

Abstract
Complement defense 59 (CD59) is a cell surface glycophosphoinositol (GPI)-anchored protein that prevents complement membrane attack complex (MAC) assembly. Here, we present evidence from ELISA assays that CD59 protein levels are significantly decreased in the frontal cortex and hippocampus of Alzheimer's disease (AD) compared with nondemented elderly (ND) patients, whereas complement component 9, a final component to form MAC, is significantly increased. To further confirm the CD59 deficit, PI-specific phospholipase C (PIPLC) was used to cleave the CD59 GPI anchor at the cell surface in intact slices from AD and ND cortex. CD59 released by PIPLC cleavage was significantly reduced in AD compared with ND samples. By the use of a ribonuclease protection technique, amyloid β-peptide was found to downregulate CD59 expression at the mRNA level, suggesting a partial explanation of CD59 deficits in the AD brain. To evaluate the pathophysiological significance of CD59 alterations in neurons, we exposed cultured NT2 cells, which normally underexpress CD59, and NT2 cells transfected to overexpress CD59 to homologous human serum. Lactic acid dehydrogenase assays revealed significant complement-induced cell lysis in CD59-underexpressing NT2 cells and significant protection from such lysis in CD59-overexpressing NT2 cells. Moreover, cells expressing normal levels of CD59 showed no evidence of MAC assembly or damage after exposure to homologous serum, whereas pretreatment of these cells with a CD59-neutralizing antibody resulted in MAC assembly at the cell surface and morphological damage. Taken together, these data suggest that CD59 deficits may play a role in the neuritic losses characteristic of AD.
Keywords: neurodegeneration, Alzheimer, neuron death, amyloid protein, complement, inflammation
The activation of complement in Alzheimer's disease (AD) seems to occur via a unique, amyloid β-peptide (Aβ)-mediated, antibody-independent mechanism (Eikelenboom and Stam, 1982; McGeer et al., 1989; Rogers et al., 1992; Shen et al., 1998) and proceeds fully to formation of the terminal complement component C5b-9 or the membrane attack complex (MAC) (Rogers et al., 1992; Webster et al., 1997; Shen et al., 1998). The MAC is a macrocomplex made up of complement components C5, C6, C7, and C8 plus multiple C9 molecules. After being formed, the ring-like structure of the MAC opens a pore in the membrane of targeted cells, permitting massive Ca2+ influx and subsequent cell lysis (Kim et al., 1987). The MAC is present in pathologically vulnerable areas of the AD brain and is highly colocalized with the Aβ deposit (McGeer et al., 1989; Rogers et al., 1992; Webster et al., 1997).
One of the major defense mechanisms against MAC attack is that most cells express complement defense 59 (CD59) (membrane inhibitor of reactive lysis), a glycophosphoinositol (GPI)-anchored membrane protein that binds C8 or C9 and thereby prevents further assembly of the poly-C9 MAC and its full insertion into the cell membrane (Zalman et al., 1989; Meri et al., 1990; Ninomiya and Sims, 1992; Stefanova et al., 1994; Nakano et al., 1995). Inhibition of MAC assembly by CD59 only occurs, however, if the MAC components are from the same species as the target cell, a phenomenon known as homologous restriction (Shen et al., 1995). Conversely, when the complement source and target cell are from different species, CD59 regulation is less effective. By these mechanisms, MAC lysis of foreign cells can occur without damage to homologous host bystander cells. Alternatively, a CD59 deficiency does permit homologous MAC attacks (Vakeva et al., 1992; Wing et al., 1992;Piddlesden and Morgan, 1993; Takeda et al., 1993; Shen et al., 1995) and can result in clinical inflammatory disorders, especially under conditions in which increased MAC formation is present (Yamahina et al., 1990; Vakeva et al., 1992; Takeda et al., 1993; Zhan et al., 1994).
Here, we demonstrate a significant deficit in CD59 expression in AD compared with nondemented elderly (ND) hippocampus and cortex plus a significant increase in the levels of C9, the critical determinant of MAC pore formation and toxicity, in the same areas. As shown by further culture studies, such deficits in CD59 expression increase the vulnerability of neuron-like cells to homologous complement attack, a mechanism that may be relevant to AD because we also observe a significant correlation between immunoreactive synaptophysin and immunoreactive CD59 in the AD brain.
MATERIALS AND METHODS
Brain samples for research. Frontal neocortex and hippocampus samples from clinically diagnosed, neuropathologically confirmed AD and ND patients were frozen at autopsy and stored in vacuum-sealed plastic bags at −80°C until assay. A board-certified neuropathologist using CERAD criteria performed the neuropathology evaluation. Postmortem intervals averaged <3 hr. We have randomly selected 42 cases that consist of 22 AD cases (including 10 females, AD history of 7.5 ± 1.13 years, and 12 males, AD history of 7.3 ± 0.68 years) and 20 aged-matched and nondemented control cases (12 males and 8 females). All of the cases we have used died from lung or kidney failure, and they did not have other CNS diseases, sudden death, or infectious diseases on the basis of clinical records and pathological examination. However, it is possible that some particular infectious diseases might affect CD59 expression. Therefore, we have excluded patients with immunological disorders or severe infectious diseases. Age (XAD = 84.6 ± 6.12 years; XND = 84.0 ± 8.12 years) and postmortem intervals (XAD = 2.65 ± 0.77 hours; XND = 2.63 ± 0.94 hours) did not differ significantly between the patient groups and, in fact, were well matched.
ELISA assays of CD59 and C9. For CD59 and C9 ELISAs, purified CD59 standard, purified C9 standard, or brain cortex homogenates were added to polyclonal anti-CD59 (Serotec, Indianapolis, IN) or anti-C9 (PharMingen, San Diego, CA) antibody-coated plates and incubated at 25°C for 2 hr to permit CD59 or C9 capture. To bind captured CD59 or C9, 100 μl of 0.3 μg/ml monoclonal anti-CD59 antibody (PharMingen) or anti-C9 antibody (Quidel, San Diego, CA) was added. Biotinylated anti-mouse IgG and avidin-conjugated HRP ando-phenylenediamine dihydrochloride were used to detect binding. The specificity of the ELISA protocols was verified by serial dilutions of purified CD59 and C9 standards. Pilot studies with the CD59 ELISA demonstrated that it could detect as little as 3–8 pg/ml CD59 (data not shown).
Western blot analysis. AD and ND brain samples were individually homogenized in 5 vol of homogenizing buffer containing 10 mm Tris-HCl, pH 7.4, 25 mm NaCl, 50 mm EDTA, 1 mm EGTA plus 0.5% Triton X-100, 10% SDS, and a protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN; 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, 5 μg/ml leupeptin, and 2 μg/ml aprotinin). For each sample, 20 μg of protein was separated on a 15% SDS Tricine gel and transferred to a polyvinylidene difluoride membrane electrophoretically for 2 hr with 0.1% Tween 20 in TBS. For CD59, a tissue membrane sample was used. The brain samples were homogenized in 5 vol of buffer containing 50 mm phosphate, pH 7.7, and a protease inhibitor cocktail. The tissue membrane sample (25000 ×g; 30 min) was collected and dissolved in the homogenizing buffer. CD59 was detected with an affinity-purified antibody (Serotec) at a 1:1000 dilution for 12 hr at 4°C, followed by incubation with an HRP-conjugated secondary antibody and processing using ECL detection (Amersham Pharmacia Biotech).
PI-specific phospholipase C cleavage. PI-specific phospholipase C (PIPLC) was purchased from Sigma (St. Louis, MO) and diluted to graded concentrations of PIPLC from 0.0375 to 0.6000 units/ml in PBS. AD and ND brain samples were cut into 130 × 130 × 350 μm slices by the use of a McIlwain tissue chopper and then incubated at 37°C for 60 min with 0.5 ml of reaction buffer containing 5 mm EDTA, a protease inhibitor cocktail (Boehringer Mannheim), and the above-listed PIPLC solution. After incubation, the samples were centrifuged for 10 min at 20,000 ×g, and the supernatant was collected. The cleaved CD59 was then measured by the use of CD59 ELISA.
Ribonuclease protection assay. Total RNA was prepared from Aβ-treated NT2 cells and from human brain tissues of AD and ND patients by the use of an RNA Trizol isolation kit (Life Technologies, Gaithersburg, MD). The human CD59 fragment, a 180 bp transgene 5′ coding sequence, was subcloned into the vector pPCRI (Invitrogen, San Diego, CA). The antisense cRNA probe was synthesized from the linearized template DNA by the use of T7 RNA polymerase in the presence of [α-32P]UTP. The ribonuclease protection assay was performed with a commercially available kit (RPAII kit; Ambion). Three micrograms of total RNA were hybridized with 0.3 ng of the cRNA probe (specific activity, 5 × 107 cpm/μg of RNA) at 48°C for 16 hr and then digested with a mixture of RNase A and T1. The protected bands were separated on a 6% denaturing acrylamide gel and detected by autoradiography. The intensity of the bands was quantified by an imaging analyzer (Chemimager 4000). A riboprobe that was directed at β-actin mRNA was included in all incubations as an internal control.
Cloning of human CD59 and C9.Poly(A+) RNA was isolated from the human brain cortex, and cDNA was synthesized with reverse transcriptase. PCR was used to amplify the coding regions of the CD59 and C9 genes. For convenience, the PCR primers were adapted to contain Xho andXba restriction enzyme sites. The PCR conditions used were 55, 72, and 94°C for 34 cycles. After running the PCR products in a low-melting agarose gel, the specific band was excised and cloned into a pcDNA3.1 expression vector. cDNA plasmid products for CD59 and C9 were sequenced by the method of Sanger, and the sequences were found to be identical to the published gene bank DNA sequences for CD59 or C9.
Cell culture. Human NT2 cells, which can be terminally differentiated into cells bearing numerous specific characteristics of neurons (Shen et al., 1997), were cultured in DMEM and heat-inactivated 10% fetal bovine serum (FBS). The cells were seeded at a density of 25,000 cells/well in 24-well plates. The culture medium was replaced every 3 d with DMEM plus 5% FBS. Human neuroblastoma SH-SY5Y cells were cultured at 25,000 cells/well in 24-well plates with 50% MEM plus 50% F12 medium, 15% heat-inactivated fetal calf serum, and 10 μm retinoic acid. The culture medium was replaced every 3 d, and the cells were differentiated for 6 d. They were seeded at a density of 5,000 cells/well in Matrigel-treated 24-well plates, with medium replacement every 3 d. By day 7 after initial plating, neurotypic cells with multiple neurites were observed.
Cell transfection. Exogenous DNA transfection followed the calcium phosphate precipitation procedure. For transient transfection, the CD59 or C9 cDNA was inserted into a pcDNA3.1 expression vector that directs its expression from both cytomegalovirus and simian virus 40 promoters. These constructs were transfected into NT2 cells.
Lactic acid dehydrogenase assay. For quantitative assessment of neuronal cell damage, the release of lactic acid dehydrogenase (LDH) from degenerating neurons was measured by the use of a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI) as described previously (Piddlesden and Morgan, 1993). The percent LDH release is calculated as the ratio of LDH contained in the supernatant relative to the total LDH contained in both the supernatant and cell lysate. This quantitative biochemical index has been shown to correlate well with morphological neurodegenerative changes observed in culture (Shen et al., 1995).
Aβ(1–42) exposure. Synthetic Aβ(1–42) or scrambled Aβ(1–42) (Bachem) was dissolved in anhydrous DMSO and diluted with serum-free N2-supplemented medium (Piddlesden and Morgan, 1993). Cell cultures were then exposed for different time courses to final concentrations of Aβ(1–42) from 10 nm to 25 μm or to a final concentration of 25 μmscrambled Aβ(1–42) as a negative control.
RESULTS
CD59 reduction in AD brains
Using the ELISA assay for CD59, we are able to measure CD59 protein levels at the picogram level. Brain homogenates were assayed in triplicate, and the mean for each sample was used in further calculations. The average yield of total protein, 40 mg/gm wet weight of tissue, was the same for both ND and AD hippocampus and frontal cortex tissue samples. The yield of total protein did not correlate with age at the time of death or with the postmortem interval for ND or AD patients (data not shown).
The CD59 ELISA quantitation of 20 ND and 22 AD hippocampus and frontal cortex tissue homogenates was performed as described in Materials and Methods. The specificity of the ELISA protocol was verified by testing purified CD59 samples in the same ELISA (Wertkin et al., 1993). We found that the CD59 protein level was >50% lower in the hippocampus and frontal cortex of AD patients compared with that of the ND (Figs.1, 2) by using ELISA. These findings were further confirmed by a 39% loss by the use of Western blot analysis (Fig. 2) and also by radiolabeled ELISA (data not shown).
Fig. 1.

Reduction of CD59 by ELISA in the AD and ND brains. The data represent the mean (± SE) quantification analysis of 20–22 cases using a specific ELISA assay showing that CD59 immunoreactivity was decreased by 52% in AD compared with ND (nondemented aged-matched controls). Results from three independent measurements displayed a significant difference between AD and ND samples (*p < 0.05).
Fig. 2.

Relationship of CD59 deficiency to neuronal or synaptic loss in AD brains. A, Western blot studies on CD59, synaptophysin, and neuron-specific enolase (NSE) in AD and ND brains are shown. B, Data represent the mean (± SD) quantification analysis using densitometry imaging (Chemimager 4000) showing that CD59 immunoreactivity was decreased by 39% in AD compared with ND, whereas synaptophysin was decreased ∼26%. There is no significant change inNSE expression levels. Results from three independent measurements displayed a significant difference between AD and ND samples (**p < 0.01). AIDV, Average integrated density value.
CD59 is a GPI-anchored membrane protein that is susceptible to PIPLC cleavage. Because CD59 is specifically bound to the cell surface, to verify further the CD59 deficit in AD brains, we have used PIPLC, an enzyme that cleaves GPI-anchored proteins from the cell surface, to cleave the GPI-anchored protein from hippocampus sections. After PIPLC was incubated with these tissue sections from AD and ND brains (n = 6) at graded concentrations (0.0375–0.6 units/ml), ELISA assays again demonstrated a significant CD59 deficit in AD compared with ND patients (p < 0.01). As seen in Table 1, after incubation with increasing concentrations of PIPLC, increasing amounts of CD59 were released. AD groups had significantly lower concentrations of CD59 than that found in ND groups at each PIPLC concentration treatment (Table1). These results are consistent with the CD59 ELISA data from the AD and ND brain homogenates and further suggest that the normal brain has much higher levels of membrane-bound CD59.
Table 1.
Effects of PIPLC treatment on the amount of CD59 released in hippocampus tissue sections from AD and ND brains
| PIPLC treatment (units/ml) | ND individuals Cleaved CD59 (medium) (pg/mg) | AD individuals Cleaved CD59 (medium) (pg/mg) |
|---|---|---|
| 0.0375 | 11.95 ± 2.06 | 8.08 ± 2.93 |
| 0.075 | 18.20 ± 2.15 | 13.60* ± 1.67 |
| 0.15 | 25.94 ± 1.74 | 18.87* ± 3.21 |
| 0.3 | 33.41 ± 2.11 | 26.02* ± 1.26 |
| 0.6 | 38.78 ± 4.15 | 31.61* ± 1.59 |
p < 0.01.
To analyze steady-state levels of CD59 mRNA in the cortical tissues, a ribonuclease protection assay was performed on brain tissues from AD and ND patients. A riboprobe that directs to the β-actin mRNA was included in all incubations as an internal control. As expected, total RNA extracted from brain tissues to express the CD59 gene protected a single band of 180 nucleotides. The level of CD59 mRNA in the AD brain was much lower than that in the ND brain (Fig.3). This finding is consistent with the CD59 protein level we described above.
Fig. 3.
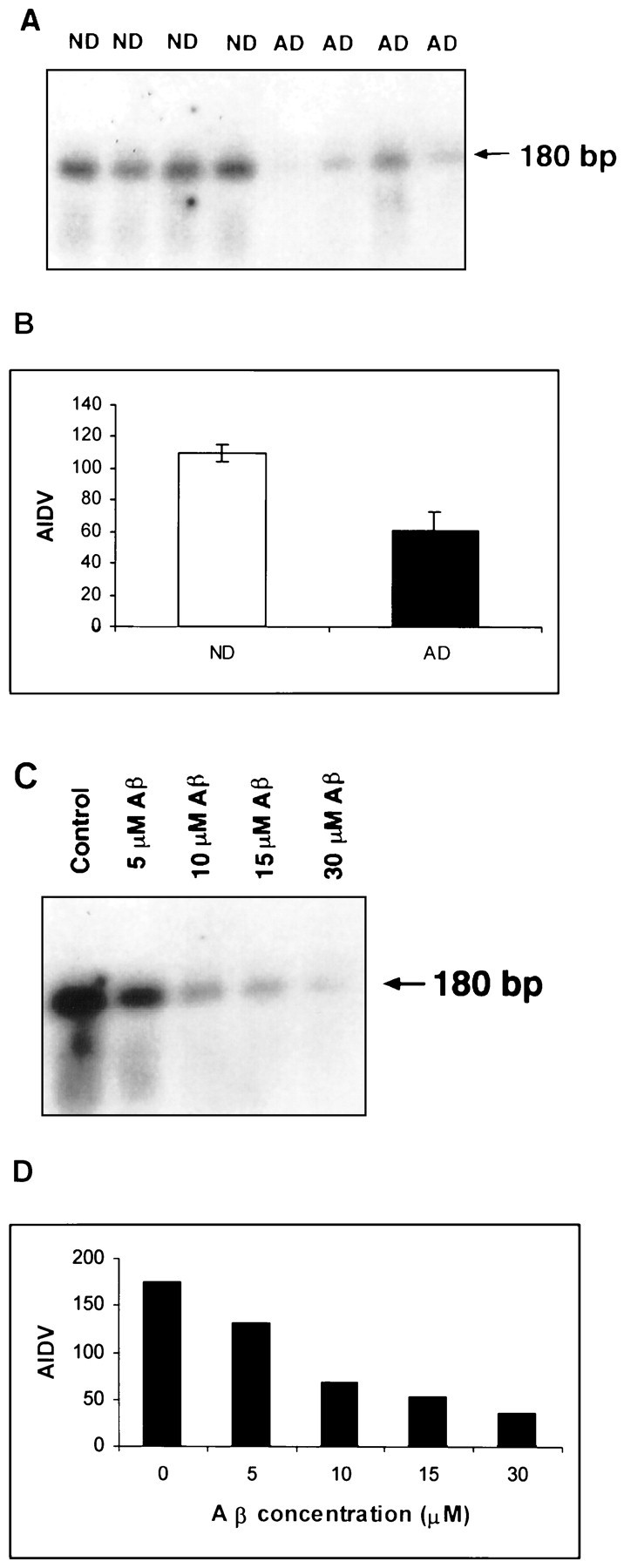
CD59 mRNA expression in the AD brain and Aβ-treated neurons by the use of the RNase protection assay. Total RNA was recovered from 0.5 gm of tissue or 2 × 106 cells after stimulation with Aβ(1–42) for 16 hr. The RNAs were hybridized with the CD59 antisense probe. After treatment with RNase A and T1, protected bands were run through an 8% polyacrylamide gel, and the dried gel was exposed to x-ray film.A, CD59 mRNA from AD and ND brains is shown.C, CD59 mRNA expression is downregulated in a dose-dependent manner in Aβ(1–42)-treated cells. Data from both experiments show that CD59 at the mRNA level is decreased in AD brains and Aβ-treated neurotypic cells. B, D,Data represent quantitative analysis results from A andC, respectively, using Chemimager 4000.
It is well established that one of the pathological hallmarks in the AD brain is the presence of plaques containing Aβ (Masters et al., 1985). It is logical to examine whether the CD59 protein level is affected by Aβ in vitro. Human neuronal SH-SY5Y cell line cells that can express normal levels of CD59 were treated with Aβ(1–42) at various concentrations. We found that, comparing cells with and without treatment, the CD59 mRNA expression level was decreased with Aβ treatments (5–30 μm) for 16 hr. All cells treated with Aβ, even at low doses, expressed significantly less CD59 than did control cells (Fig. 3).
C9 production in AD brains
Because the complement C9 component is a final component to be added into the C5b-8 subcomplex of MAC, it would be important to examine whether the C9 expression level is altered in the same regions where CD59 protein is deficient in AD brains. To address this issue, C9 component levels in frontal cortex and hippocampus brain homogenates were measured by C9 ELISA. We found that C9 protein levels were significantly elevated in both the frontal cortex (p < 0.05) and hippocampus (p < 0.01) of AD compared with ND patients (Fig. 4). Within subjects, C9 and CD59 protein levels were significantly correlated across all patients (r = 0.88; p < 0.01) and in the AD group alone (r = 0.81; p < 0.01).
Fig. 4.

C9 production in AD brains. Data represent the mean (± SE) quantification analysis of 20–22 cases using a specific ELISA assay showing that C9 immunoreactivity was increased by 35% in AD compared with ND. Results from repeated measurements displayed a significant difference between AD and ND samples (*p < 0.05; **p < 0.01).
Relationship of CD59 deficiency to neuronal or synaptic loss in AD brains
Densitometry of CD59-immunoreactive bands in CD59 Western blots and of NSE- or synaptophysin-immunoreactive bands in Western blots from the same patients and the same brain regions showed a 26% reduction in synaptophysin and a 39% decrease in CD59 in AD frontal tissue compared with that found in ND tissue (Fig. 2). Furthermore, this analysis revealed a significant correlation between CD59 and synapse decrements (r = 0.81; p < 0.05). We also note that the decreased level (52%) of CD59 from ELISA data is lower than that from the Western blot study (39%), suggesting that our CD59 ELISA is more sensitive than regular Western analysis. Meanwhile, we have also used NSE, a neuronal cell body marker, to evaluate neurodegeneration in AD brains (Fig. 2). Our results demonstrate that NSE has little change between AD and ND brains. These results suggest that CD59 deficiency may cause neuronal vulnerability and that the CD59 deficiency may not be entirely caused by neuronal loss.
CD59 deficiency and overexpression in in vitro models
To determine further the possible activity of CD59 on neurons, cultured NT2 cells, which normally underexpress CD59, and NT2 cells, transfected to overexpress CD59, were exposed to 1–3% homologous human complement-containing serum. LDH assays revealed significant complement-induced cell lysis in CD59-underexpressing NT2 cells and significant protection from such lysis in CD59-overexpressing NT2 cells (Fig. 5).
Fig. 5.

Overexpression of CD59 protects neurons against cell lysis by complement. Human neuronal NT2 cells were transfected with human CD59 cDNA and then exposed to 1 and 3% human complement for 24 hr. Values represent the mean (± SE) of four separate determinations (**p < 0.01 when compared with transfected and nontransfected cells by Student's pairedt test).
C9 overexpression and MAC induction in vitro
In AD brains, we found a CD59 deficit and C9 elevation. To examine further whether the upregulated complement C9 component would functionally induce MAC formation in vitro, silver enhancement of the peroxidase–diaminobenzidine (DAB) immunocytochemistry technique was used for MAC detection. MAC was detected on cells only after C9 transfection before C9-deficient serum treatment in NT2 cells (Fig.6D). No MAC DAB–silver deposit was observed with C9-deficient serum alone, in C9-transfected cells alone, or in nontransfected cells (Fig. 6A–C). Dramatic morphological changes, e.g., broken neurites, in C9-transfected NT2 cells were noted at 18 hr after treatment with C9-deficient serum (Fig. 6D).
Fig. 6.
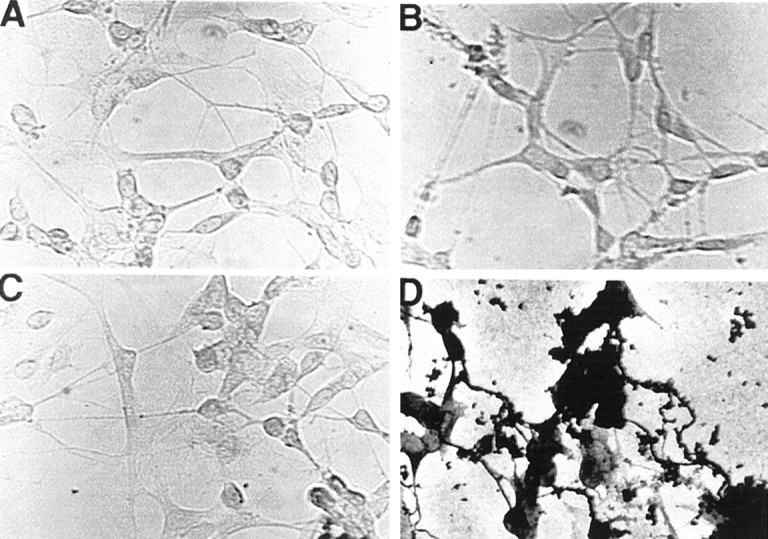
Detection of MAC from human neuronal cells by silver-staining enhancement for peroxidase–DAB. A, Cells after supplementation with human C9-deficient serum yielded minimal MAC detection in cells expressing CD59. B–D,Anti-CD59 antibody treatment alone for 18 hr (B), Aβ(1–42) treatment alone for 18 hr (C), and C9 transfection plus C9-deficient serum treatment for 18 hr after CD59-anchored protein was blocked by anti-CD59 antibody (D) are shown.B, C, Thus, treatment with the antibody of CD59 or Aβ alone produced little MAC detection in cells expressing CD59.D, Conversely, when CD59-anchored protein was blocked by anti-CD59 antibody, cells overexpressing C9 with C9-deficient serum treatment show that MAC detection was prevalent.
DISCUSSION
This is the first report of CD59 deficits in the AD brain. The absence of membrane-bound CD59 has been suggested to be causally related to the increased vulnerability of AD neurons to complement attack (Shen et al., 1998). Multiple lines of evidence demonstrate that MAC is upregulated in the AD brain (Eikelenboom and Stam, 1982; McGeer et al., 1989, 1991; Rogers et al., 1992; Piddlesden and Morgan, 1993;Itagaki et al., 1994; Webster et al., 1997; Shen et al., 1998). Logically, MAC assembly should be significantly inhibited by CD59 homologous restriction. AD deficits in the brain expression of CD59 provide at least a partial resolution of this problem. The abundance of CD59 in neurons of the brain has been related to the need for protection against complement-mediated attack (Kim et al., 1987;Yamahina et al., 1990; Wertkin et al., 1993; Shen et al., 1997, 1998;Webster et al., 1997). Conversely, CD59 levels appear to be reduced in cells with less access to complement such as oligodendrocyes in the brain (McGeer et al., 1991; Itagaki et al., 1994). Our hypothesis is that the exposure of AD cells that have an absence of or little CD59 expression to complement might result in their destruction.
We have developed an ELISA method to measure the CD59 protein at the picogram level. This assay demonstrates specific CD59 detection by the use of purified CD59 as an internal control. Moreover, we have characterized CD59 protein expression using Western blotting and found that a single band was detectable using each anti-CD59 antibody used in the ELISA (data not shown), suggesting that the antibodies are specific to CD59 detection. Because CD59 is expressed in all areas of the brain, one would expect that when PIPLC cleaves GPI-anchored CD59 proteins from ND brain slices, the released soluble CD59 in the media would be increased. However, if CD59 is previously lost in the AD brain, the cleaved CD59 in the media by PIPLC from AD brain slices would continue to be detected at the low level. Our experimental results support this hypothesis.
The level of CD59 released by PIPLC from both AD and ND brain slices was proportionally increased as the PIPLC concentration rose (Table 1), and we also found no mutation in the CD59 polymorphism (data not shown), suggesting that PIPLC cleaves the same site of CD59 in both AD and ND cases.
Although deficiency of CD59 might render cells vulnerable to MAC-mediated attack, it is possible that exposure to complement also might stimulate and increase CD59 expression in AD. Two studies (McGeer et al., 1991; Yasojima et al., 1999) have reported the presence of CD59 immunoreactivity in AD and ND brains, with particularly clear labeling of neurons. Although these two studies suggested that CD59 might be upregulated in the AD cortex at mRNA and protein levels, it is important to recognize that these studies were based entirely on reverse transcription-PCR. These immunohistochemical observations from those studies were based on unquantified data. Moreover, these same authors have reported increased MAC assembly on AD neurites (McGeer et al., 1991), which is incompatible with normal, much less elevated, CD59 expression in AD. Our results also demonstrate that synaptophysin was only reduced 26% whereas CD59 was significantly decreased 39–52% in AD brains. As our results demonstrate, measurement of CD59 by ELISA is much more sensitive than that by Western blot analysis. Surprisingly, we do not see significant change in NSE expression levels in the AD brain (Fig. 2) compared with the ND brain or observe any significant changes in microtubulin-associated protein-2 or β-tubulin III (data not shown). One possibility is that these neuronal cell body markers are not sensitive enough to detect neuron loss in the AD brain compared with synaptophysin detection. The other possibility is that synaptic loss may be faster than neuron loss in the AD brain. Taken together, these data further suggest that CD59 deficiency may not, at least entirely, be caused by synaptic loss or neuron cell body loss in the AD brain. Meanwhile, we cannot exclude the possibility that NSE may not be that sensitive to reflect neuron loss.
In the present study, analyses of CD59 mRNA and protein levels were performed using the same tissue samples from cortex regions of AD and nondemented aged-matched controls. All the tissues were collected, stored, and processed similarly. By the use of the RNase protection assay, examination of the CD59 mRNA expression in both AD and ND brain tissues and the effect of Aβ(1–42) on transcription of the human CD59 revealed that the signal indicating the CD59 mRNA level was weaker in AD brains as well as in Aβ-treated cells, but similar amounts of β-actin mRNA were found in all samples. The reduction in CD59 mRNA that occurs in the AD brain or in Aβ-treated cells strongly suggests that the CD59 gene product plays an important role in the regulation of human complement activation. A proportion of the decrease in CD59 protein in the AD vulnerable areas was a result of a decrease in CD59 mRNA. Furthermore, a decrease in CD59 protein may be a consequence of a loss of phospholipid from CD59 by PIPLC-like enzymes upregulated in the AD brain. This would result in release of the GPI-anchored CD59 protein from cell membranes. It is also noted that the scope of our studies in gene expression is not broad enough for us to exclude the possibility that endogenous Aβ may regulate CD59 gene transcription in ways other than those that may occur naturally. We propose four possibilities for how Aβ might regulate CD59 gene transcription. (1) The amyloid precursor protein (APP) gene may require post-translational modification to mediate CD59 gene expression. (2) The exogenous APP gene products, including soluble Aβ(1–40), act after protein processing because a large amount of APP mRNA is constitutively transcribed in the cells. (3) Secreted extracellular Aβ might activate some enzymes like PIPLC that cleave GPI-anchored proteins, including CD59, and make cells vulnerable to complement activation and other toxic inflammatory molecules. (4) Recent reports indicate that Aβ may form and aggregate intracellularly (Gouras et al., 2000) and that the endogenous intracellular Aβ in the AD brain might be enough to downregulate CD59 expression. Nonetheless, the present findings support recent data suggesting that Aβ in the AD brain may trigger a series of inflammatory responses and downregulate certain defense molecules, including CD59, all of which not only activate microglia and astrocytes but also compromise neuron integrity, ultimately leading to neuronal loss.
Footnotes
This work was supported by grants from the Alzheimer's Association and the Arizona Alzheimer's Research Center and by National Institute of Aging Grant NIAAGO7367. We thank Dr. T. Beach for the neuropathology evaluation and L. Sue, S. Guest, K. Lindholm, M. Gurule, and R. Lee for technical assistance.
Correspondence should be addressed to Dr. Yong Shen, L. J. Roberts Center for Alzheimer's Research, Sun Health Research Institute, P.O. Box 1278, Sun City, AZ 85351. E-mail: yshen@mail.sunhealth.org.
REFERENCES
- 1.Eikelenboom P, Stam FC. Immunoglobulins and complement factors in senile plaques. Acta Neuropathol (Berl) 1982;57:239–242. doi: 10.1007/BF00685397. [DOI] [PubMed] [Google Scholar]
- 2.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itagaki S, Akiyama H, Saito H, McGeer PL. Ultrastructural localization of complement membrane attack complex (MAC)-like immunoreactivity in brains of patients with Alzheimer's disease. Brain Res. 1994;645:78–84. doi: 10.1016/0006-8993(94)91640-3. [DOI] [PubMed] [Google Scholar]
- 4.Kim SH, Darney DF, Hammer CH, Shin ML. Nucleated cell killing by complement: effects of C5b-9 channel size and extracellular Ca2+ on the lytic process. J Immunol. 1987;138:1530–1536. [PubMed] [Google Scholar]
- 5.Masters CL, Simms G, Weiman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. 1989;107:341–346. doi: 10.1016/0304-3940(89)90843-4. [DOI] [PubMed] [Google Scholar]
- 7.McGeer PL, Walker DG, Akiyama H, Kawamata AL, Guan AL, Parker CJ, Okada N, McGeer EG. Detection of the membrane inhibitor of reactive lysis (CD59) in diseased neurons of Alzheimer's disease. Brain Res. 1991;544:315–319. doi: 10.1016/0006-8993(91)90071-3. [DOI] [PubMed] [Google Scholar]
- 8.Meri S, Morgan BP, Davies A, Danials RH, Olavesen MG, Waldmann H, Lachmann PJ. Human protein (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;72:1–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Nakano Y, Tozaki T, Kikuta N, Tobe T, Oda E, Miura N, Sakamoto T, Tomita M. Determination of the active site of CD59 with synthetic peptides. Mol Immunol. 1995;32:241–247. doi: 10.1016/0161-5890(94)00154-s. [DOI] [PubMed] [Google Scholar]
- 10.Ninomiya H, Sims PJ. The human complement regulatory protein CD59 binds to the α-chain of C8 and to the “b” domain of C9. J Biol Chem. 1992;267:13675–13680. [PubMed] [Google Scholar]
- 11.Piddlesden SJ, Morgan BP. Killing of rat glial cells by complement: deficiency of the rat analogue of CD59 is the cause of oligodendrocyte susceptibility to lysis. J Neuroimmunol. 1993;48:169–176. doi: 10.1016/0165-5728(93)90189-6. [DOI] [PubMed] [Google Scholar]
- 12.Rogers J, Cooper N, Webster S, Schultz J, McGeer P, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I. Complement activation by β-amyloid in Alzheimer's disease. Proc Natl Acad Sci USA. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen Y, Halperin JA, Lee C-M. Complement-mediated neurotoxicity is regulated by homologous restriction. Brain Res. 1995;671:282–292. doi: 10.1016/0006-8993(94)01264-i. [DOI] [PubMed] [Google Scholar]
- 14.Shen Y, Li R, McGeer E, McGeer P. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer's brain. Brain Res. 1997;769:391–395. doi: 10.1016/s0006-8993(97)00850-0. [DOI] [PubMed] [Google Scholar]
- 15.Shen Y, Sullivan T, Shiosaki K, Lin CW. Induced expression of neuronal membrane attack complex (MAC) and cell death by β-amyloid peptide. Brain Res. 1998;796:187–197. doi: 10.1016/s0006-8993(98)00346-1. [DOI] [PubMed] [Google Scholar]
- 16.Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H. GPI anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 1994;254:1016–1019. doi: 10.1126/science.1719635. [DOI] [PubMed] [Google Scholar]
- 17.Takeda J, Miyata T, Kawagoe K, Iida Y, Fujita T, Takahashi M, Kitani T, Kinoshi T. Deficiency of the GPI anchor caused by somatic mutation of the pig-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 18.Vakeva A, Laurila P, Meri S. Loss of protectin (CD59) is associated with deposition of the complement membrane attack complex in myocardial infarction. Lab Invest. 1992;67:608–616. [PubMed] [Google Scholar]
- 19.Webster S, Lue L-F, Brachova L, Tenner AJ, McGeer PL, Terei K, Walker DG, Bradt B, Cooper NR, Rogers J. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer's disease. Neurobiol Aging. 1997;48:415–421. doi: 10.1016/s0197-4580(97)00042-0. [DOI] [PubMed] [Google Scholar]
- 20.Wertkin AM, Turner RS, Pleasure SJ, Golde TE, Younkin SG, Trojanowski JQ, Lee VMY. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular-amyloid or A4 peptides. Proc Natl Acad Sci USA. 1993;90:9513–9517. doi: 10.1073/pnas.90.20.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wing MG, Zajicek J, Sceilly DJ, Compston DAS, Lachmann PJ. Oligodendrocytes lack glycoprotein anchored proteins which protect them against complement lysis. Restoration of resistance to lysis by incorporation of CD59. Immunology. 1992;76:140–145. [PMC free article] [PubMed] [Google Scholar]
- 22.Yamahina M, Ueda E, Kinoshita T, Takan T, Ojima A, Ono H, Inoue K, Kitani T. Inherited complete deficiency of 20-k homologous restriction factor (CD59) as cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1990;323:1184–1189. doi: 10.1056/NEJM199010253231707. [DOI] [PubMed] [Google Scholar]
- 23.Yasojima K, McGeer EG, McGeer PL. Complement regulators C1 inhibitor and CD59 do not significantly inhibit complement activation in Alzheimer disease. Brain Res. 1999;833:297–301. doi: 10.1016/s0006-8993(99)01514-0. [DOI] [PubMed] [Google Scholar]
- 24.Zalman LS, Brothers MS, Muller-Eberhard HJ. Isolation of homologous restriction factor from human urine. Immunochemical properties and biologic activity. J Immunol. 1989;143:1943–1947. [PubMed] [Google Scholar]
- 25.Zhan S-S, Veerhuis R, Janssen I, Kamphorst W, Eikelenboom P. Immunohistochemical distribution of the inhibitors of the terminal complement complex in Alzheimer's disease. Neurodegeneration. 1994;3:111–117. [Google Scholar]