects of Ca2+ on facilitation and inactivation is provided by differences in Ca2+ binding to the N- and C-terminal lobes of calmodulin (James et al., 1995). Ca2+ binding to calmodulin is highly cooperative with Ca2+ binding first to the C-terminal EF-hands, which have the highest intrinsic affinity for Ca2+, followed by Ca2+ binding to lower affinity sites in the N-terminal lobe (Wang, 1985). Thus,ICa facilitation and inactivation could result from conformational changes in calmodulin after Ca2+ binding to the C- and N-terminal lobes, respectively. That the two lobes of calmodulin can differentially regulate ion channel function is evident from analyses of calmodulin mutants in Paramecium (Kink et al., 1990), Ca2+ activation of K+ channels (Keen et al., 1999), and Ca2+-dependent inactivation of L-type Ca2+ channels (Peterson et al., 1999).
Finally, paired-pulse protocols reveal some prepulse-induced facilitation, even when intracellular Ca2+is strongly buffered with BAPTA (Figs. 4, 6, 7) and when extracellular Ca2+ is replaced by Ba2+ (Fig. 4). This residual, Ca2+-independent facilitation may result from a voltage-dependent enhancement of channel activation. Membrane depolarization might cause the initial facilitation of the current because of activation of one or more voltage sensors. The association of Ca2+ and calmodulin with α1A could promote and stabilize the activated conformation of the sensor or sensors such that these partially activated channels would activate more rapidly and at lower voltages, causing facilitation in response to subsequent depolarizations. This mechanism would explain the longer lifetime of the facilitated state of the channel in the presence of Ca2+ and calmodulin (Fig. 7) and the accumulation and maintenance of Ca2+- and calmodulin-dependent facilitation of P/Q-type channels during trains of pulses (Fig.3C).
Ca2+-dependent modulation of P/Q-type Ca2+ channels and synaptic plasticity
Inactivation and facilitation of Ca2+currents through P/Q-type channels expressed in tsA-201 closely resemble the behavior of presynaptic P/Q-type channels such as those recorded at the calyx of Held synapse in the rat brainstem (Borst and Sakmann, 1998; Cuttle et al., 1998; Forsythe et al., 1998). In both cases, trains of depolarizations cause an initial facilitation followed by inactivation of the Ca2+ current over time, recovery from inactivation is relatively slow, and facilitation is strongly dependent on incoming Ca2+ions. At the calyx of Held, inactivation of P/Q-type channels, along with other presynaptic mechanisms (Wang and Kaczmarek, 1998; Borst and Sakmann, 1999; Wu and Borst, 1999), contributes to post-tetanic depression of synaptic transmission (Forsythe et al., 1998). However, the effects of extracellular Ba2+and intracellular BAPTA in slowing inactivation are significantly weaker for native than for recombinant P/Q-type channels. This could be explained by the existence of multiple β subunits in rat auditory neurons that would produce Ca2+ channels that are modulated by both Ca2+-dependent and -independent mechanisms. In addition, a Ca2+-independent regulation of native Ca2+ channels could be mediated by signaling molecules not present in the tsA-201 cells recorded in our study. Nevertheless, inactivation of P/Q-type channels at the calyx of Held is strongest for Ca2+ currents in the absence of strong intracellular Ca2+buffers (Forsythe et al., 1998), suggesting a role for Ca2+ and calmodulin in the negative feedback of P/Q-type channels at this and other synapses.
Considering its rapid onset (Figs. 3, 6), Ca2+-dependent facilitation of P/Q-type channels may contribute to the short-term enhancement of synaptic transmission that depends on elevated intracellular Ca2+ during the second of two depolarizing pulses (Zucker, 1999). Although synaptic enhancement occurs in the absence of measurable increases in presynaptic Ca2+ influx (Tank et al., 1995; Zucker, 1999), fluorometric techniques typically used for measuring intracellular Ca2+ signals are relatively insensitive and may not detect very high but local changes in Ca2+ caused by facilitation of Ca2+ channel opening. At the calyx of Held synapse, where the contribution of presynaptic Ca2+ currents to neurotransmitter release can be assessed by simultaneous presynaptic and postsynaptic recording, paired-pulse stimulation causes facilitation of presynaptic P/Q-type channels and, occasionally, the enhancement of the postsynaptic response when the effects of synaptic depression are minimized (Borst and Sakmann, 1998). Facilitation of presynaptic Ca2+ channels at this synapse is attenuated when extracellular Ca2+ is replaced by Ba2+ and by presynaptic dialysis with BAPTA, but not by GTPγS, GDPβS, or pharmacological blockade of Ca2+-dependent kinases and phosphatases (Cuttle et al., 1998). These results are consistent with the facilitation of P/Q-type channels caused by the Ca2+-dependent association of calmodulin with the α1A subunit. Because P/Q-type channels are important in the regulation of neurotransmitter release at numerous synapses, Ca2+-dependent facilitation of these channels may fundamentally contribute to the enhancement of synaptic function in the nervous system.
Ca2+ and calmodulin have long been implicated in mechanisms of synaptic plasticity. That P/Q-type Ca2+ channels are significantly regulated by these molecules is unexpected, in part because current knowledge is primarily limited to the behavior of Ba2+currents through these channels. However, the importance of P/Q-type channel modulation by Ca2+ and calmodulin is underscored by the physiological consequences of Ca2+-dependent inactivation and facilitation of presynaptic P/Q-type channels in neurons. Elucidating how Ca2+/calmodulin, G-proteins, and other regulatory influences coordinately control Ca2+ influx through P/Q-type channels promises further insight into mechanisms leading to altered synaptic function.
Footnotes
This work was supported by National Institutes of Health Grant NS 22625 to W.A.C. and postdoctoral National Research Service Award NS 10645 to A.L.
Correspondence should be addressed to Dr. William A. Catterall, Department of Pharmacology, Box 357280, University of Washington, Seattle, WA 98195-7280. E-mail:wcatt@u.washington.edu.
REFERENCES
- 1.Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol (Lond) 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borst JG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol (Lond) 1999;521:123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 4.Branchaw JL, Banks MI, Jackson MB. Ca2+- and voltage-dependent inactivation of Ca2+ channels in nerve terminals of the neurohypophysis. J Neurosci. 1997;17:5772–5781. doi: 10.1523/JNEUROSCI.17-15-05772.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brody DL, Yue DT. Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. J Neurosci. 2000;20:889–898. doi: 10.1523/JNEUROSCI.20-03-00889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brody DL, Patil PG, Mulle JG, Snutch TP, Yue DT. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J Physiol (Lond) 1997;499:637–644. doi: 10.1113/jphysiol.1997.sp021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol (Lond) 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Leon M, Wang Y, Jones L, Perez-Reyes E, Wei XY, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- 9.De Waard M, Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. J Physiol (Lond) 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodge FA, Jr, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J Physiol (Lond) 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- 12.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 13.Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- 14.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 16.James P, Vorherr T, Carafoli E. Calmodulin-binding domains: just two faced or multi-faceted? Trends Biochem Sci. 1995;20:38–42. doi: 10.1016/s0968-0004(00)88949-5. [DOI] [PubMed] [Google Scholar]
- 17.Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, Janowsky A, Fakler B, Adelman JP, Maylie J. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J Neurosci. 1999;19:8830–8838. doi: 10.1523/JNEUROSCI.19-20-08830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kink JA, Maley ME, Preston RR, Ling KY, Wallen-Friedman MA, Saimi Y, Kung C. Mutations in paramecium calmodulin indicate functional differences between the C-terminal and N-terminal lobes in vivo. Cell. 1990;62:165–174. doi: 10.1016/0092-8674(90)90250-i. [DOI] [PubMed] [Google Scholar]
- 19.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;339:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 20.Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- 21.Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 22.Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 23.Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 24.Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. The localization and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc Natl Acad Sci USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka O, Sakagami H, Kondo H. Localization of mRNAs of voltage-dependent Ca2+-channels: four subtypes of α1- and β-subunits in developing and mature rat brain. Mol Brain Res. 1995;30:1–16. doi: 10.1016/0169-328x(94)00265-g. [DOI] [PubMed] [Google Scholar]
- 27.Tank DW, Regehr WG, Delaney KR. A quantitative analysis of presynaptic calcium dynamics that contribute to short-term enhancement. J Neurosci. 1995;15:7940–7952. doi: 10.1523/JNEUROSCI.15-12-07940.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Usowicz MM, Sugimori M, Cherksey B, Llinás R. P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron. 1992;9:1185–1199. doi: 10.1016/0896-6273(92)90076-p. [DOI] [PubMed] [Google Scholar]
- 29.Wang CL. A note on Ca2+ binding to calmodulin. Biochem Biophys Res Commun. 1985;130:426–430. doi: 10.1016/0006-291x(85)90434-6. [DOI] [PubMed] [Google Scholar]
- 30.Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- 31.Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TVB, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 33.Wu Z, Wong ST, Storm DR. Modification of the calcium and calmodulin sensitivity of the type I adenylyl cyclase by mutagenesis of its calmodulin binding domain. J Biol Chem. 1993;268:23766–23768. [PubMed] [Google Scholar]
- 34.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]