

Abstract
Long-term potentiation (LTP) in perforant path-granule cell synapses is decreased in aged rats, stressed rats, and rats injected intracerebroventricularly with the proinflammatory cytokine interleukin-1β (IL-1β). One factor that is common to these experimental conditions is an increase in the concentration of IL-1β in the dentate gyrus, suggesting a causal relationship between the compromise in LTP and increased IL-1β concentration. In this study, we have investigated the downstream consequences of an increase in IL-1β concentration and report that the reduced LTP in rats injected intracerebroventricularly with IL-1β was accompanied by a decrease in KCl-stimulated glutamate release in synaptosomes prepared from dentate gyrus, although unstimulated glutamate release was increased. These changes were paralleled by increased activity of the stress-activated kinases, c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase. Intracerebroventricular injection of IL-1β increased reactive oxygen species production in hippocampal tissue, whereas IL-1β and H2O2 increased activities of both JNK and p38 in vitro. Dietary manipulation with antioxidant vitamins E and C blocked the increase in reactive oxygen species production, the stimulation of JNK and p38 activity, the attenuation of glutamate release, and the IL-1β-induced inhibitory of LTP. We propose that IL-1β stimulates activity of stress-activated kinases, which in turn may inhibit glutamate release and result in compromised LTP and that these actions are a consequence of increased production of reactive oxygen species.
Keywords: LTP, dentate gyrus, IL-1β, stress-activated kinases, glutamate release, reactive oxygen species
Consistent with the high expression of IL-1 receptors in the hippocampus (Lechan et al., 1990; Ban et al., 1991; Parnet et al., 1994) are several observed effects of exogenous IL-1 in this brain area. For example, IL-1β exerts an inhibitory effect on (1) long-term potentiation (LTP) in CA1 (Bellinger et al., 1993), CA3 (Katsuki et al., 1990), and dentate gyrus (Cunningham et al., 1996; Murray and Lynch, 1998a,b), (2) release of acetylcholine (Rada et al., 1991) and glutamate (Murray et al., 1997) in hippocampal synaptosomes, (3) calcium influx in hippocampal synaptosomes (Murray et al., 1997), and (4) Ca2+ channel currents in hippocampal neurons (Plata-Salaman and ffrench-Mullen, 1994).
The mechanism by which IL-1β inhibits LTP remains to be established. Because maintenance of LTP has been associated with increased glutamate release (Bliss and Collingridge, 1993; Canevari et al., 1994; McGahon and Lynch, 1996; McGahon et al., 1997), one factor that may contribute to inhibition of LTP is the inhibitory effect of IL-1β on glutamate release. However, it has been recently reported that the IL-1β-induced attenuation of LTP in dentate gyrus in vitrois blocked by SB203580 (Coogan et al., 1997), an inhibitor of p38 that is one member of the family of mitogen-activated protein (MAP) kinases. The MAP kinase family has been identified as a major player in cellular signaling and markedly influences such diverse processes as cell proliferation, cell differentiation, and cell death. Consistent with the evidence that nerve growth factor and other growth factors stimulate extracellular signal-regulated protein kinase (ERK), is the generally held view that activation of this particular pathway results in neurite outgrowth, cell proliferation, or differentiation (Seger and Krebs, 1995; Xia et al., 1995; Creedon et al., 1996). In contrast, c-Jun N-terminal kinase (JNK) and p38 are activated by environmental stress, including oxidative stress (Raingeaud et al., 1995;Uciechowski et al., 1996; Junger et al., 1997), and activation leads to growth arrest or even cell death (Park et al., 1996; Maroney et al., 1998). JNK and p38 are stimulated by IL-1β (Derijard et al., 1994; Raingeaud et al., 1995; Rizzo and Carlo-Stella, 1996; Uciechowski et al., 1996; Lu et al., 1997), which is consistent with the observation that IL-1β has been implicated in cell death (Rothwell, 1999). In general, the evidence that indicates that IL-1β activates JNK and p38 and that activation of these pathways induces cell damage or cell death has been obtained in various circulating and cultured cells, whereas evidence for similar changes in neuronal tissue is lacking.
In this study we have investigated the downstream consequences of an increase in IL-1β in hippocampal tissue and report that this cytokine increases activity of both JNK and p38. We also provide evidence that IL-1β-induced activation of JNK and p38 leads to a decrease in glutamate release and might be responsible for the attenuation in both the early and later components of LTP observed in rats which received an intracerebroventricular injection of IL-1β.
MATERIALS AND METHODS
Animals. Groups of male Wistar rats (300–350 gm) were used in these experiments. Animals were housed in groups of two to four under a 12 hr light/dark schedule. Ambient temperature was controlled between 22 and 23°C. Food and water were availablead libitum. In some experiments food and water intake was measured daily for 1 week, and at the end of this period rats were randomly subdivided into two groups. One group received normal laboratory chow with added vitamin E (250 mg ofdl-α-tocopheryl acetate per rat per day, dissolved in corn oil; Beeline Healthcare, Dublin, Ireland). The laboratory chow contained 3.5% crude oil and 55 mg/kg vitamin E; thus average daily intake of vitamin E from this diet was 2.75 mg/rat. Ascorbic acid (250 mg/rat per day) was added to the water given to these rats. The second group received normal laboratory chow with corn oil added to ensure isocaloric intake with the first group. Rats were offered 100% of their average daily food and average water intakes so that the full daily allowances of vitamins would be ingested. Diet was prepared freshly each day. Food and water intake did not vary between groups, and there was no significant difference in daily food and water intake before and after dietary modifications were made. Rats were fed on the respective control or supplemented diet for 5 d and were under veterinary supervision for the duration of this experiment.
Induction of LTP in perforant path-granule cell synapses in vivo. LTP was induced as described previously (McGahon and Lynch, 1996). Rats were anesthetized by intraperitoneal injection of urethane (1.5 gm/kg), placed in a head holder in a stereotaxic frame, and injected intracerebroventricularly with either IL-1β (5 μl; 3.5 ng/ml; human recombinant; 5 × 10−7 U/mg; The Biological Response Modifiers Program, National Cancer Institute, Bethesda, MD) or saline (5 μl). [We measured deep body temperature immediately before injection and at 15 min intervals after injection for 90 min. The temperature increased slightly from 35.18°C (±1.29, SEM, n = 5) to 36.05°C (± 2.08) in saline-injected rats and 35.08°C (±0.32) to 35.22°C (±0.61) in IL-1β-treated rats during the course of the treatments.] A window of skull was removed to allow placement of recording and stimulating electrodes in the molecular layer of the dentate gyrus (2.5 mm lateral and 3.9 mm posterior to bregma) and perforant path, respectively (angular bundle, 4.4 mm lateral to lambda). The depth of the electrodes was adjusted to obtain maximal responses in the cell body region. Stable baseline recordings were recorded for ∼15 min, and electrophysiological recording commenced 30 min after intracerebroventricular injection; this was ∼60 min after administration of urethane. Test shocks at the rate of 1/30 sec were delivered for 10 min before and 40 min after tetanic stimulation (three trains of stimuli; 250 Hz for 200 msec; intertrain interval 30 sec). In a separate series of experiments, a group of four rats received tetanic stimulation, and another group of four received the same total number of stimuli, but no high-frequency train of stimuli, i.e., one stimulus every 12 sec. At the end of the electrophysiological recording period, rats were killed by decapitation, the hippocampus was removed, and the tetanized and untetanized dentate gyri, as well as the hippocampus proper, were dissected on ice and cross-chopped into slices (350 × 350 μm) using a McIlwain tissue chopper. The time taken to prepare slices from the time of death was 2.5–3.5 min. All samples were frozen separately in 1 ml of Krebs' solution (composition of Krebs' in mm: NaCl 136, KCl 2.54, KH2PO4 1.18, MgSO4.7H2O 1.18, NaHCO3 16, glucose 10, and CaCl2 1.13) containing 10% dimethylsulfoxide according to the method of Haan and Bowen (1981). For analysis, thawed slices of untetanized and tetanized dentate gyrus were rinsed three times in fresh ice-cold Krebs' solution and homogenized in 1 ml of ice-cold sucrose (0.32 m) for preparation of P2 (McGahon and Lynch, 1996), which was used for analysis of glutamate release and activity of p38 and JNK. Slices of hippocampus were rinsed and homogenized in either 200 μl of fresh Krebs' solution for analysis of vitamins E and C and superoxide dismutase activity or 40 mm Tris-HCl, pH 7.4, for analysis of reactive oxygen species production.
Release of glutamate. The impure synaptosomal preparation P2 was resuspended in oxygenated Krebs' solution containing 2 mm CaCl2 (McGahon and Lynch, 1996), and glutamate release was assessed as described previously (McGahon et al., 1999). Briefly, synaptosomal tissue was aliquoted onto Millipore (Bedford, MA) filters (0.45 μm), rinsed under vacuum, and the filtrate was discarded. Synaptosomes were then incubated in 250 μl of oxygenated Krebs' solution at 37°C for 3 min, in the presence or absence of 40 mm KCl, and filtrate was collected and stored for analysis as described (Ordronneau et al., 1991). Triplicate samples (50 μl) or glutamate standards (50 μl; 25 nm to 1 μm prepared in 100 mmNa2HPO4 buffer, pH 8.0) were added to glutaraldehyde-coated 96-well plates, incubated for 60 min at 37°C, and washed with 100 mmNaH2PO4 buffer. Ethanolamine (250 μl; 0.1 m in 100 mmNa2HPO4 buffer) was used to bind unreacted aldehydes and donkey serum (200 μl; 3% in PBS-T) was added to block nonspecific binding. Samples were incubated overnight at 4°C in the presence of antiglutamate antibody (raised in rabbit; 100 μl; 1:5000 in PBS-T; Sigma, Poole, UK), washed and reacted with secondary antibody [anti-rabbit horseradish peroxidase (HRP)-linked secondary antibody; 100 μl; 1:10,000 in PBS-T; Amersham, UK] for 60 min at room temperature. 3,3′,5,5′-Tetramethylbenzidine liquid substrate was added as chromogen, and incubation continued for exactly 60 min at room temperature, at which time the reaction was stopped by H2SO4 (4 m; 30 μl). Optical densities were determined at 450 nm using a multiwell plate reader, and values were calculated with reference to the standard curve, corrected for protein (Bradford, 1976) and expressed as micromoles of glutamate per milligram of protein.
Analysis of the activity of the MAP kinases. The activities of JNK and p38 were analyzed in P2 preparations obtained from frozen hippocampal slices obtained from saline-pretreated and IL-1β-pretreated rats, some of which were fed on control and experimental diets. Activity of the kinases was also assessed by preincubating samples for 20 min in the absence and presence of IL-1β (10 pg/ml) and in the absence and presence of H2O2 (5 mm); these experiments were performed in P2 obtained from freshly prepared hippocampus. In all experiments, samples were analyzed for protein, diluted to equalize for protein concentration, and these samples of synaptosomal protein (10 μl, 1 mg/ml) were added to 10 μl of sample buffer (Tris-HCl, 0.5 mm, pH 6.8; glycerol 10%; SDS, 10%; β-mercaptoethanol, 5%; bromophenol blue, 0.05% w/v), boiled for 5 min, and loaded onto gels (10% SDS for p38 and 12% for JNK). Proteins were separated by application of 30 mA of constant current for 25–30 min, transferred onto nitrocellulose strips (225 mA for 75 min), and immunoblotted with the appropriate antibody. Proteins were immunoblotted with antibodies that specifically target phosphorylated JNK [Santa Cruz Biotechnology, Santa Cruz, CA; 1:500 in PBS–Tween (0.1% Tween 20) containing 2% nonfat dried milk] or phosphorylated p38 [Santa Cruz Biotechnology; 1:500 in PBS–Tween (0.1% Tween 20) containing 2% nonfat dried milk] by incubating for 2 hr at room temperature. Nitrocellulose strips were washed and incubated for 2 hr at room temperature with secondary antibody [peroxidase-linked anti-mouse IgG; 1:1000 dilution (Sigma) in the case of JNK and peroxidase-linked anti-mouse IgM; 1:1,000 dilution (Amersham) in the case of p38]. Visualization was achieved by ECL detection (Amersham); immunoblots were exposed to film overnight and processed using a Fuji x-ray processor. Quantification of protein bands was achieved by densitometric analysis using two software packages, Grab It (Grab It Annotating Grabber, version 2.04.7, Synotics; UVP Ltd) and Gelworks (Gelworks ID, version 2.51; UVP Ltd) for photography and densitometry, respectively. Gelworks provides a single value (in arbitrary units) representing the density of each blot, and the values presented here are means of data generated from at least four separate experiments. The antibodies used in these experiments were specific as judged by the fact that only one band was observed after ECL detection.
Analysis of reactive oxygen species formation. Formation of reactive oxygen species was assessed by the method of Lebel and Bondy (1990), which relies on the measurement of 2′7′-dichlorofluorescein (DCF), the oxidized, fluorescent product of 2′7′-dichlorofluorescin diacetate (DCFH-DA). Assessments were made in synaptosomes prepared from hippocampus of saline-injected and IL-1β-injected rats and also in synaptosomes prepared from freshly dissected hippocampus (in which the effects of incubating in the presence/absence of IL-1β and glutamate were assessed). Synaptosomes were incubated at 37°C for 15 min in the presence of DCFH-DA (10 μl; 5 μm, from a stock of 500 μm in methanol). To terminate the reaction, the dye-loaded suspensions were centrifuged at 13,000 ×g for 8 min at 4°C, and the pellets were resuspended in 2 ml of ice-cold 40 mm Tris buffer, pH 7.4, and monitored for fluorescence at 37°C with the excitation wavelength at 488 nm and the emission wavelength at 525 nm. In some experiments IL-1β (1 ng/ml) or glutamate (50 μm or 250 μm) was included in the incubation to medium to assess its effect of reactive oxygen species production. Results were expressed as micromoles of DCF formed per milligram of protein from a DCF standard curve (0.05–1 μm).
Analysis of superoxide dismutase activity. Superoxide dismutase activity was determined according to the method described bySpitz and Oberley (1989). Aliquots (800 μl) of incubation buffer [50 mm potassium buffer, pH 7.8, containing 1.8 mmxanthine, 2.24 mm nitroblue tetrazolium (NBT), 40 U of catalase, 7 μl/ml xanthine oxidase, and 1.33 mmdiethylenetriaminepentacetic acid] were added to 1.5 ml microfuge tubes containing samples of supernatant prepared from hippocampal tissue (100 μl) at different dilutions (1:2, 1:5, 1:10, 1:20, 1:50, and 1:100) and analyzed by UV spectroscopy at 560 nm. Slices were homogenized, and enzyme activity was assessed as the rate of reduction of NBT, which was inhibited with increasing concentrations of protein. One unit of activity was defined as the amount of protein necessary to decrease the rate of the reduction of NBT by 50%.
Analysis of vitamin C. Vitamin C concentrations were determined as previously described (Omaye et al., 1979). Briefly, duplicate aliquots of supernatant prepared from hippocampus (100 μl) were added to a 2,4-dinitrophenylhydrazine/thiourea/copper (DTC) solution (in mm: 50 thiourea, 2 copper sulfate, and 150 dinitrophenylhydrazine in 9 N H2SO4; 20 μl) and incubated for 3 hr at 37°C. Ice-cold H2SO4 (65%; 150 μl) was added to stop the reaction, and samples were vortex-mixed and incubated at room temperature for 30 min before aliquots (100 μl) were transferred to 96-well plates for assessment by UV spectroscopy at 545 nm. Results were expressed as micromoles per gram of tissue. Ascorbic acid standards were prepared in 5% trichloroacetic acid.
Analysis of vitamin E. Vitamin E was analyzed according to the method of Vatassery (1994). Briefly, aliquots of homogenate prepared from hippocampus (150 μl) were incubated in the presence of ethanol containing 0.025% butylhydroxytoluene (150 μl), 25% ascorbic acid (70 μl), and 10% potassium hydroxide (135 μl) for 30 min at 60°C. Hexane (540 μl) containing 0.025% butylhydroxytoluene was added, samples were vortex-mixed for 1 min, and they were centrifuged at 1500 rpm for 6 min. The hexane phase was removed and evaporated to dryness under nitrogen; the recovery of vitamin E using this procedure was between 70 and 80%. For HPLC analysis, dried samples were resuspended in methanol (150 μl) containing 0.025% butylhydroxytoluene, and 30 μl volumes were injected onto an Intersil C18 column. Separation of α-tocopherol was achieved using a mobile phase of 75% acetonitrile: 25% methanol at a flow rate of 1.2 ml/min, and samples were detected by UV spectroscopy at 292 nm. Vitamin E concentration was estimated by the external standard method and expressed as nanomoles per gram of tissue.
Statistical analysis. A one-way ANOVA was performed to determine whether there were significant differences between conditions. When this analysis indicated significance (at the 0.05 level), a post hoc Student-Newman-Keuls test analysis was used to determine which conditions were significantly different from each other. The Student's t test was used to establish statistical significance in some cases; for example, when analyses were performed on aliquots of the same tissue incubated in the presence or absence of IL-1β. The use of the Student's t test for paired values was also appropriate when analyses were performed on tissue prepared from untetanized and tetanized tissue from the same rat.
RESULTS
Intracerebroventricular injection of IL-1β inhibited LTP in perforant path-granule cell synapses. The mean percentage increases in EPSP slope in the 2 min immediately after tetanic stimulation (compared with the mean value in the 5 min immediately before tetanic stimulation) were 158.6 ± 6.2 and 120.3 ± 3.1 in the saline-pretreated and IL-1β-pretreated groups, respectively (n = 6 in each case). The mean percentage increases in EPSP slope in the last 5 min of the experiment were 139.3 ± 1.6 and 106.2 ± 1.3 in the saline-pretreated and IL-1β-pretreated groups, respectively (Fig.1A).
Fig. 1.

Intracerebroventricular injection of IL-1β inhibited LTP and the associated increase in glutamate release.A, Tetanic stimulation induced an immediate increase in EPSP slope in both saline-injected and IL-1β-injected rats, although this was attenuated after IL-1β injection. Mean EPSP slope decreased in IL-1β-treated rats so that the value was close to baseline at the end of the 40 min recording period. Sample recordings in the 5 min immediately before tetanic stimulation and in the last 5 min of the experiment are superimposed for saline-injected (left-hand records) and IL-1β-injected (right-hand record) rats. SEM values are included for every 10th response. B, Endogenous glutamate release was significantly increased in synaptosomes prepared from untetanized dentate gyrus (Untet) of saline-injected rats by addition of 40 mm KCl to incubation medium (*p < 0.05; ANOVA), but this was enhanced to a greater degree in synaptosomes prepared from tetanized dentate gyrus (Tet; **p < 0.01; ANOVA). Injection of IL-1β increased unstimulated release in synaptosomes prepared from both untetanized and tetanized dentate gyrus (+p < 0.05; ANOVA; compared with the values obtained from saline-injected rats), but addition of 40 mm KCl to the incubation failed to enhance glutamate release in these preparations. The data are means (± SEM) of six individual experiments.
Analysis of endogenous glutamate release in synaptosomes prepared from tetanized and untetanized tissue obtained from these rats (six rats per group) revealed significant effects of IL-1β injection. Figure1B shows that addition of 40 mmKCl to synaptosomes prepared from untetanized dentate gyrus obtained from saline-treated rats, significantly increased glutamate release (p < 0.05; ANOVA), but a further enhancement of release was observed in synaptosomes prepared from tetanized tissue (p < 0.01; ANOVA). In contrast, KCl failed to stimulate glutamate release in synaptosomes prepared from both untetanized and tetanized tissue obtained from IL-1β-pretreated rats (Fig. 1B). In this experiment, we observed that unstimulated glutamate release was significantly greater in tissue prepared from IL-1β-pretreated rats compared with saline-treated animals (p < 0.05; ANOVA). However, although an increase in unstimulated glutamate release was also observed in a separate experiment (see Fig. 7A), the increase did not reach statistical significance on this second occasion.
Fig. 7.

Dietary manipulation reversed some of the effects of IL-1β. A, Glutamate release was significantly increased by addition of 40 mm KCl to synaptosomes of dentate gyrus prepared from saline-injected rats fed on both the control and experimental diets (*p < 0.05; Student's t test for paired values). This effect was blocked in synaptosomes prepared from dentate gyrus of IL-1β-injected rats that were fed on the control diet, but the attenuation was reversed in synaptosomes prepared from IL-1β-injected rats that were fed on the experimental diet (*p < 0.05; Student's t test for paired values). Activities of JNK (B) and p38 (C) were assessed in aliquots of hippocampal synaptosomes prepared from the same rats. Dietary manipulation did not significantly alter activity of either enzyme in samples prepared from saline-injected rats. However, activities of both JNK and p38 were significantly increased in synaptosomes of IL-1β-injected rats (compare lanes 3 and4 with lanes 1 and 2), as shown by the sample immunoblots and mean data derived from densitometric analysis; (*p < 0.05; Student's t test for paired values). The IL-1β-induced changes in activities of both enzymes were blocked by dietary manipulation (lanes 2 and4). The values are means (± SEM) of six observations in all experiments.
Activities of JNK and p38 were assessed in aliquots of synaptosomal tissue prepared from the hippocampus of the same group of rats. Figure2 demonstrates that tetanic stimulation did not affect JNK (A) or p38 (B) activity in tissue prepared from saline-pretreated rats. Both the sample immunoblot and the mean data obtained from densitometric analysis indicate that the activities of both kinases were enhanced in tissue prepared from IL-1β-pretreated rats (*p < 0.05; †p < 0.01; ANOVA). In the case of p38 activity, the increase was similar in untetanized and tetanized tissue, but in the case of JNK, the increase was more marked in untetanized, compared with tetanized, tissue (*p < 0.05; ANOVA). To confirm that the changes observed in glutamate release and activities of JNK and p38 were, in fact, LTP-associated, we compared these measures in ipsilateral and contralateral dentate gyri prepared from rats that received tetanic stimulation (as described above) or that received the same total number of stimuli to the perforant path but without the high-frequency train of stimuli. Figure3A shows that stimulation at a rate of one shock per 12 sec (low-frequency stimulation) for 50 min did not significantly affect EPSP slope. Figure 3B indicates that addition of KCl stimulated glutamate release to a similar extent in synaptosomes prepared from dentate gyrus of rats that received low-frequency stimulation (both sides) and from the untetanized tissue of rats that received tetanic stimulation (p < 0.05 in all cases; n = 4; ANOVA); however, release was enhanced in synaptosomes prepared from tetanized dentate gyrus (p < 0.01; ANOVA). Figure 3, C andD, indicates that activation of JNK and p38 were similar in all preparations.
Fig. 2.
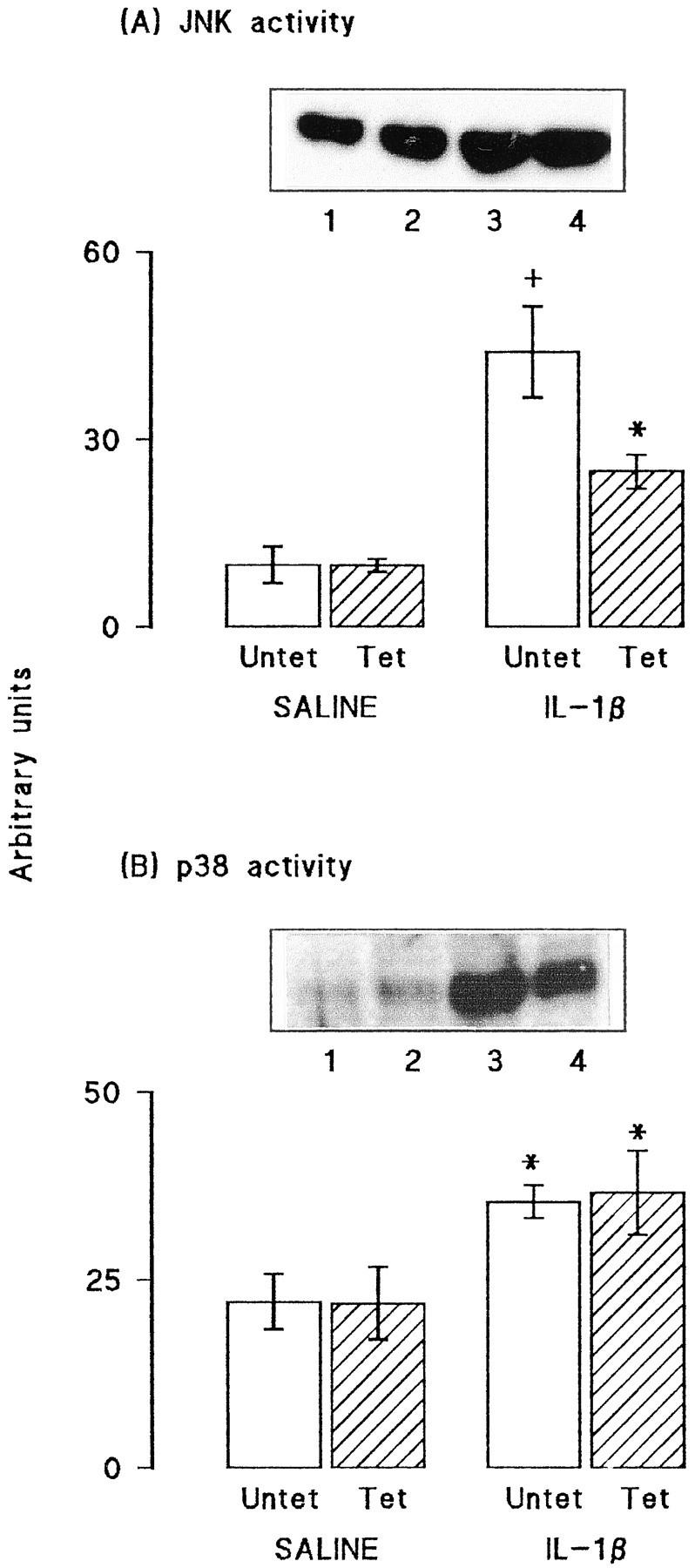
IL-1β increased activities of JNK and p38 in synaptosomes prepared from dentate gyrus. A, JNK activity was significantly increased in synaptosomes prepared from untetanized and tetanized (*p < 0.05; +p < 0.01; ANOVA) dentate gyrus of IL-1β-injected rats compared with either untetanized or tetanized tissue prepared from saline-injected rats. JNK activity was reduced in tetanized, compared with untetanized, tissue after IL-1β injection but was similar in the two preparations obtained from saline-injected rats. B, Activity of p38 tissue was significantly increased in synaptosomes prepared from untetanized and tetanized dentate gyrus of IL-1β-injected rats compared with either untetanized and tetanized tissue obtained from saline-injected rats (*p < 0.05; ANOVA). Tetanic stimulation did not affect enzyme activity in tissue prepared from saline- or IL-1β-treated rats. The data are means of six (± SEM) individual experiments. Sample immunoblots in A andB demonstrate kinase activities in untetanized and tetanized tissue prepared from saline-treated (lanes 1and 2) and IL-1β-treated (lanes 3 and4) rats.
Fig. 3.

Glutamate release and activation of JNK and p38 were not affected by low-frequency stimulation. A, Mean EPSP slope was unchanged by stimulation at a rate of one shock per 12 sec for a 50 min recording period, whereas tetanic stimulation increased mean EPSP slope. B, Addition of KCl significantly increased glutamate release in synaptosomes prepared from dentate gyrus of rats that received low-frequency stimulation (both sides; p < 0.05; ANOVA) and from the untetanized (p < 0.05; ANOVA) and tetanized (p < 0.01; ANOVA) tissue of rats that received tetanic stimulation. C, D, Activation of JNK and p38 were similar in all preparations. Values are means (± SEM) of four separate experiments for JNK and three for p38.
Previous studies have suggested that IL-1β might induce an increase in reactive oxygen species production, and because activities of JNK and p38 have been shown to be increased by both reactive oxygen species and IL-1β in some cell types, we investigated these changes in hippocampal tissue. Figure4A shows that incubation of hippocampal synaptosomes in the presence of IL-1β (1 ng/ml) in vitro significantly increased reactive oxygen species production (p < 0.05; Student'st test for paired values). Because IL-1β may increase unstimulated glutamate release, we assessed the effect of glutamate (50 and 250 μm) on reactive oxygen species production in vitro and found that it was without effect (data not shown). Figure 4B–E shows sample immunoblots and the mean data derived from densitometric analysis in seven separate experiments in which the effects of IL-1β (10 pg/ml) and H2O2 (5 mm) were assessed on activity of JNK and four separate experiments in which effects on p38 activity were assessed. Analysis of the mean data indicated that IL-1β significantly increased activities of both JNK and p38 (p < 0.05 in each case; Student's t test for paired values; Fig.4B,C). Although in the case of p38, SEM values in control and IL-1β-treated tissue overlapped, the difference between the mean values was significant because IL-1β increased p38 phosphorylation in all experiments. H2O2, like IL-1β, significantly increased activities of both kinases (p < 0.05; Student's t test for paired values; Fig. 4D,E).
Fig. 4.

Activities of JNK and p38 were increased by IL-1β and reactive oxygen species. A, IL-1β significantly increased reactive oxygen species (ROS) production in hippocampal synaptosomes prepared from untreated rats (*p < 0.05; Student's t test for paired samples; n = 6). IL-1β (10 pg/ml) significantly increased activities of both JNK (B) and p38 (C), as shown in the sample immunoblots (compare lane 2 with lane 1) and in the mean data that was derived from densitometric analysis of seven separate immunoblots for JNK and four for p38 (p < 0.05 in each case; Student'st test for paired samples). H2O2(5 mm) also significantly increased activities of JNK (D) and p38 (E), as shown in the sample immunoblots (compare lane 2 with lane 1) and in the mean data (± SEM) that was derived from densitometric analysis of six separate immunoblots (p < 0.05 in each case; Student'st test for paired samples).
These data suggested that at least some of the effects of IL-1β might arise from its stimulatory effect on reactive oxygen species production; if this is the case, it might be predicted that the effect of IL-1β will be inhibited by antioxidants. We therefore assessed the effect of intracerebroventricular injection of IL-1β on LTP in rats fed on control diet and on a diet enriched in antioxidant vitamins E and C. Figure 5 demonstrates that the IL-1β-induced impairment in LTP was absent in rats which were fed on the vitamin-enriched diet. In these experiments, the mean percentage increases in EPSP slope in the 2 min immediately after tetanic stimulation (compared with the mean value in the 5 min immediately before to tetanic stimulation) were 143.2 ± 5.6 and 127.1 ± 2.0 in the saline-pretreated and IL-1β-pretreated groups fed on the control diet and 141.0 ± 3.7 and 172.7 ± 9.7 in the saline-pretreated and IL-1β-pretreated groups fed on the vitamin-enriched diet. The corresponding mean percentage increases in EPSP slope in the last 5 min of the experiment were 120.1 ± 1.2 and 107.5 ± 1.6 and 119.3 ± 3.5 and 125.8 ± 2.3 in the saline- and IL-1β-pretreated groups fed on control and vitamin-enriched diets, respectively (Fig. 5); the difference between the values in the two groups fed on the vitamin-enriched diet did not reach statistical significance.
Fig. 5.

The IL-1β-induced impairment in LTP was reversed by dietary manipulation with antioxidant vitamins E and C. LTP was inhibited in IL-1β-injected rats fed on the control diet, but supplementation with a diet enriched in vitamins E and C (see Materials and Methods for details) reversed this effect. Dietary manipulation did not affect LTP in saline-treated rats. The data are derived from six observations in each treatment group, and SEM values are included for every 10th response.
To assess the changes that accompanied dietary manipulation, we analyzed the activity of superoxide dismutase, the formation of reactive oxygen species, and the concentrations of vitamins E and C in hippocampal tissue prepared from saline- and IL-1β-injected rats that were fed on control and vitamin-enriched diets. Figure6 shows that IL-1β significantly increased activity of superoxide dismutase in tissue prepared from rats fed on both control and experimental diets (p < 0.05; Student's t test for paired samples; Fig.6A). We observed that intracerebroventricular injection of IL-1β resulted in a significant increase in reactive oxygen species production in hippocampal synaptosomes prepared from rats fed on the control diet (p < 0.05; Student's t test for independent means) but that dietary manipulation reversed this effect (Fig. 6B). There was a significant increase in vitamin C concentration in hippocampus of rats fed on the experimental, compared with the control, diet (p < 0.05; Student's t test for independent means; Fig. 6C), but vitamin E concentration was unaffected by dietary manipulation (Fig. 6D); IL-1β pretreatment did not alter the concentration of either vitamin.
Fig. 6.

Dietary manipulation reversed the IL-1β-induced increase in reactive oxygen species formation. Intracerebroventricular injection of IL-1β significantly increased activity of superoxide dismutase (SOD; p < 0.05; Student's t test for paired means); dietary manipulation did not affect activity of the enzyme (A). Reactive oxygen species formation (ROS) was significantly increased in hippocampal synaptosomes prepared from IL-1β-injected rats compared with synaptosomes prepared from saline-injected rats (p 0.05; Student's t test for paired means). Dietary manipulation reversed this effect (B). The concentrations of vitamin C (C) and vitamin E (D) were similar in hippocampal tissue prepared from saline-injected and IL-1β-injected rats, but dietary manipulation significantly increased vitamin C concentration in tissue prepared from both groups of rats (p < 0.05; Student's t test for paired means). Vitamin E concentration was unaffected by diet. The values are means (± SEM) of six observations in each case.
Glutamate release and the activities of JNK and p38 were assessed in synaptosomes prepared from dentate gyrus of saline- and IL-1β-treated rats that were fed on the control and vitamin-enriched diet. Figure7A demonstrates that addition of 40 mm KCl significantly increased glutamate release in tissue prepared from saline-injected rats fed on the control and experimental diets (p < 0.05; ANOVA). The KCl-induced enhancement of release was inhibited in tissue prepared from IL-1β-injected rats that were fed on the control diet, but this inhibition was reversed in rats that received the experimental diet, such that KCl-stimulated release was significantly greater than unstimulated release (p < 0.05; ANOVA).
Analysis of the effect of these treatments on activity of JNK revealed a significant increase in enzyme activity in IL-1β-treated rats that were fed on the control diet compared with enzyme activity in saline-treated rats in either dietary group or IL-1β treated rats fed on the experimental diet (p < 0.05; Student'st test; Fig. 7B). In parallel with these effects on JNK activity, we observed that p38 activity was similar in tissue prepared from saline-pretreated rats fed on either diet, whereas enzyme activity was significantly increased in IL-1β-pretreated rats fed on the control diet (p < 0.05; Student'st test for paired values; Fig. 7C). Assessment of p38 activity was made in each of the four conditions (saline-pretreated rats and IL-1β-pretreated rats fed on control and experimental diets) on six separate samples and, in each case, enzyme activity was enhanced in tissue prepared from IL-1β-pretreated rats fed on control diet compared with saline-pretreated rats. Thus, dietary manipulation reversed the effects of IL-1β injection. Sample immunoblots indicate these trends in activities of JNK (Fig. 7B) and p38 (Fig.7C).
DISCUSSION
We set out to investigate the downstream consequences of an increase in IL-1β in hippocampus, with the objective of increasing our understanding of the mechanisms that might contribute to impairment of LTP in dentate gyrus. The data indicate that among the cellular consequences of increased IL-1β concentration that accompany the impairment in LTP is an increase in reactive oxygen species production with the consequent increased activity of JNK and p38. On the basis of the evidence presented we propose that the IL-1β-induced increase in reactive oxygen species production increases activation of JNK and p38 which, in turn, inhibits LTP. IL-1β-induced inhibition of LTP may also result from the observed inhibition of glutamate release. This working hypothesis is presented in schematic form in Figure8.
Fig. 8.

Scheme suggesting cascade of events leading to the IL-1β-induced impairment in LTP. Intracerebroventricular injection of IL-1β leads to an increase in reactive oxygen species production that increases activity of JNK and p38. We propose that glutamate release is compromised by activation of IL-1 receptors, one consequence of which is activation of these kinases, and that this inhibition of glutamate release significantly contributes to the IL-1β-induced impairment in LTP.
Consistent with this hypothesis, we report that the inhibitory effect of IL-1β on LTP in perforant path-granule cell synapses was accompanied by increased activity of both JNK and p38. The IL-1β-induced attenuation of LTP confirms our previous observations (Murray and Lynch, 1998a) and also confirms the results ofin vitro experiments that demonstrated that IL-1β inhibited LTP in dentate gyrus (Cunningham et al., 1996), CA1 (Bellinger et al., 1993), and CA3 (Katsuki et al., 1990). Coupled with the IL-1β-induced inhibition of LTP, we observed an IL-1β-induced inhibition of KCl-induced glutamate release. Thus, whereas an increase in KCl-stimulated release was observed in synaptosomes prepared from tetanized tissue obtained from saline-treated rats, KCl-stimulated release was markedly decreased in synaptosomes prepared from both untetanized and tetanized tissue obtained from IL-1β-treated rats. These data support the hypothesis that LTP in dentate gyrus is tightly coupled with an increase in glutamate release at perforant path-granule cell synapses (Canevari et al., 1994; McGahon and Lynch, 1996; McGahon et al., 1999). We observed that IL-1β increased unstimulated release of glutamate, however this change did not always reach statistical significance.
We observed that the inhibitory effect of IL-1β on LTP was coupled with a stimulatory effect on the activities of JNK and p38. Thus, whereas activity of both kinases was unaffected by tetanic stimulation, both were markedly increased in synaptosomes prepared from hippocampus obtained from rats that were injected with IL-1β. One interpretation of this finding is that IL-1β activates these kinases and that this action may result in inhibition of LTP; this is consistent with the reported inhibitory effect of the p38 inhibitor SB203580 on IL-1β-induced attenuation of LTP (Coogan et al., 1997). To address this question directly, we analyzed the effect of IL-1β on activity of JNK and p38 in vitro and found that activities of both kinases were increased by incubation in the presence of the cytokine. A number of groups using different cell types have reported such an action. For example, IL-1β has been reported to increase activity of JNK in human glomerular mesangial (Uciechowski et al., 1996) and HeLa (Raingeaud et al., 1995) cells, whereas IL-1β-induced activation of p38 has been reported in Chinese hamster CCl39 (Guay et al., 1997) and HeLa (Raingeaud et al., 1995) cells.
It has been suggested that certain effects of IL-1β might be mediated through an increase in reactive oxygen species production (Sumoski et al., 1989; Raingeaud et al., 1995; Murray and Lynch, 1998b), and in this study we provide further evidence to support this view by demonstrating that (1) IL-1β increases reactive oxygen species formation in hippocampal tissue and (2) hydrogen peroxide, which leads to formation of the hydroxyl radical (Qin et al., 1999), mimics the effect of IL-1β by stimulating JNK and p38 in vitro. These findings are consistent with the observation that UV radiation (Zhang et al., 1997) and osmotic stress (Qin et al., 1999), which also induce formation of reactive oxygen species, stimulate activities of both JNK and p38 (Raingeaud et al., 1995) in HeLa cells. These results support our working hypothesis; if IL-1β concentration in hippocampus is increased, reactive oxygen species formation is increased, stress-activated kinases are activated, and expression of LTP is blocked. Although the data, in particular the in vitroanalyses, are consistent with the idea that these effects may be sequential, this remains to be unequivocally established.
To challenge this hypothesis, it is reasonable to propose that if IL-1β acts by stimulating reactive oxygen species production, then subsequent actions of IL-1β will be inhibited by antioxidants. We attempted to address this question by feeding groups of rats with the antioxidant vitamins E and C before IL-1β injection. The data show that the IL-1β-induced inhibition of LTP was blocked by dietary manipulation, leading us to the conclusion that the IL-1β effect is mediated through reactive oxygen species production. This is supported by the work of Pellmar et al. (1991), who reported that hydrogen peroxide inhibits LTP in guinea pig CA1 in vitro. The data presented here suggest that the increase in reactive oxygen species production in hippocampus of IL-1β-treated rats is a consequence of increased superoxide dismutase activity, which is consistent with previous reports of an IL-1β-induced upregulation of Mn-superoxide dismutase gene expression (Antras-Ferry et al., 1997), mRNA (Borg et al., 1992), and enzyme activities in various cell preparations (Borg et al., 1992). Analysis of hippocampal tissue prepared from rats fed on control and vitamin-enriched diets revealed that reactive oxygen species production was increased in tissue prepared from IL-1β-treated rats that were given the control diet, but that this IL-1β-induced change was blocked in vitamin-treated rats. No change in tissue concentration of vitamin E was observed after dietary manipulation, but this is not surprising because it has been shown that long-term treatment with this lipid-soluble vitamin is necessary for its incorporation into the membrane (Halliwell 1992; Murray and Lynch, 1998b). These findings are consistent with previous observations from this laboratory in which we found that IL-1β concentration and reactive oxygen species production were increased in hippocampal tissue prepared from aged rats, which demonstrated an impaired ability to sustain LTP (McGahon et al., 1997; Murray and Lynch, 1998a,b). They are also consistent with the observation that dietary manipulation with vitamins E and C for 3 months restored ability of aged rats to sustain LTP (Murray and Lynch, 1998b). Predictably, dietary manipulation did not effect IL-1β-induced stimulation of superoxide dismutase, therefore we propose that the enhanced tissue concentration of vitamin C that was observed after dietary manipulation was responsible for reversing the IL-1β-induced increase in reactive oxygen species production.
To obtain further evidence in support of the hypothesis that antioxidant treatment reverses the effects of IL-1β on hippocampal function, we analyzed glutamate release and activity of JNK and p38 in synaptosomes prepared from saline-treated and IL-1β-treated rats fed on the control and vitamin-enriched diets. The data showed that the inhibitory effect of IL-1β on release was blocked by dietary manipulation. Coupled with this change, we observed that dietary manipulation reversed the IL-1β-induced increases in activities of JNK and p38. These results suggest that the increase in tissue vitamin C induced by dietary manipulation prevents the stimulatory effect of IL-1β and its subsequent inhibitory effect on glutamate release. If the argument that the inhibitory effect of IL-1β on LTP is a consequence of activation of JNK and/or p38, then it must be predicted that LTP will be inhibited, and activities of JNK and/or p38 will be increased when endogenous IL-1β concentration in hippocampus is increased, for example in hippocampal tissue prepared from aged rats (Murray and Lynch, 1998a,b). We have recently observed that JNK activity and p38 activity are increased in hippocampal tissue prepared from aged rats (O'Donnell et al., 2000), in which IL-1β concentration and reactive oxygen species production are increased (Lynch, 1998).
Increased activation of both JNK and p38 have been associated with cell death (Yang et al., 1997; Luo et al., 1998), and evidence suggests that IL-1β plays a role in cell death induced by ischemia (Rothwell, 1999). It might be speculated that, under the current experimental conditions, IL-1β stimulates JNK and/or p38 and that cell damage might result from this action. This proposal concurs with our recent finding that parenteral administration of lipopolysaccharide increased IL-1β concentration and cell degeneration in entorhinal cortex and that these changes were inhibited by intracerebroventricular injection of a caspase-1 inhibitor indicating a key role for IL-1β (Campbell et al., 2000).
We propose that the inhibitory effect of IL-1β on LTP in perforant path-granule cell synapses is a consequence of an increase in reactive oxygen species formation, and the evidence suggests that the primary effect of IL-1β is to stimulate activity of superoxide dismutase. Our evidence suggests that increased activation of JNK and p38 represent critical downstream events of the increase in reactive oxygen species, leading to the inhibitory effect of IL-1β.
Footnotes
This work was supported by the Health Research Board (Ireland).
Correspondence should be addressed to Marina A. Lynch at the above address. E-mail: lynchma@tcd.ie.
REFERENCES
- 1.Antras-Ferry J, Maheo K, Morel F, Guillouzo A, Cillard P, Cillard J. Dexamethasone differently modulates TNF-alpha- and IL-1 beta-induced transcription of the hepatic Mn-superoxide dismutase gene. FEBS Lett. 1997;403:100–104. doi: 10.1016/s0014-5793(97)00033-1. [DOI] [PubMed] [Google Scholar]
- 2.Ban E, Milon G, Prudhomme N, Fillion G, Haour F. Receptors for interleukin-1 (α and β) in mouse brain: mapping and neuronal localization in hippocampus. Neuroscience. 1991;43:21–30. doi: 10.1016/0306-4522(91)90412-h. [DOI] [PubMed] [Google Scholar]
- 3.Bellinger FP, Madamba S, Siggins GR. Interleukin-1β inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 4.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 5.Borg LA, Cagliero E, Sandler S, Welsh N, Eizirik DL. Interleukin-1β increases the activity of superoxide dismutase in rat pancreatic islets. Endocrinology. 1992;130:2851–2857. doi: 10.1210/endo.130.5.1533363. [DOI] [PubMed] [Google Scholar]
- 6.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Campbell V, Roche E, Lynch MA. Intraperitoneal administration of lipopolysaccharide induces apoptosis in rat entorhinal cortex. J Physiol (Lond) 2000;523:189. [Google Scholar]
- 8.Canevari L, Richter-Levin G, Bliss TVP. LTP in the dentate gyrus is associated with a persistent NMDA receptor-dependent enhancement of synaptosomal glutamate release. Brain Res. 1994;667:115–117. doi: 10.1016/0006-8993(94)91720-5. [DOI] [PubMed] [Google Scholar]
- 9.Coogan A, Lynch MA, O'Connor JJ. The p38 MAP kinase inhibitor, SB203580, antagonises the effect of IL-1β on LTP in the rat dentate gyrus. Soc Neurosci Abstr. 1997;2:1511. [Google Scholar]
- 10.Creedon DJ, Johnson EM, Jr, Lawrence JC., Jr Mitogen-activated protein kinase-independent pathways mediate the effect of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham AJ, Murray CA, O'Neill LAJ, Lynch MA, O'Connor JJ. Interleukin-1β (IL-1β) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:1–4. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- 12.Derijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 13.Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110:357–368. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- 14.Haan EA, Bowen DM. Protection of neocortical prisms from freeze-thaw injury by dimethylsulphhoxide. J Neurochem. 1981;37:243–246. doi: 10.1111/j.1471-4159.1981.tb05315.x. [DOI] [PubMed] [Google Scholar]
- 15.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 16.Junger WG, Hoyt DB, Hamreus M, Liu FC, Herdon-Remelius C, Junger W, Altman A. Hypertonic saline activates protein tyrosine kinases and mitogen-activated protein kinase p38 in T cells. J Trauma Injury Infect Crit Care. 1997;42:437–445. doi: 10.1097/00005373-199703000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M. Interleukin-1β inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol. 1990;181:323–326. doi: 10.1016/0014-2999(90)90099-r. [DOI] [PubMed] [Google Scholar]
- 18.Lechan RM, Toni R, Clark BD, Cannon JG, Shaw AR, Dinarello CA, Reichlin S. Immunoreactive interleukin-1β localization in the rat forebrain. Brain Res. 1990;514:135–140. doi: 10.1016/0006-8993(90)90445-h. [DOI] [PubMed] [Google Scholar]
- 19.Lebel CP, Bondy SC. Sensitive and rapid quantitation of oxygen reactive species formation in rat synaptosomes. Neurochem Int. 1990;17:435–440. doi: 10.1016/0197-0186(90)90025-o. [DOI] [PubMed] [Google Scholar]
- 20.Lu G, Beuerman RW, Zhao S, Sun G, Nguyen DH, Ma S, Kline DG. Tumour necrosis factor-alpha and interleukin-1 induce activation of MAP kinase and SAP kinase in human neuroma fibroblasts. Neurochem Int. 1997;30:401–410. doi: 10.1016/s0197-0186(96)00075-7. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y, Umegaki H, Wang X, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- 22.Lynch MA. Age-related impairment in long-term potentiation in hippocampus: a role for the cytokine, interleukin-1β? Prog Neurobiol. 1998;56:1–19. doi: 10.1016/s0301-0082(98)00054-9. [DOI] [PubMed] [Google Scholar]
- 23.Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signalling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGahon BM, Lynch MA. The synergism between metabotropic glutamate receptor activation and arachidonic acid on glutamate release is occluded by induction of long-term potentiation in dentate gyrus. Neuroscience. 1996;72:847–855. doi: 10.1016/0306-4522(95)00579-x. [DOI] [PubMed] [Google Scholar]
- 25.McGahon B, Clements MP, Lynch MA. The ability of aged rats to sustain long-term potentiation is restored when the age-related decrease in membrane arachidonic acid concentration is reversed. Neuroscience. 1997;81:9–16. doi: 10.1016/s0306-4522(97)00116-4. [DOI] [PubMed] [Google Scholar]
- 26.McGahon BM, Martin DSD, Horrobin DF, Lynch MA. Age-related changes in synaptic function: analysis of the effect of dietary supplementation with ω-3 fatty acids. Neuroscience. 1999;94:305–314. doi: 10.1016/s0306-4522(99)00219-5. [DOI] [PubMed] [Google Scholar]
- 27.Murray CA, McGahon B, McBennett S, Lynch M. Interleukin-1β inhibits glutamate release in hippocampal synaptosomes prepared from young, but not aged, rats. Neurobiol Aging. 1997;18:343–348. doi: 10.1016/s0197-4580(97)80317-x. [DOI] [PubMed] [Google Scholar]
- 28.Murray C, Lynch MA. Analysis of the mechanism by which dietary supplementation with vitamin E and vitamin C restores ability of aged animals to sustain long-term potentiation in dentate gyrus. J Biol Chem. 1998a;273:12161–12168. doi: 10.1074/jbc.273.20.12161. [DOI] [PubMed] [Google Scholar]
- 29.Murray C, Lynch MA. Evidence that increased hippocampal expression of the cytokine, IL-1β, is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci. 1998b;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Donnell E, Vereker E, Lynch MA. Age-related impairment in LTP is accompanied by enhanced activity of stress-activated protein kinases: Analysis of underlying mechanisms. Eur J Neurosci. 2000;12:345–352. doi: 10.1046/j.1460-9568.2000.00900.x. [DOI] [PubMed] [Google Scholar]
- 31.Omaye ST, Turnbull JD, Sauberlich HE. Selected methods for the determination of ascorbic acid in animal cells, tissues and fluids. In: McCormick DB, Wright LD, editors. Methods in enzymology, Vol 62. Academic; New York: 1979. pp. 3–13. [DOI] [PubMed] [Google Scholar]
- 32.Ordronneau P, Abdullah L, Petruse P. An efficient enzyme immunoassay for glutamate using glutaraldehyde coupling of the hapten to microtiter plates. J Immunol Meth. 1991;142:169–176. doi: 10.1016/0022-1759(91)90103-m. [DOI] [PubMed] [Google Scholar]
- 33.Park DS, Stefanis L, Yan CYI, Farinelli SE, Greene LA. Ordering the cell death pathway. J Biol Chem. 1996;271:36:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- 34.Parnet PM, Amindari S, Wu C, Brunke-Reese D, Goujon E, Weyhenmeyer JA, Danzer R, Kelley KW. Expression of type I and type II interleukin-1 receptors in mouse brain. Brain Res Mol Brain Res. 1994;27:63–70. doi: 10.1016/0169-328x(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 35.Pellmar TC, Hollinden GE, Sarvey JM. Free radicals accelerate the decay of long-term potentiation in field CA1 of guinea pig hippocampus. Neuroscience. 1991;44:353–359. doi: 10.1016/0306-4522(91)90060-2. [DOI] [PubMed] [Google Scholar]
- 36.Plata-Salaman CR, ffrench-Mullen JMH. Interleukin-1β inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur J Pharmacol. 1994;266:1–10. doi: 10.1016/0922-4106(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 37.Qin S, Ding J, Takano T, Yamamura H. Involvement of receptor aggregation and reactive oxygen species in osmotic stress-induced Syk activation in B cells. Biochem Biophys Res Comm. 1999;262:231–236. doi: 10.1006/bbrc.1999.1079. [DOI] [PubMed] [Google Scholar]
- 38.Rada P, Mark GP, Vitek MP, Manago RM, Blume AJ, Beer B, Hoebel BG. Interleukin-1β decreases acetylcholine measured by microdialysis in the hippocampus of freely moving rats. Brain Res. 1991;550:287–290. doi: 10.1016/0006-8993(91)91330-4. [DOI] [PubMed] [Google Scholar]
- 39.Raingeaud J, Gutpa S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Proinflammatory cytokines and environmental stress cause p38 mitogen activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 40.Rizzo MT, Carlo-Stella C. Arachidonic acid mediates interleukin-1 and tumour necrosis factor-α-induced activation of the c-jun amino-terminal kinases in stromal cells. Blood. 1996;88:3792–3800. [PubMed] [Google Scholar]
- 41.Rothwell NJ. Cytokines: killers in the brain? J Physiol (Lond) 1999;514:3–17. doi: 10.1111/j.1469-7793.1999.003af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seger R, Krebs EG. The MAPK signalling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 43.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 44.Sumoski W, Baquerizo H, Rabinovitch A. Oxygen free radical scavengers protect rat islet cells from damage by cytokines. Diabetologia. 1989;32:792–796. doi: 10.1007/BF00264909. [DOI] [PubMed] [Google Scholar]
- 45.Uciechowski P, Saklatvala J, von der Ohe J, Resch K, Szamel M, Kracht M. Interleukin-1 activates jun-N-terminal kinases JNK1 and JNK2 but not extracellular-regulated MAP kinase (ERK) in human glomerular mesangial cells. FEBS Lett. 1996;394:273–278. doi: 10.1016/0014-5793(96)00967-2. [DOI] [PubMed] [Google Scholar]
- 46.Vatassery GT. In vitro oxidation of vitamins C and E, cholesterol and thiols in rat brain synaptosomes. Lipids. 1994;30:1007–1013. doi: 10.1007/BF02536285. [DOI] [PubMed] [Google Scholar]
- 47.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 48.Yang DD, Kuan C-Y, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–869. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Rosenstein BS, Wang Y, Lebwohl M, Wei H. Identification of possible reactive oxygen species involved in ultraviolet radiation-induced oxidative DNA damage. Free Radic Biol Med. 1997;23:980–985. doi: 10.1016/s0891-5849(97)00126-3. [DOI] [PubMed] [Google Scholar]