

Abstract
The nonopioid actions of spinal dynorphin may promote aspects of abnormal pain after nerve injury. Mechanistic similarities have been suggested between opioid tolerance and neuropathic pain. Here, the hypothesis that spinal dynorphin might mediate effects of sustained spinal opioids was explored. Possible abnormal pain and spinal antinociceptive tolerance were evaluated after intrathecal administration of [d-Ala2,N-Me-Phe4, Gly-ol5]enkephalin (DAMGO), an opioid μ agonist. Rats infused with DAMGO, but not saline, demonstrated tactile allodynia and thermal hyperalgesia of the hindpaws (during the DAMGO infusion) and a decrease in antinociceptive potency and efficacy of spinal opioids (tolerance), signs also characteristic of nerve injury. Spinal DAMGO elicited an increase in lumbar dynorphin content and a decrease in the μ receptor immunoreactivity in the spinal dorsal horn, signs also seen in the postnerve-injury state. Intrathecal administration of dynorphin A(1–17) antiserum blocked tactile allodynia and reversed thermal hyperalgesia to above baseline levels (i.e., antinociception). Spinal dynorphin antiserum, but not control serum, also reestablished the antinociceptive potency and efficacy of spinal morphine. Neither dynorphin antiserum nor control serum administration altered baseline non-noxious or noxious thresholds or affected the intrathecal morphine antinociceptive response in saline-infused rats. These data suggest that spinal dynorphin promotes abnormal pain and acts to reduce the antinociceptive efficacy of spinal opioids (i.e., tolerance). The data also identify a possible mechanism for previously unexplained clinical observations and offer a novel approach for the development of strategies that could improve the long-term use of opioids for pain.
Keywords: dynorphin, abnormal pain, morphine tolerance, spinal, dynorphin antiserum, μ-opioid receptors
An important clinical observation is that sustained spinal administration of μ-opioids produces abnormal pain that is reminiscent of neuropathic pain (Woolf, 1981; Ali, 1986;Arner et al., 1988; Yaksh and Harty, 1988; De Conno et al., 1991;Trujillo and Akil, 1991; Devulder, 1997). Paradoxical opioid-induced novel pain often occurs in areas isolated from the site of the original pain complaint (De Conno et al., 1991). Additionally, decreased analgesia after sustained spinal μ-opioids is similar to the diminished effects of spinal μ-opioids in animal models of neuropathy. Thus, both opioid tolerance and neuropathic conditions share features of diminished μ-opioid analgesia with abnormal pain (thermal hyperalgesia and tactile allodynia). These common features have led to suggestions of common mechanisms in nerve-injury and in spinal μ-opioid tolerance states (Mao et al., 1994, 1995a,b).
A similarity linking abnormal pain from opioids or nerve injury is the sensitivity to NMDA antagonists. NMDA antagonists reverse thermal hyperalgesia after nerve injury (Mao et al., 1995a,b; Wegert et al., 1997; Bian et al., 1999). Additionally, the loss of morphine antinociception in rats with spinal nerve injury was restored by intrathecal MK-801, an NMDA antagonist (Ossipov et al., 1995a,b). Similarly, tolerance and hyperalgesia produced by spinal injections and/or infusions of morphine were prevented by MK-801 (Mao et al., 1994) or dextromethorphan (Trujillo and Akil, 1991; Tiseo and Inturrisi, 1993; Mao et al., 1994; Tiseo et al., 1994; Manning et al., 1996); MK-801 did not produce antinociception alone and did not increase morphine antinociception in nontolerant rats.
Although the blockade of nerve-injury pain by NMDA antagonists may be caused by the blockade of afferent discharge from injured nerves, the mechanism by which NMDA antagonists block or reverse opioid tolerance is not understood because the source of the NMDA receptor activation during chronic opioid exposure is uncertain. NMDA receptors may coexist on cells expressing opioid receptors, including primary afferent fibers (Liu et al., 1994, 1997). Primary afferent neurons do not tend to discharge spontaneously and are probably not essential to the development of opioid tolerance (Price, 1988), suggesting the importance of intracellular pathways as a potential mechanism by which NMDA antagonists might affect tolerance. Another possibility may be that an intermediary might mechanistically link opioid tolerance and nerve injury directly or indirectly via the NMDA receptor.
A feature of both nerve-injury pain and opioid treatment is increased spinal dynorphin (Cho and Basbaum, 1989; Kajander et al., 1990; Draisci et al., 1991; Dubner, 1991; Dubner and Ruda, 1992; Rattan and Tejwani, 1997). Dynorphin might be pronociceptive in chronic pain states (Bian et al., 1999; Malan et al., 2000). Possible pronociceptive actions by spinal dynorphin are supported because dynorphin antiserum, but not control serum, (1) blocks nerve-injury hyperalgesia (Wegert et al., 1997), (2) restores spinal morphine antinociception after nerve injury (Nichols et al., 1997; Wegert et al., 1997), (3) elicits spinal morphine antiallodynia, and (4) restores the expected synergy of spinal/supraspinal morphine that is lost after nerve injury (Bian et al., 1999). The effects of dynorphin A(1–17) antiserum parallel those seen with MK-801 and suggest that dynorphin may interact, directly or indirectly, with the NMDA receptor (Wegert et al., 1997; Bian et al., 1999; Tang et al., 1999).
Here, the hypothesis that abnormal pain and decreased spinal opioid antinociception seen after sustained opioid exposure might be mediated by spinal dynorphin was tested.
MATERIALS AND METHODS
Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN), 200–300 gm at the time of testing, were maintained in a climate-controlled room on a 12 hr light/dark cycle (lights on at 06:00 A.M.) with food and water available ad libitum. All of the testing was performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines for the handling and use of laboratory animals and received approval from the Animal Care and Use Committee of the University of Arizona. Groups of 5–10 rats were used in all experiments.
Cannulation and drug administration. While under halothane anesthesia, all of the rats were implanted with intrathecal catheters (polyethylene-10 tubing; 7.5 cm) as described previously (Yaksh and Rudy, 1976) for drug administration at the level of the lumbar spinal cord. Chronic spinal infusions were performed with osmotic minipumps (Alza, Mountain View, CA). The osmotic pumps delivered saline at 1 μl/hr or [d-Ala2,N-Me-Phe4, Gly-ol5]enkephalin (DAMGO) at 1 nmol · μl−1 · hr−1for 7 d. The minipumps were attached to the indwelling intrathecal catheters and placed in the subcutaneous space. On day 6 all animals were tested for tactile allodynia and thermal hyperalgesia while the infusions continued. On day 7 the minipumps were disconnected from the infusion catheter while the animals were briefly anesthetized with halothane and the infusion catheter was exteriorized. Morphine, control serum, or antiserum to dynorphin A(1–17) (200 μg per injection) was injected through the intrathecal catheter in a volume of 5 μl followed by a 9 μl saline flush.
Nociceptive/allodynic testing. Paw withdrawal thresholds were determined in response to probing with von Frey filaments in a range normally not found to be noxious (i.e., 0.40, 0.70, 1.20, 2.00, 3.63, 5.50, 8.50, and 15.1 gm). Each filament was applied perpendicularly to the plantar surface of the right paw of rats kept in suspended wire-mesh cages. The paw withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (“up-and-down” method) and analyzed with a Dixon nonparametric test (Dixon, 1980; Chaplan et al., 1994). Tactile allodynia was indicated by a significant (p ≤ 0.05; Student'st test) reduction in the paw withdrawal threshold when compared with that obtained before any manipulations. Tactile allodynia was measured on day 6 of DAMGO infusion while the DAMGO infusion was maintained.
Thermal hyperalgesia was determined by focusing a radiant heat source onto the plantar aspect of a hindpaw of the rat. After paw withdrawal, a photodetection device interrupted both the stimulus and the timer. A maximal cutoff of 40 sec was used to prevent tissue damage. Paw withdrawal latencies were determined to the nearest 0.1 sec. Hyperalgesia was indicated by a significantly (p ≤ 0.05; Student's t test) shorter paw withdrawal latency than that detected before any manipulations (i.e., preinfusion). Thermal hyperalgesia was measured on day 6 of DAMGO infusion while the DAMGO infusion was maintained. Antihyperalgesia was indicated by a return of the response latencies to preinfusion baseline values, and antinociception was indicated by a significant (p ≤ 0.05) increase in withdrawal latencies above the normal baseline values. In both tactile allodynia and thermal hyperalgesia testing, animals were acclimated to their surroundings for 30 min and tested only once.
Nociceptive testing was performed by placing the distal third of the tail of a rat in a water bath maintained at 52°C. The latency to withdrawal was measured to 0.1 sec, and a cutoff latency of 10 sec was used to prevent tissue injury. The tail-flick test was used to determine the antinociceptive A90 dose (the dose estimated to produce 90% antinociception) of either intrathecal DAMGO or morphine in rats before and after a 7 d DAMGO infusion. Tolerance to the antinociceptive effect of opioids was indicated by a significant reduction in the tail-flick latency after challenge with an A90 dose. Data were converted to percent antinociception to generate dose–response curves by the following formula: (response latency − baseline latency)/(cutoff − baseline latency) × 100. Animals received either intrathecal morphine alone or morphine 10 min after intrathecal pretreatment with either control serum or antiserum to dynorphin A(1–17) (200 μg/5 μl) and were tested 10 min later by the use of the tail-flick test (i.e., dynorphin antiserum given 20 min before the test).
Dynorphin A immunoassay. Rats were deeply anesthetized with ether and decapitated on day 7 of DAMGO infusion. The spinal cord was injected with ice-cold saline and placed on an iced glass Petri dish, and the lumbar cord was rapidly dissected. These tissue samples were immediately frozen on dry ice and stored at −70°C. Thawed tissue was placed in 1N acetic acid, disrupted with a Polytron homogenizer, and incubated for 20 min at 95°C. After centrifugation at 10,000 ×g for 20 min (4°C), the supernatant was lyophilized and stored at −70°C. Protein concentrations were determined by the use of the bicinchoninic acid method with bovine serum albumin as a standard. Immunoassay was performed by the use of a commercial enzyme immunoassay kit with an antibody specific for dynorphin A(1–17) (Peninsula Laboratories, Belmont, CA). Standard curves were constructed and the dynorphin content was determined with Graph Pad Prism (San Diego, CA). Pairwise comparisons between treatments were detected by Student's t test. Significance was determined at thep < 0.05 level.
Immunohistochemistry. The naive, saline-infused, and DAMGO-infused rats were deeply anesthetized with ketamine and perfused transcardially with 200 ml of PBS, pH 7.4, containing heparin (1500 IU/l), followed by 500 ml of cold 4% paraformaldehyde. After perfusion the spinal cords were isolated and post-fixed for 4 hr in 4% paraformaldehyde and then cryoprotected with 30% sucrose in PBS overnight at 4°C.
Frontal frozen sections (40 μm) were prepared from the lumbar enlargement of the spinal cord. These sections were immunolabeled either with a guinea pig antiserum against prodynorphin or with a rabbit antiserum against the rat μ-opioid receptor (MOR; antisera were kindly provided by Dr. Robert Elde, University of Minnesota). Briefly, the spinal cord sections were rinsed twice for 5 min each in PBS and then preincubated with PBS containing 4% normal goat serum, 0.3% Triton X-100, and 1% bovine serum albumin for 30 min at room temperature. The sections were then incubated with the primary antiserum diluted in the preincubation buffer overnight at 4°C (prodynorphin antiserum at 1:40,000 dilution; MOR antiserum at 1:20,000 dilution). The sections were washed three times for 10 min each in PBS, followed by incubation with a biotinylated secondary antibody (goat anti-guinea pig IgG or goat anti-rabbit IgG; Vector Laboratories, Burlingame, CA) at 1:1000 in PBS with 0.25% bovine serum albumin and 0.1% Triton X-100 (PBS-BT) for 60 min at room temperature. Sections were washed three times for 10 min each in PBS and stained with the avidin–biotin complex (ABC kit; Vector Laboratories); sections were incubated with an avidin-biotinylated horseradish peroxidase complex diluted 1:500 in PBS-BT for 2 hr at room temperature, washed three times for 10 min each in PBS, and developed with a solution of diaminobenzidine and H2O2(FAST DAB SETS; Sigma, St. Louis, MO) maintained uniformly throughout the experiments. Sections from control and treated animals were processed in parallel under identical experimental conditions. The sections were washed and mounted on glass slides, air-dried overnight, rinsed in histological clearing solvent, and coverslipped with DPX.
Chemicals. Morphine sulfate was purchased from Sigma. DAMGO was purchased from Research Biochemicals (Natick, MA). Antiserum to dynorphin A(1–17) was obtained from Peninsula Laboratories. This rabbit antiserum was 100% cross-reactive with dynorphin A(1–13) and dynorphin A(1–8). Control serum was serum collected from the same species of rabbits without injection of antigen but not from the same rabbit used to generate dynorphin antiserum. All chemicals were dissolved in normal saline.
Data analysis. The data were converted to the percent of maximal possible effect (% MPE) by the formula: % MPE = (WT − CT)/(CO − CT) × 100, where WT is the withdrawal threshold or latency obtained experimentally, CT is the baseline control value before drug administration, and CO is the cutoff value (i.e., 15 gm for tactile allodynia, 40 sec for the paw-flick test, and 10 sec for the tail-flick test). Dose–response curves were generated where possible, and the A50 value (the dose estimated to produce 50% MPE) and 95% confidence intervals were determined for additional statistical calculations (Tallarida and Murray, 1987).
RESULTS
Spinal infusion of the μ-opioid agonist DAMGO over a 7 d period elicited antinociceptive tolerance to a subsequent challenge with intrathecal DAMGO (1 μg) and cross-tolerance to challenge with intrathecal morphine (10 μg) (Fig. 1). The effect produced by DAMGO in the tail-flick test before the start of the DAMGO infusion was 92 ± 4% antinociception. The same dose of DAMGO given immediately after disconnection of the intrathecal catheter from the infusion pump on day 7 produced a significantly reduced antinociceptive response of 12 ± 4% antinociception in the tail-flick test, indicating the development of tolerance to the antinociceptive actions of DAMGO (p ≤ 0.05; Student's t test). Similarly, intrathecal morphine given before the intrathecal DAMGO infusion produced 91 ± 6% antinociception in the tail-flick test, whereas the same morphine dose given to animals receiving spinal DAMGO infusion over a 7 d period resulted in a significantly reduced antinociceptive effect of 25 ± 6% antinociception, indicating cross-tolerance between DAMGO and morphine (p ≤ 0.05; Student's ttest). The antinociceptive effect of intrathecal morphine was not altered in animals receiving chronic saline via minipumps over a 7 d period; intrathecal morphine (10 μg) produced 88 ± 8.6 and 89 ± 11.4% antinociception before and after saline infusion, respectively.
Fig. 1.

Left, Male Sprague Dawley rats received an intrathecal infusion of saline or of DAMGO (1 nmol · μl−1 · hr−1) for 7 d. The spinal cords were removed and assayed for dynorphin content with an enzyme immunoassay. The rats with DAMGO infusions showed a significant (*p ≤ 0.05; Student'st test) increase in spinal dynorphin content when compared with saline-infused rats. Right, Antinociceptive tolerance on day 8 to intrathecal DAMGO (1 μg/5 μl;n = 8; top) or cross-tolerance to intrathecal morphine (10 μg/5 μl; n = 8;bottom) in the 52°C water tail-flick test is demonstrated.
The content of dynorphin was quantitated by immunoprecipitation using an antiserum raised against dynorphin A(1–17). In animals that had received an intrathecal infusion of DAMGO, the level of dynorphin in the dorsal half of the lumbar spinal cord showed a significant increase when compared with that in saline-infused animals (1700 ± 120 pg/mg of protein vs 1297 ± 95 pg/mg of protein in DAMGO-treated and saline control animals, respectively; p ≤ 0.05 by Student's t test). These data were based on an average of six to eight rats. To characterize this apparent elevation of spinal dynorphin in DAMGO-infused rats further, the distribution of its precursor peptide prodynorphin was also examined by immunohistochemistry using an antiserum specific for prodynorphin. The immunoreactivity (-IR) of prodynorphin was found mainly in the superficial laminae, laminae IV and V, and lamina X of the dorsal horn of the lumbar spinal cord (Fig.2). In the superficial laminae, discrete cell bodies and numerous fibers were labeled (Fig.2A,B), whereas in laminae V and VI (Fig.2C,D) and lamina X (Fig. 2E,F), prodynorphin-IR was predominantly associated with fibers. Lumbar spinal sections from DAMGO-infused rats exhibited a reduction of prodynorphin-IR in all the laminae stated above when compared with the saline-infused control (Fig. 2). This reduction appeared to be primarily caused by a loss of fiber staining in these laminae (Fig.2B,D,F). In the superficial laminae, staining was still clearly present in cell bodies; the density of immunolabeled cell bodies was similar between control and DAMGO-infused rats (Fig.2A,B).
Fig. 2.
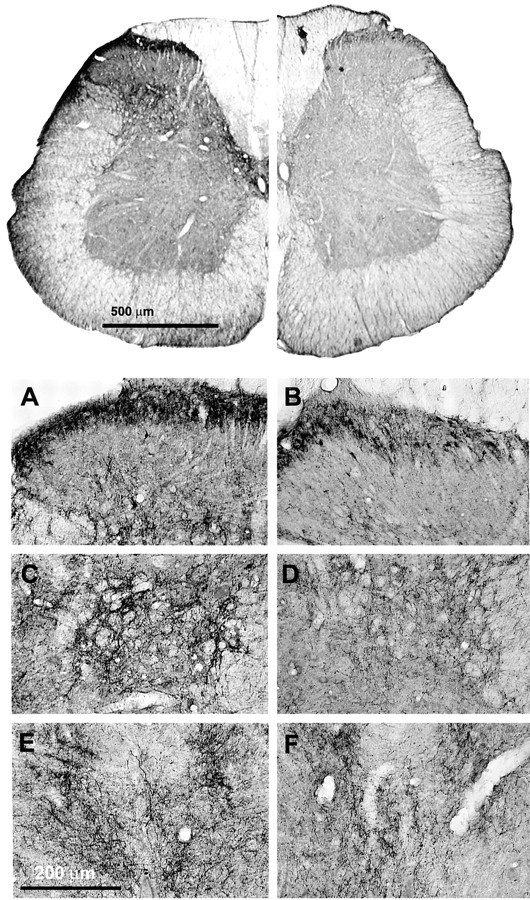
Immunohistochemical analysis of prodynorphin in lumbar spinal sections. Rats have been infused with either saline (control) or DAMGO for 7 d. Lumbar spinal cross sections (40 μm) were labeled with the antiserum for prodynorphin (1:40,000) and processed for DAB staining by the ABC method. Spinal cord halves are represented in close juxtaposition to allow visual comparison.Top, The lefthalfis obtained from the saline-infused control, and therighthalf is obtained from the DAMGO-infused rat. The micrographs were acquired via a Hamamatsu digital-imaging system with a Nikon microscope. Bottom, As seen under higher magnification, the superficial laminae (A, B), discrete cell bodies, and numerous fibers were labeled, whereas in laminae V and VI (C, D) and in lamina X (E, F), prodynorphin-IR was predominantly associated with fibers. Lumbar spinal sections from DAMGO-infused (B, D,F) rats exhibited a reduction of prodynorphin-IR in all the laminae stated above when compared with the saline-infused control (A, C, E). This reduction appears to be primarily caused by a loss of fiber staining in these laminae. In the superficial laminae, staining was still clearly present in cell bodies; the density of immunolabeled cell bodies was similar between control and DAMGO-infused rats.
Lumbar spinal cord sections from control and DAMGO-infused rats were also labeled with μ-opioid receptor-specific antibodies. μ-Opioid receptor-IR was associated predominantly with the superficial laminae of the dorsal horn of the spinal cord (Fig.3). When compared with tissue sections from control rats, sections from the DAMGO-infused rats exhibited a significant reduction in MOR-IR in the superficial laminae (Fig.3).
Fig. 3.

Immunohistochemical analysis of the μ-opioid receptor in lumbar spinal sections. Rats have been infused with either saline (control) or DAMGO for 7 d. Lumbar spinal cross sections (40 μm) were labeled with the antiserum for rat MOR (1:20,000) and processed for DAB staining by the ABC method. Spinal cord halves are represented in close juxtaposition to allow visual comparison. Thelefthalf is obtained from the saline-infused control, and the righthalf is obtained from the DAMGO-infused rat. Substantially higher MOR immunoreactivity is observed in the superficial laminae (I and II) of the control cord, indicating a greater concentration of μ-opioid receptors, than in that of the DAMGO-treated group. The micrographs were acquired via a Hamamatsu digital-imaging system with a Nikon microscope.
The sensory threshold for a response to a non-noxious stimulus was significantly reduced in rats receiving an intrathecal infusion of DAMGO (1 nmol · μl−1 · hr−1for 7 d). When tested on the sixth day of DAMGO infusion (i.e., while the infusion was continuing), the hindpaw response to probing with von Frey filaments decreased from a baseline of 15 ± 0.0 gm (before initiation of infusion) to 4.1 ± 0.5 gm (Fig.4), indicating the development of tactile allodynia during the DAMGO infusion. Animals receiving an intrathecal infusion of saline by osmotic minipump showed no change between their preinfusion baseline (i.e., 15 ± 0.0 gm) and the response threshold at the sixth day of infusion (i.e., 15 ± 0.0 gm). The intrathecal administration of an antiserum to dynorphin A(1–17) at the termination of the intrathecal DAMGO infusion on day 7, however, significantly reversed tactile allodynia; the paw withdrawal threshold was 11.5 ± 2.2 gm (Fig. 4).The intrathecal administration of control (preimmune) serum at the termination of the intrathecal DAMGO infusion on day 7 had no effect on tactile allodynia; the von Frey response after control serum was 4.2 ± 0.5 gm (n= 10), a value that was not significantly different from that determined on day 6 during the DAMGO infusion. Neither control serum nor antiserum to dynorphin A(1–17) alone had any effect on the paw withdrawal thresholds after the intrathecal saline infusion (baseline and postserum values were 15 ± 0.0 gm; n = 10).
Fig. 4.

Male Sprague Dawley rats received intrathecal infusions of saline or of DAMGO (1 nmol · μl−1 · hr−1) for 7 d. Antinociceptive tolerance after chronic infusion of DAMGO was accompanied by tactile allodynia, indicated by a significant (*p ≤ 0.05; Student's t test;n = 10) decrease in paw withdrawal thresholds to probing with von Frey filaments on the sixth day of infusion. The acute intrathecal injection of 200 μg of antiserum to dynorphin A(1–17) on day 7 blocked tactile allodynia when given 20 min before testing in DAMGO-tolerant rats (n = 10). DYN A/S, Dynorphin antiserum.
The latencies to withdrawal of the hindpaw from a noxious thermal stimulus were also significantly reduced in rats receiving an intrathecal infusion of DAMGO. When tested on the sixth day of DAMGO infusion (i.e., while the infusion was continuing), the hindpaw response to noxious heat decreased from a preinfusion baseline of 17.3 ± 1.5 to 10.8 ± 1.2 sec (Fig.5), indicating the development of thermal hyperalgesia during the DAMGO infusion. Animals receiving an intrathecal infusion of saline showed no change between their preinfusion baseline paw withdrawal latencies (i.e., 18.9 ± 1.1 sec) and the response latencies at the sixth day of infusion (i.e.,18.7 ± 0.9 sec). The intrathecal administration of an antiserum to dynorphin A(1–17) at the termination of the spinal DAMGO infusion on day 7, however, significantly reversed thermal hyperalgesia; the paw-flick response after dynorphin antiserum was 35.6 ± 3.6 sec, which was significantly greater than the preinfusion baseline latency (Fig. 5). This response indicates not only a reversal of hyperalgesia but also the production of antinociception. The intrathecal administration of control serum at the termination of the DAMGO infusion on day 7 had no effect on thermal hyperalgesia; the mean paw withdrawal latency obtained after the intrathecal injection of control serum was 12.7 ± 0.8 sec, which was not significantly different (p > 0.05) from the response latency determined on day 6 during the spinal infusion of DAMGO (see above). Neither control serum nor antiserum to dynorphin A(1–17) had any effect on paw withdrawal latencies after the spinal infusion of saline (i.e., preinfusion baseline values were 20 ± 0.7 sec, and postcontrol serum or dynorphin A(1–17) antiserum values were 20.5 ± 0.7 and 19.7 ± 0.6 sec, respectively).
Fig. 5.

Male Sprague Dawley rats received intrathecal infusions of saline or of DAMGO (1 nmol · μl−1 · hr−1) for 7 d. Antinociceptive tolerance was accompanied by thermal hyperalgesia after chronic infusion of DAMGO, indicated by a significant (*p ≤ 0.05; Student'st test; n = 10) decrease in paw withdrawal latencies to radiant heat applied to the plantar aspect of the hindpaw on the sixth day of infusion. The acute intrathecal injection of 200 μg of antiserum to dynorphin A(1–17) on day 7 reversed thermal hyperalgesia when given 20 min before testing in DAMGO-tolerant rats (‡ p ≤ 0.05; Student'st test; n = 10).
Dose–response curves for the antinociceptive effect of intrathecal morphine in the 52°C water tail-flick test were generated in naive, saline-infused, and DAMGO-infused rats. The A50values (and 95% confidence limits) for intrathecal morphine were 3.1 μg (2.4–4.1 μg), 3.3 μg (2.9–3.8 μg), and >10 μg, respectively (Fig. 6). Administration of control serum did not alter the antinociceptive effect of a single intrathecal morphine dose (10 μg/5 μl; n = 8) in animals infused with intrathecal saline; this dose of morphine elicited 91.6 ± 6 and 93.5 ± 5% antinociception before and after control serum, respectively. Antiserum to dynorphin A(1–17) also did not alter the antinociceptive effect of intrathecal morphine in animals infused with intrathecal saline, with the A50(and 95% confidence limits) for intrathecal morphine being 3.7 μg (3.1–4.4 μg) after dynorphin A(1–17) antiserum. However, the decreased morphine antinociceptive response seen in animals infused with intrathecal DAMGO was reversed by the antiserum to dynorphin A(1–17) (Fig. 6). After intrathecal dynorphin A(1–17) antiserum, the A50 for intrathecal morphine in DAMGO-infused rats was 3.6 μg (2.9–4.3 μg), a value that did not differ significantly from that of saline-infused rats (see above). Control serum (200 μg/5 μl, intrathecally; 20 min before the test) had no effect on the antinociceptive effect of intrathecal morphine in animals infused with intrathecal DAMGO. The effect of a single intrathecal dose of morphine (10 μg) in DAMGO-infused rats before and after control serum was 3.3 ± 2.1 and 2.8 ± 1.9% antinociception (n = 4), respectively, indicating significant cross-tolerance between DAMGO and morphine. Control serum had no effect on the antinociceptive effect of intrathecal morphine in animals infused with intrathecal saline. The effect of a single intrathecal dose of morphine (10 μg) in saline-infused rats before and after control serum was 88 ± 8.6 and 93.0 ± 5.3% antinociception (n = 4), respectively.
Fig. 6.

Male Sprague Dawley rats received intrathecal infusions of saline or of DAMGO (1 nmol · μl−1 · hr−1) for 7 d. Antinociceptive dose–response functions for intrathecal morphine were generated in the 52°C water tail-flick test 10 min after treatment. The saline- or DAMGO-infused rats received either intrathecal morphine alone or morphine 10 min after intrathecal pretreatment with antiserum to dynorphin A(1–17) (i.e., dynorphin antiserum given 20 min before the test). The following groups were used: naive rats (○), rats infused with saline and challenged with morphine (■), rats infused with saline and challenged with morphine after pretreatment with dynorphin antiserum (▵), rats infused with DAMGO and challenged with morphine (●), and rats infused with DAMGO and challenged with morphine after pretreatment with dynorphin antiserum (▪). Five to 10 separate animals were used for eachpoint on the dose–response curve. i.th., Intrathecal.
DISCUSSION
The data from the present study, along with previous data from nerve-injury studies, support the hypothesis of mechanistic similarities at the spinal level between states of opioid tolerance and nerve injury. A novel concept introduced here is that spinal dynorphin acts as a common mediator promoting these conditions. Both the spinal infusion of an opioid μ agonist or peripheral nerve injury elicit tactile allodynia, thermal hyperalgesia, and decreased antinociceptive responsiveness to spinal morphine (opioid tolerance), as well as increases in the spinal content of dynorphin. In both cases, thermal hyperalgesia and opioid tolerance can be reversed by antiserum to dynorphin. These data suggest that spinal dynorphin acts as an endogenous mediator promoting opioid-induced abnormal pain as well as opioid antinociceptive tolerance.
The finding that continuous intrathecal infusion of the μ agonist DAMGO produces abnormal pain is consistent with multiple clinical observations that indicate that chronic perispinal (intrathecal or epidural) opioid administration is associated with paradoxical hyperalgesia and/or allodynia. A review of the clinical literature found that many patients who receive long-term spinal opioids developed hyperesthesia and allodynia, especially with high doses of morphine (Arner et al., 1988). Abnormal pain that is qualitatively different and somatically unrelated to the original pain complaint has been reported in several clinical studies after prolonged opioid exposure (Ali, 1986;Stillman et al., 1987; De Conno et al., 1991; Devulder, 1997).
These clinical reports are consistent with many experimental observations in which hyperalgesia was reported after prolonged spinal opioid exposure in rats (Yaksh and Harty, 1988; Trujillo and Akil, 1991; Mao et al., 1994, 1995a,c). Large doses of intrathecal morphine have been associated with paradoxical algesia and hyperesthesias, including intermittent bouts of biting and scratching at the dermatomes near the catheter tip and aggressive responses to light brushing of the flanks (Woolf, 1981; Yaksh et al., 1986; Yaksh and Harty, 1988). These effects of morphine were not sensitive to naloxone, nor did they diminish with tolerance (Yaksh and Harty, 1988). In spite of these many preclinical and clinical reports of abnormal pain elicited by opioids, the mechanism(s) by which such pain might occur is obscure. The present study suggests an important role for spinal dynorphin in such abnormal pain because (1) blockade of the effects of dynorphin with antiserum clearly blocks opioid-induced allodynia and hyperalgesia and (2) continuous intrathecal infusion (this study) or subcutaneous administration (Rattan and Tejwani, 1997) of the μ agonist morphine as well as spinal nerve ligation (Cho and Basbaum, 1989; Kajander et al., 1990; Draisci et al., 1991; Dubner, 1991; Dubner and Ruda, 1992) results in the increase in spinal dynorphin content.
In addition to its role in abnormal pain, spinal dynorphin appears to promote opioid tolerance. As expected, the dose effect of morphine antinociception after a 7 d intrathecal DAMGO infusion is shifted to the right, accompanied by a decreased maximum effect that is typical of opioid tolerance. The efficacy of spinal morphine was restored by the administration of antiserum to dynorphin, but not by control serum. There are two important aspects to these findings. The first is that the effects of the antiserum are specific because control serum has no effect on either saline- or DAMGO-infused rats. The second is that the antiserum to dynorphin has no effect on morphine-induced antinociception in saline-infused rats, suggesting that dynorphin does not play an important part in normal sensory thresholds.
These data further substantiate the parallel actions of the antiserum to dynorphin A(1–17) and MK-801 in cases of pain and opioid tolerance, although some reports suggest the need for multiple doses of MK-801 to reverse established opioid tolerance (Trujillo and Akil, 1991; Lee and Yaksh, 1995; Chaplan et al., 1997). An important feature of the present experimental design is that allodynia and hyperalgesia were demonstrated during the infusion of DAMGO (on day 6) and that antiallodynic and antihyperalgesic effects of dynorphin antiserum were measured immediately after the cessation of DAMGO infusion (on day 7), in both cases when the peptide is still present in the spinal cord. This procedure was designed to ensure that the observed abnormal pain was not caused by the development of a state of opioid withdrawal. Under these conditions, dynorphin antiserum blocks allodynia and not only reverses hyperalgesia but is actually antinociceptive. The antiallodynic and antinociceptive effects are probably a combined result of blocking dynorphin activity and unmasking the agonist activity of DAMGO. This is consistent with previous observations that the antiallodynic, antihyperalgesic, and antinociceptive effects of spinal morphine were all increased in the presence of antiserum to dynorphin A(1–17) in the nerve-injured rat, suggesting a restoration of the actions of spinal opioids after normalization of the sensitized state of the spinal cord (Nichols et al., 1997; Wegert et al., 1997;Bian et al., 1999).
The most likely mechanism for the acute effects of dynorphin antiserum is by sequestering dynorphin after its release from spinal interneurons, suggesting that abnormal pain and opioid tolerance may be caused by direct or indirect actions of dynorphin. This possibility is supported by the observation that spinal dynorphin content was elevated after DAMGO infusion, suggesting that spinal opioid administration can regulate the expression of spinal dynorphin, probably as a result of opioid receptor occupation. The involvement of opioid receptors in regulating dynorphin expression seems likely because subcutaneous implantation of morphine pellets or spinal delivery of opioid δ agonists such as [d-Ala2, Glu4]deltorphin also increases levels of spinal dynorphin (L. R. Gardell, E. J. Bilsky, and F. Porreca, unpublished observations). However, the increase in dynorphin content was concomitant with an apparent reduction in the level of its precursor, prodynorphin, when compared with spinal cord tissues from saline-infused rats. These findings suggest that spinal opioid infusion may enhance the processing and subsequent release of dynorphin while having modest effects on prodynorphin synthesis.
In addition to the changes in the content of dynorphin, the data show that spinal opioid infusion elicits a clear downregulation of μ receptors in the spinal dorsal horn. Because receptor downregulation is one of the mechanisms for opioid tolerance in a variety of in vitro systems, a reduction in μ receptor density would be consistent with a diminished response to a subsequent challenge of morphine after DAMGO infusion. However, because dynorphin antiserum can effectively restore morphine efficacy, these data also indicate that the loss of receptors is not sufficient for the manifestation of opioid tolerance and that homologous receptor desensitization and downregulation are not likely to be the primary mediators of opioid tolerance in vivo.
Although the mechanism(s) by which dynorphin promotes abnormal pain is not entirely clear, it seems reasonable to speculate that decreased μ receptor expression on primary afferent fibers might promote abnormal pain via an increase in the release of excitatory neurotransmitters because of possibly diminished endogenous opioid tone. In this regard, μ-opioid receptor knock-out mice were shown to be hyperalgesic by the use of a warm-water tail-flick and hot-plate test (Sora et al., 1997) and further demonstrated tactile allodynia by the use of von Frey filaments (T. W. Vanderah and F. Porreca, unpublished observations). Furthermore, peripheral nerve injury also results in a decrease in μ-opioid receptor expression, supporting the possibility of diminished opioid tone as a factor in abnormal pain (Porreca et al., 1998). The overexpression of spinal dynorphin is also likely to have consequences on the release of excitatory neurotransmitters. Recent observations have shown that dynorphin A(1–17) can increase the release of capsaicin-stimulated substance P (Arcaya et al., 1999) and that the des-Tyr fragments of dynorphin can increase capsaicin-stimulated calcitonin gene-related peptide release (Claude et al., 1999) (Vanderah and Porreca, unpublished observations). These observations indicate a common mechanism in both the nerve-injury and opioid-tolerant states by which the nonopioid actions of this peptide might act to promote a state of “spinal sensitization.”
Elevated spinal dynorphin may represent an endogenous mechanism that promotes opioid tolerance and spinal opioid-associated pain as well as the consequences of nerve injury. Such observations suggest the possibility of therapeutic interventions such as the development of a specific dynorphin antibody for clinical use or approaches that can block the actions of, or facilitate the degradation of, spinal dynorphin. These possibilities may be of considerable significance because NMDA antagonists, which produce actions similar to those of the antiserum to dynorphin, may ultimately be limited in their clinical utility because of their significant side-effect profile. Inhibiting the pathological actions resulting from overexpression of an endogenous substance, such as dynorphin, may prove to provide attainable clinical benefit for opioid tolerance and pain without the side effects associated with the blockade of NMDA receptors.
Footnotes
Correspondence should be addressed to Dr. Frank Porreca, Department of Pharmacology, College of Medicine, University of Arizona Health Sciences Center, Tucson, AZ 85724. E-mail: frankp@u.arizona.edu.
REFERENCES
- 1.Ali NM. Hyperalgesic response in a patient receiving high concentrations of spinal morphine. Anesthesiology. 1986;65:449. doi: 10.1097/00000542-198610000-00028. [DOI] [PubMed] [Google Scholar]
- 2.Arcaya JL, Cano G, Gomez G, Maixner W, Suarez-Roca H. Dynorphin A increases substance P release from trigeminal primary afferent C-fibers. Eur J Pharmacol. 1999;366:27–34. doi: 10.1016/s0014-2999(98)00897-8. [DOI] [PubMed] [Google Scholar]
- 3.Arner S, Rawal N, Gustafsson LL. Clinical experience of long-term treatment with epidural and intrathecal opioids—a nationwide survey. Acta Anaesthesiol Scand. 1988;32:253–259. doi: 10.1111/j.1399-6576.1988.tb02725.x. [DOI] [PubMed] [Google Scholar]
- 4.Bian D, Ossipov MH, Ibrahim M, Raffa RB, Tallarida RJ, Malan TP, Jr, Lai J, Porreca F. Loss of antiallodynic and antinociceptive spinal/supraspinal morphine synergy in nerve-injured rats: restoration by MK-801 or dynorphin antiserum. Brain Res. 1999;831:55–63. doi: 10.1016/s0006-8993(99)01393-1. [DOI] [PubMed] [Google Scholar]
- 5.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 6.Chaplan SR, Malmberg AB, Yaksh TL. Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia in the rat. J Pharmacol Exp Ther. 1997;280:829–838. [PubMed] [Google Scholar]
- 7.Cho HJ, Basbaum AI. Ultrastructural analysis of dynorphin B-immunoreactive cells and terminals in the superficial dorsal horn of the deafferented spinal cord of the rat. J Comp Neurol. 1989;281:193–205. doi: 10.1002/cne.902810204. [DOI] [PubMed] [Google Scholar]
- 8.Claude P, Gracia N, Wagner L, Hargreaves KM. Ninth World Congress on Pain. IASP Press; Vienna: 1999. Effect of dynorphin on ICGRP release from capsaicin-sensitive fibers. p. 262. [Google Scholar]
- 9.De Conno F, Caraceni A, Martini C, Spoldi E, Salvetti M, Ventafridda V. Hyperalgesia and myoclonus with intrathecal infusion of high-dose morphine. Pain. 1991;47:337–339. doi: 10.1016/0304-3959(91)90225-M. [DOI] [PubMed] [Google Scholar]
- 10.Devulder J. Hyperalgesia induced by high-dose intrathecal sufentanil in neuropathic pain. J Neurosurg Anesthesiol. 1997;9:146–148. doi: 10.1097/00008506-199704000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 12.Draisci G, Kajander KC, Dubner R, Bennett GJ, Iadarola MJ. Up-regulation of opioid gene expression in spinal cord evoked by experimental nerve injuries and inflammation. Brain Res. 1991;560:186–192. doi: 10.1016/0006-8993(91)91231-o. [DOI] [PubMed] [Google Scholar]
- 13.Dubner R. Neuronal plasticity and pain following peripheral tissue inflammation or nerve injury. In: Bond MR, Charlton JE, Woolf CJ, editors. Proceedings of the VIth World Congress on Pain. Elsevier; Amsterdam: 1991. pp. 263–276. [Google Scholar]
- 14.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 15.Kajander KC, Sahara Y, Iadarola MJ, Bennett GJ. Dynorphin increases in the dorsal spinal cord in rats with a painful peripheral neuropathy. Peptides. 1990;11:719–728. doi: 10.1016/0196-9781(90)90187-a. [DOI] [PubMed] [Google Scholar]
- 16.Lee YW, Yaksh TL. Analysis of drug interaction between intrathecal clonidine and MK-801 in peripheral neuropathic pain rat model. Anesthesiology. 1995;82:741–748. doi: 10.1097/00000542-199503000-00016. [DOI] [PubMed] [Google Scholar]
- 17.Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI. Evidence for presynaptic N-methyl-d-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci USA. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- 19.Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F. Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain. 2000;86:185–194. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 20.Manning BH, Mao J, Frenk H, Price DD, Mayer DJ. Continuous co-administration of dextromethorphan or MK-801 with morphine: attenuation of morphine dependence and naloxone-reversible attenuation of morphine tolerance. Pain. 1996;67:79–88. doi: 10.1016/0304-3959(96)81972-5. [DOI] [PubMed] [Google Scholar]
- 21.Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao J, Price DD, Mayer DJ. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995a;61:353–364. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- 23.Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995b;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- 24.Mao J, Price DD, Phillips LL, Lu J, Mayer DJ. Increases in protein kinase C gamma immunoreactivity in the spinal cord of rats associated with tolerance to the analgesic effects of morphine. Brain Res. 1995c;677:257–267. doi: 10.1016/0006-8993(95)00161-i. [DOI] [PubMed] [Google Scholar]
- 25.Nichols ML, Lopez Y, Ossipov MH, Bian D, Porreca F. Enhancement of the antiallodynic and antinociceptive efficacy of spinal morphine by antisera to dynorphin A(1–13) or MK-801 in a nerve-ligation model of peripheral neuropathy. Pain. 1997;69:317–322. doi: 10.1016/S0304-3959(96)03282-4. [DOI] [PubMed] [Google Scholar]
- 26.Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. Inhibition by spinal morphine of the tail-flick response is attenuated in rats with nerve ligation injury. Neurosci Lett. 1995a;199:83–86. doi: 10.1016/0304-3940(95)12026-z. [DOI] [PubMed] [Google Scholar]
- 27.Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci Lett. 1995b;199:87–90. doi: 10.1016/0304-3940(95)12022-v. [DOI] [PubMed] [Google Scholar]
- 28.Porreca F, Tang QB, Bian D, Riedl M, Elde R, Lai J. Spinal opioid mu receptor expression in lumbar spinal cord of rats following nerve injury. Brain Res. 1998;795:197–203. doi: 10.1016/s0006-8993(98)00292-3. [DOI] [PubMed] [Google Scholar]
- 29.Price DD. Psychological and neural mechanisms of pain. Raven; New York: 1988. [DOI] [PubMed] [Google Scholar]
- 30.Rattan AK, Tejwani GA. Effect of chronic treatment with morphine, midazolam and both together on dynorphin(1–13) levels in the rat. Brain Res. 1997;754:239–244. doi: 10.1016/s0006-8993(97)00084-x. [DOI] [PubMed] [Google Scholar]
- 31.Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, Miner LL, Uhl GR. Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci USA. 1997;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stillman MJ, Moulin DE, Foley KM. Paradoxical pain following high-dose spinal morphine. Pain. 1987;4:85. [Google Scholar]
- 33.Tallarida RJ, Murray RB. Manual of pharmacologic calculations with computer programs, Second edition, 297 pp. Springer; New York: 1987. [Google Scholar]
- 34.Tang Q, Gandhoke R, Burritt A, Hruby VJ, Porreca F, Lai J. High-affinity interaction of (des-tyrosyl)dynorphin A(2–17) with NMDA receptors. J Pharmacol Exp Ther. 1999;291:760–765. [PubMed] [Google Scholar]
- 35.Tiseo PJ, Inturrisi CE. Attenuation and reversal of morphine tolerance by the competitive N-methyl-d-aspartate receptor antagonist, LY274614. J Pharmacol Exp Ther. 1993;264:1090–1096. [PubMed] [Google Scholar]
- 36.Tiseo PJ, Cheng J, Pasternak GW, Inturrisi CE. Modulation of morphine tolerance by the competitive N-methyl-d-aspartate receptor antagonist LY274614: assessment of opioid receptor changes. J Pharmacol Exp Ther. 1994;268:195–201. [PubMed] [Google Scholar]
- 37.Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- 38.Wegert S, Ossipov MH, Nichols ML, Bian D, Vanderah TW, Malan TP, Jr, Porreca F. Differential activities of intrathecal MK-801 or morphine to alter responses to thermal and mechanical stimuli in normal or nerve-injured rats. Pain. 1997;71:57–64. doi: 10.1016/s0304-3959(97)03337-x. [DOI] [PubMed] [Google Scholar]
- 39.Woolf CJ. Intrathecal high dose morphine produces hyperalgesia in the rat. Brain Res. 1981;209:491–495. doi: 10.1016/0006-8993(81)90176-1. [DOI] [PubMed] [Google Scholar]
- 40.Yaksh TL, Harty GJ. Pharmacology of the allodynia in rats evoked by high dose intrathecal morphine. J Pharmacol Exp Ther. 1988;244:501–507. [PubMed] [Google Scholar]
- 41.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 42.Yaksh TL, Harty GJ, Onofrio BM. High dose of spinal morphine produces a nonopiate receptor-mediated hyperesthesia: clinical and theoretic implications. Anesthesiology. 1986;64:590–597. doi: 10.1097/00000542-198605000-00008. [DOI] [PubMed] [Google Scholar]