

Abstract
Nav2/NaG is a putative sodium channel, whose physiological role has long been an enigma. We generatedNav2 gene-deficient mice by inserting the lacZ gene. Analysis of the targeted mice allowed us to identify Nav2-producing cells by examining the lacZ expression. Besides in the lung, heart, dorsal root ganglia, and Schwann cells in the peripheral nervous system, Nav2 was expressed in neurons and ependymal cells in restricted areas of the CNS, particularly in the circumventricular organs, which are involved in body-fluid homeostasis. Under water-depleted conditions,c-fos expression was markedly elevated in neurons in the subfornical organ and organum vasculosum laminae terminalis compared with wild-type animals, suggesting a hyperactive state in theNav2-null mice. Moreover, the null mutants showed abnormal intakes of hypertonic saline under both water- and salt-depleted conditions. These findings suggest that the Nav2 channel plays an important role in the central sensing of body-fluid sodium level and regulation of salt intake behavior.
Keywords: sodium channel, knock-out mouse, circumventricular organs, salt appetite, body-fluid homeostasis, osmoreceptor
Voltage-gated sodium channels (NaChs) generate the early inward current of action potentials in neurons, muscles, and related electrically excitable cells, and are thus essential for a variety of physiological processes (Noda, 1993; Catterall, 1995). The α-subunit of NaCh is the major subunit that constitutes an ion-selective channel with voltage sensor, and it is associated with one or two distinct smaller β-subunits. Since α-subunit cDNAs for brain types I, II, and III were identified first by us (Noda et al., 1986; Kayano et al., 1988), multiple structurally related isoforms of the α-subunit have been cloned from various mammalian tissues, forming a multigene family (Goldin, 1999). In addition to the excitable cells, it has recently been found that glial cells also express voltage-sensitive sodium currents (Ritchie, 1992; Sontheimer, 1994; Sontheimer et al., 1996). However, the functional roles of these NaChs in so-called “electrically inexcitable cells” have not yet been delineated.
Several years ago, a partial cDNA for the NaCh α-subunit, designated NaG, was cloned from a cDNA library derived from cultured rat astrocytes (Gautron et al., 1992). Subsequently, similar α-subunit isoforms were independently cloned from various animal species: Nav2.1 from human heart (George et al., 1992), Nav2.3 from a mouse atrial tumor cell line (Felipe et al., 1994), and SCL11 from rat dorsal root ganglia (Akopian et al., 1997); among them, SCL11 corresponded to an alternative splicing variant of NaG. From the sequence homology, it is possible to assume that they are species orthologs and/or to classify them into another α-subunit subfamily of NaCh, subfamily 2 NaCh (Nav2). The amino acid sequences were highly divergent (<50% overall identity) from those of the previously cloned voltage-gated NaChs even in the regions associated with ion selectivity and voltage-dependent activation and inactivation, suggesting that the Nav2 has specific channel properties. However, all the attempts to express functional Nav2 channels using heterologous expression systems such as Xenopus oocytes, Chinese hamster ovary cells, and human embryonic kidney 293 cells, (Felipe et al., 1994; Akopian et al., 1997) have been unsuccessful.
NaG/SCL11 was originally thought to be one of the NaChs expressed in astrocytes because it was cloned from cultured astrocytes, but subsequent in situ hybridization studies revealed that NaG is expressed not in astrocytes but in Schwann cells and the spinal sensory neurons in vivo (Felts et al., 1997). Relatively high levels of NaG mRNA are also detected outside of the nervous system, particularly in lung, kidney, and heart (Gautron et al., 1992;Akopian et al., 1997).
To examine the functional role of NaG in vivo, we generatedNaG-deficient mice, in which NaG is expressed as a fusion protein with β-galactosidase at the N terminus. In this study, we found that mouse NaG corresponds to Nav2.3. Analysis of β-galactosidase activity in the heterozygous (and homozygous) mutants revealed the localization of the NaG channel with a high spatiotemporal resolution. This showed for the first time that mouse NaG was restricted to neurons and ependymal cells in a subset of nuclei and organs in the CNS. In addition, behavioral studies of the null mutants revealed a selective involvement of the NaG channel in central control of salt intake behavior.
MATERIALS AND METHODS
Construction of the targeting vector. Nine independent genomic clones were isolated from mouse genomic libraries (kindly donated by Dr. Masahiko Mori, Osaka University, Suita, Japan) prepared from a cell line, R1, by hybridization with a 446 bp fragment of rat NaG cDNA (nucleotide residues 11–456 encompassing the first three coding exons: GenBank accession number Y09164). By Southern blot analysis using several restriction enzymes, all these overlapping clones were confirmed to be derived from a single genomic locus. The hybridization-positive 3.2 and 3.7 kb HindIII fragments were subcloned into pBluescript II SK(−) (Stratagene, La Jolla, CA) and subjected to DNA sequencing (the sequence is in GenBank under accession number AF190472). The 3.2 kb fragment contained protein-coding exon 1 (13 bases of the 5′ untranslated region and the first 238 bases in the mNav2 protein-coding sequence; nucleotide residues 238–490 of GenBank accession number L36179), and the 3.7 kb fragment contained exons 2 (491–609) and 3 (610–701). To construct the targeting vector, the 12.5 kb SalI fragment containing the three exons was inserted into the XhoI site of pDT-A (Yagi et al., 1993) (Fig. 1a). The SalI-XhoI fragment of the lacZ-neocassette was introduced into the endogenous XhoI site located in exon 1. An EcoRI linker sequence was inserted beforehand into the KpnI site, which is located on the 3′ side of the SalI site in the 5′ terminus of thelacZ-neo cassette, to join the first 20 amino acids of the mNav2 protein to the N terminus of β-galactosidase and to use it as an exogenous restriction site in Southern blot screening analysis.
Fig. 1.

Targeted disruption ofmNav2 gene. a, Restriction maps of the targeting vector (top), endogenousmNav2 gene locus (middle), and recombinant gene locus (bottom). The protein-coding exons are indicated asclosed boxes. Targeted insertion of thelacZ-neo cassette into the first protein-coding exon was accomplished using the targeting vector. Restriction sites shown are as follows: B, BamHI; Bg,BglII; E, EcoRI;H, HindIII; and X,XhoI. b, Southern blot analysis of genomic DNA digested with EcoRI. Samples are derived from tails of wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice. Blotted membranes were hybridized with probe 1 located outside of the 5′ terminus of the targeting vector. The sizes for the wild-type (18 kb) and recombinant (10 kb) genotypes are shown on theright. The insertion was verified by using probe 2 located inside the targeting vector. c, Genomic PCR analysis of wild-type, heterozygous, and homozygous mutant mice. The sizes for the wild-type (200 bp) and recombinant (400 bp) genotypes are shown on the right. d, Western blot analysis using anti-mNav2 polyclonal antibody. Samples were prepared from the lungs of wild-type, heterozygous, and homozygous mutant mice. The position of the channel protein (220 kDa) is indicated on the right. The sodium channel gave a broad signal, because the protein is highly glycosylated and readily aggregates even in the SDS-containing buffer.
Generation of Nav2/NaG deficient mice.Culture of embryonic stem cells (R1 cell line of 129/Sv mouse) with G418 and screening of the targeted clones were performed as described previously (Shintani et al., 1998). In brief, homologous recombination was verified by Southern blot analysis using EcoRI digestion with probe 1 (Fig. 1a; the 0.3 kbEcoRI-XbaI fragment located ∼8 kb upstream from the XhoI site in exon 1). The selected clones were checked using probe 2 (the 0.6 kb PstI fragment derived from theneo gene). The targeted locus was also verified by genomic PCR using one sense primer (primer 1, ATGTTGACTTCCCCAGAGCC in the 5′ terminal region of exon 1) and two antisense primers (primer 2, AACCAGGCAAAGCGCCATTC in the 5′-terminal region of lacZ; primer 3, CATCTTCCAAGGGCTCTGACA in the 3′-terminal region of exon 1). Amplification was performed in two stages with EX-Taq DNA polymerase (Takara, Shiga, Japan) according to the manufacturer's protocol using a programmable thermal cycler. One of the two targeted cell lines (of 98 ES clones) was used to generate the mutant mice through germline transmission. Generation of mutant mice was performed as described previously (Shintani et al., 1998). All the animal experiments were performed according to the guidelines of the National Institute for Basic Biology (Okazaki, Japan).
Western blot. The mNav2-protein expression in the mutant mice was examined by Western blot analysis. Lung tissue samples were prepared according to the methods of Knittle et al. (1996). SDS-PAGE and immunoblotting were performed as described previously (Shintani et al., 1998). Anti-mNav2.3 antiserum (a generous gift from Dr. M. Tumkun, Colorado State University, Fort Collins, CO) was used at a 1:500 dilution in PBS. The immunoblots were incubated also with several nonimmune sera to verify the fidelity of the antibody.
X-Gal staining. Embryos were fixed by immersion in 3.5% formaldehyde in PBS for 1 hr at room temperature and then cut midsagittally with a razor. The postnatal animals were perfused transcardially under deep pentobarbital anesthesia first with PBS and then with the fixative. The fixed brains were cut coronally at 2-mm-thick or sagittally at the midline level with a razor. Samples were rinsed twice with PBS and incubated overnight in PBS containing 1 mg/ml X-Gal, 5 mmK3Fe(CN)6, 5 mmK4Fe(CN)6, 2 mmMgCl2, and 0.2% NP-40 at 37°C. For immunostaining, some X-Gal-stained slices were cut further into coronal sections at 14-μm-thick with a cryostat microtome and mounted onto gelatin-coated slides. Immunostaining was performed with rabbit polyclonal antibodies to neurofilament 200 (N-4142; Sigma, St. Louis, MO) or glial fibrillary acidic protein (GFAP) (sc-6170; Santa Cruz Biotechnology, Santa Cruz, CA) as described previously (Shintani et al., 1998).
Fos-immunohistochemistry. Animals were deprived of water for 0 hr (n = 4 formNav2+/+;n = 4 formNav2−/−), 12 hr (n = 5 and 5), 24 hr (n = 6 and 7), or 48 hr (n = 6 and 5). The animals were then perfused with the fixative as described above, and their brains were immersed in the same fixative at 4°C overnight. Brains were cut coronally into serial sections at 50-μm-thick on a vibratome (VT1000S; Leica, Heerbrugg, Switzerland). Immunostaining was performed by free floating with a goat anti-Fos polyclonal antibody (sc-52-G; Santa Cruz Biotechnology) at a dilution of 1:1000 in PBS. Sections containing regions of interest were chosen, and the Fos-immunopositive nuclei were enumerated. Each area was measured by using an image analysis system (KS400 attached to Axiophoto2; Zeiss, Hallbergmoos, Germany). The number of nuclei present per square millimeter was determined in five regions of the brain: organum vasculosum laminae terminalis (OVLT), subfornical organ (SFO), median preoptic nucleus (MnPO), supraoptic nucleus (SON), and paraventricular nucleus (PVN).
Behavioral analyses. For all behavioral studies, male mice at 12–24 weeks of age were used. They were individually housed under constant room temperature, humidity, and 12 hr light/dark cycle (lights on at 7:00 A.M.). Protocols: (1) Preference-aversion behavior was measured with a 48 hr two-bottle preference test (see Fig. 5). Mice were presented with a choice between distilled water and a tasting solution for 48 hr in their home cage: the positions of the two bottles were switched every 24 hr to avoid side preference. The total intake for each animal in 48 hr was measured and used to calculate a preference ratio according to the following formula: preference ratio = volume of tasting solution (in milliliters)/total intake volume of tasting solution and water (in milliliters). (2) Dehydration-induced aversion to hypertonic saline was assessed with a 6 hr two-bottle preference test in the home cages (see Fig. 7). Before testing, mice were trained to drink water from two bottles for 1 week. On the day before dehydration, mice were presented with a choice between water and 0.3 m NaCl at 10:00 A.M. and then measured for fluid intake at 4:00 P.M. At 10:00 A.M. on the next day, the bottles were removed. Dry food was placed throughout the period of water deprivation. After 24 hr dehydration, the two bottles were returned, and fluid intakes were measured at 4:00 P.M. In some cases, blood was recovered by decapitation from animals before or after dehydration, and then the concentrations of plasma electrolytes were measured by using an electrolyte analyzer (9180; AVL Scientific, Roswell, GA). (3) The sodium depletion-induced salt appetite test was performed in the following way (see Fig. 8). Before testing, control measurements of water and 0.3 m NaCl intake were performed for several days. At 10:00 A.M., mice were injected intraperitoneally with 0.12 ml of normal saline (0.9% NaCl). The bottle of 0.3m NaCl was withdrawn, and sodium-depleted food was supplied in place of a normal diet. A second injection of normal saline was given at 4:00 P.M. On the following day, water and 0.3 mNaCl were presented at 10:00 A.M., and intakes of 0.3 mNaCl and water were measured at 12:00, 2:00, and 4:00 P.M. After that, a similar protocol with furosemide injection (0.6 mg in 0.12 ml of normal saline) was performed with a sodium-depleted food in the same mice (acute salt-appetite condition). Finally, the same protocol, except that normal sodium-containing food was supplied, was performed to evaluate the effect of sodium-depleted food.
Fig. 5.

The null mutants showed normal preferences to various tastants under the condition satiated with water and salt. Preference ratios for NaCl solutions with a series of concentrations (a) or three fundamental tastants with fixed concentrations (b) were examined by a 48 hr two-bottle preference test. n = 5 (+/+), 5 (+/−), and 5 (−/−). Vertical bars indicate SE.
Fig. 7.

The null mutants showed an abnormal ingestion of hypertonic saline under the thirst condition. Preference ratio for 0.3m NaCl solution (a) and total fluid intake (b) per 6 hr was measured before and after 24 hr dehydration; n = 6 (+/+), 6 (+/−), and 6(−/−). Vertical bars indicate SE. *t test analyses revealed a significant difference (p < 0.05) betweenmNav2−/−and mNav2+/+mice.
Fig. 8.

The null mutants showed an excessive ingestion of hypertonic saline under the acute salt appetite condition. An acute salt appetite was induced by feeding a sodium-depleted diet combined with furosemide injection intraperitoneally. As a control, normal saline was injected intraperitoneally instead of the furosemide solution. As another control, a sodium-containing diet was given instead of the sodium-depleted diet. The behavioral study was sequentially performed on alternate days as follows: sodium-depleted diet combined with normal saline injection (top), sodium-depleted diet combined with furosemide injection (middle), and sodium-repleted diet combined with furosemide injection (bottom). Mean cumulative intakes of 0.3m NaCl (right) and water (left) per 2 hr on the day just after each experimental procedure were plotted; n = 10 (+/+), 10 (+/−), and 10 (−/−). Vertical bars indicate SE. *t test analyses revealed a significant difference (p < 0.05) betweenmNav2−/−and mNav2+/+mice.
Electrophysiology. Male mice at 12–24 weeks of age were used (four wild-type and five homozygous mice for the normal condition and three and five mice for the acute salt-appetite condition as described above). The animals were deeply anesthetized by an intraperitoneal injection of sodium pentobarbital (60 mg/kg). Each animal was tracheotomized and secured with a head holder. The right chorda tympani nerve was exposed, freed from surrounding tissues, and cut at the point of its entry to the bulla. The whole bundle of the nerve was dissected and lifted on a platinum wire recording electrode (0.1 mm in diameter). An indifferent electrode was attached to nearby tissue. The nerve activities were amplified, displayed on an oscilloscope, and monitored with an audioamplifier. The amplified signal was passed through an integrator with a time constant of 0.3 sec and was displayed on a slipchart recorder. The taste solutions were (inm): 0.1 NH4Cl, 0.1 NaCl, 0.5 sucrose, 0.01 HCl, 0.02 Q-HCl, 0.1 KCl, and 0.1 CH3COONa. These solutions were made up with distilled water or 0.1 mmamiloride solution. Each solution and rinsing water were applied to the anterior part of the tongue at room temperature (25 ± 2°C). The tongue was rinsed for at least 45 sec between successive stimulations. The magnitude of the whole nerve response was measured as the height of the integrated response from the baseline at 10 sec after onset of stimulation. Responses to taste stimuli were expressed as relative values with the magnitude of the response to 0.1 mNH4Cl taken as the standard.
RESULTS
Mouse Nav2.3 is the ortholog of rat NaG/SCL11
To construct the targeting vector (Fig. 1a), we cloned mouse genomic fragments containing protein-coding exons 1, 2, and 3 with a rat NaG/SCL11 probe. The DNA sequence encoding the three exons was identical to that of mouseNav2.3 cDNA cloned by Tamkun and coworkers (Felipe et al., 1994). The cloned genomic fragments showed identical restriction maps with mouse genomic DNA on Southern blot analysis with four restriction enzymes (BamHI,BglII, EcoRI, and HindIII; data not shown). This finding indicates that mouse Nav2.3 is the ortholog of rat NaG. In this paper, we designate Nav2.3/NaG/SCL11 as Nav2.
Targeted disruption of mNav2 gene
The lacZ gene was designed to be inserted in-frame into the protein-coding exon 1 of the mNav2gene: the N-terminal 20 amino-acid sequence of mNav2 is fused with β-galactosidase. The original genomic structure of mNav2 gene was not modified in the targeting vector except for the insertion of the lacZ-neo cassette (Fig. 1a) to make sure that the lacZ gene is expressed in place of themNav2 gene in the targeted mice. The gene replacement was confirmed by genomic Southern blot hybridization (Fig.1b) and PCR (Fig. 1c) analysis and the absence of mNav2 protein expression by immunoblot analysis with anti-mNav2 antibody (Fig. 1d). Approximately half of the normal amount of mNav2 protein in heterozygous mutant (mNav2+/−) mice and no mNav2 protein in homozygous mutant (mNav2−/−) mice was detected in lung membrane preparations, indicating that the allele was a null mutation.
Figure 2a shows X-Gal staining of a whole-mountmNav2+/− embryo at embryonic day 15 (E15). Intensive β-galactosidase activity was observed in the trigeminal and dorsal root ganglia (DRG). In E15 mice,lacZ expression was evident also in the lung. In these organs, the expression of lacZ persisted into adulthood. When DRGs were cut into thin sections after X-Gal staining, β-galactosidase activity was detected in spinal sensory neurons with various cellular diameters (Fig. 2b). Signals were confined to the somata of neurons within the DRG, and they were not detected in axons. A similar pattern of lacZ expression was observed in tissue sections of the trigeminal ganglia. Positive expression was also observed in Schwann cells in the adult sympathetic trunk (Fig.2c), cardiac autonomic nerves, lingual nerves, nonmyelinated nerve fibers beneath the taste buds, and uterine nerves (data not shown). These patterns of lacZ expression agreed well with the results of Northern blot, RT-PCR, RNase protection, andin situ hybridization studies onrNav2 andmNav2 expressions (Gautron et al., 1992;Akopian et al., 1997; Felts et al., 1997), indicating that thelacZ gene expression is duly under the control of the regulatory elements of the mNav2gene.
Fig. 2.

The distribution pattern of lacZexpression in the peripheral organs. a,LacZ expression in whole-mount E15 embryos ofmNav2+/− mice. E15 embryo was cut midsagittally and then stained with X-Gal. Theblue signals represent the site expressinglacZ. The arrow points to DRGs, thearrowhead points to a trigeminal ganglion, and theasterisk shows the lung. b, An X-Gal-stained cryostat tissue section of dorsal root ganglion of postnatal day 2mNav2+/−mice. Nerve tracts are shown by asterisks.c, A cryostat section of adult sympathetic nerve trunk in the thoracic region. Based on the appearance, distribution, and size of the cell bodies, the numerous intensely stained cells are likely to be Schwann cells. Arrowheads identify the somata of Schwann cells. Scale bar, 50 μm.
The null mutant animals (mNav2−/−) were healthy, fertile, and apparently normal. The genotypic analysis of 458 4-week-old offspring obtained from breeding heterozygous animals showed an approximately Mendelian ratio between wild-type (29.5%,n = 135), heterozygous mutant (48.2%,n = 221), and homozygous mutant (22.3%,n = 102) animals. This suggests that embryonic development and body functions were not significantly impaired in either heterozygous or homozygous mutant animals. It was reported thatmNav2 shows acute and transient expression in the perinatal uterine smooth muscle (Felipe et al., 1994; Knittle et al., 1996). In fact, myometrium of the pregnant uterus at days 18 and 20 was significantly positive for lacZ (data not shown). However, noteworthily, pups were delivered normally inmNav2−/−. The pattern of lacZ expression in the null mutants was identical to that in the heterozygous mutants except for the intensity of the expression, suggesting that deficiency ofmNav2 does not affect the differentiation or viability of the mNav2-expressing cells.
Expression pattern of mNav2 gene in the CNS
For designing an experimental strategy to elucidate the physiological roles of mNav2, we surveyed thelacZ expression throughout the CNS using fixed brains ofmNav2+/− andmNav2−/−mice. Interestingly, clusters of lacZ expression were limited to specific loci in the CNS (Fig.3a–f): the medial preoptic, anterior, and dorsomedial part of the hypothalamic area (MPO, AH, and DMH, respectively), dorsomedial part of the interpeduncular nucleus (IPDM), medial part of the median raphe (MMR, so-called rhabdoid nucleus), mesencephalic nucleus of V (MeV), medial habenular nucleus (MH), median eminence (ME), SFO, OVLT, and neurohypophysis (NHP). The four midline structures, ME, SFO, OVLT, and NHP, are known as the circumventricular organs (CVOs), having unusual dense and permeable capillary networks that facilitate secretion or tissue penetration of circulating substances (Johnson and Gross, 1993). Relatively weak lacZ expression was detected in the cerebral cortex (CX) in layer IV of the lateral area (from the most anterior portion to the end of the posterior portion of the cortex) and the basolateral amygdala (BLA) inmNav2+/− mice (data not shown). The intensity of the lacZ expression in these areas was more evident inmNav2−/−mice, expectedly (Fig. 3f).
Fig. 3.

mNav2 was expressed in specialized neurons and ependymal cells in the adult CNS.LacZ expression in the CNS ofmNav2+/−(a–e) andmNav2−/−(f) mutant mice. Fixed adult brains were cut coronally at 2 mm (a, b, d–f) or into halves midsagittally (c) and then stained with X-Gal. Inc, the skull under the brain was not removed. Ine, homozygous mutant mice were used for the analysis to detect the locus of low-level expression. AH, Anterior hypothalamic area; MH, medial habenular nucleus;ME, median eminence; OVLT, organum vasculosum laminae terminalis; MPO, medial preoptic area;DMH, dorsomedial hypothalamus; IPDM, interpeduncular nucleus of the dorsomedial part; MMR, medial part of the median raphe; NHP, neurohypophysis;SFO, subfornical organ; CX, cerebral cortex;BLA, basolateral amygdala. In c, OVLT was removed from the CNS and attached to the skull. The coronal semi-whole-mount brains were cut 50-μm-thick using cryostat microtome and then stained with anti-neurofilament (g, h), anti-GFAP (i) polyclonal antibodies, or cresyl violet (j). Brown signals are the site that reacted with the antibodies. The samples are AH (g), SFO (h,i), and ME (j). Arrowheads indicate double-positive neurons. The asterisk in j indicates the third ventricle. The dorsal side is toward the top of the panels. Scale bar: g–i, 30 μm; j, 100 μm.
To examine the cell types expressing lacZ, the semi-whole-mount brains stained with X-Gal were cut into tissue sections with a cryostat microtome and subsequently immunostained with anti-neurofilament or anti-GFAP polyclonal antibodies or stained with cresyl violet. Most of the cells expressing lacZ were positive for neurofilament in the PO, AH (Fig. 3g), IPDM, MMR, MH, and MeV. GFAP-positive cells were negative for thelacZ expression, suggesting that astrocytes are negative formNav2. The distribution oflacZ-expressing cells in the CVOs was of particular interest to us. In the ME, the lacZ-expressing cells lined the floor of the third ventricle (Fig. 3j). This distribution corresponds to the location of nonciliated ependymal cells, tanycytes, which are characteristic cells providing a morphological connection between CSF, nerve cells and blood vessels (Pilgrim, 1978). They are thought to be involved in exchange of substances between the CSF and pericapillary space. The lacZ-positive cells were sparsely distributed all over the SFO, and most of them were colocalized with neurofilaments (Fig. 3h) and negative for GFAP (Fig. 3i). Intensive lacZ-positive cells also populated the lining of the entire third ventricle, suggesting that they are ependymal cells. The profiles in the OVLT were very similar to those observed in the SFO. In the NHP, the X-Gal signals were densely clustered. We could not identify the cell types, however, they are likely to correspond to so-called pituicytes (Hatton, 1988), which are a mixture of several unidentified cell types.
Abnormal increase of Fos-immunoreactivity in the CVOs
Analysis of the lacZ expression clearly demonstrated that mNav2 was expressed in the four CVOs and several minor nuclei in the CNS. Because themNav2-expressing cells were thus diverse not only in tissue distribution but also in cell types, it is difficult to obtain a unified view of channel function or property simply from the distribution. However, among them, the four CVOs are thought to be involved in body-fluid homeostasis (Andersson, 1978; Johnson and Gross, 1993; Ferguson and Bains, 1996; Bourque and Oliet, 1997; Fitzsimons, 1997; Johnson et al., 1999). If the mNav2 channel functions in the sensory circuits for body-fluid osmolarity, it was expected that the activity and gene expression in these organs would be affected in the mNav2 mutant mice. Therefore, we next examined the effects of water deprivation on the central expression of a nuclear protein, Fos, the product of thec-fos proto-oncogene. Fos is a marker of changes in neural activity in response to the extracellular fluid balance in mice and rats (Ueta et al., 1995; Chae and Heideman, 1998; Morien et al., 1999). Figure 4 shows the time course of changes in Fos-immunopositive cell density in five regions of the brain under water-satiated and dehydrated conditions. In the water-satiated condition, Fos-immunopositive cells were not detected in any region examined (the MnPO, OVLT, SFO, PVN, and SON) inmNav2+/+ andmNav2−/−mice (Fig. 4a). At 12, 24, and 48 hr after water deprivation, cell numbers with Fos-immunopositive nuclei were remarkably increased in these regions in both mice. However, in the SFO and OVLT, approximately twofold increases in Fos-immunopositive nuclei were observed inmNav2−/−mice as compared with inmNav2+/+ mice. In the MnPO, PVN, and SFO, on the other hand, the rates of increase in Fos-immunopositive cells were comparable between the two groups (Fig.4b).
Fig. 4.
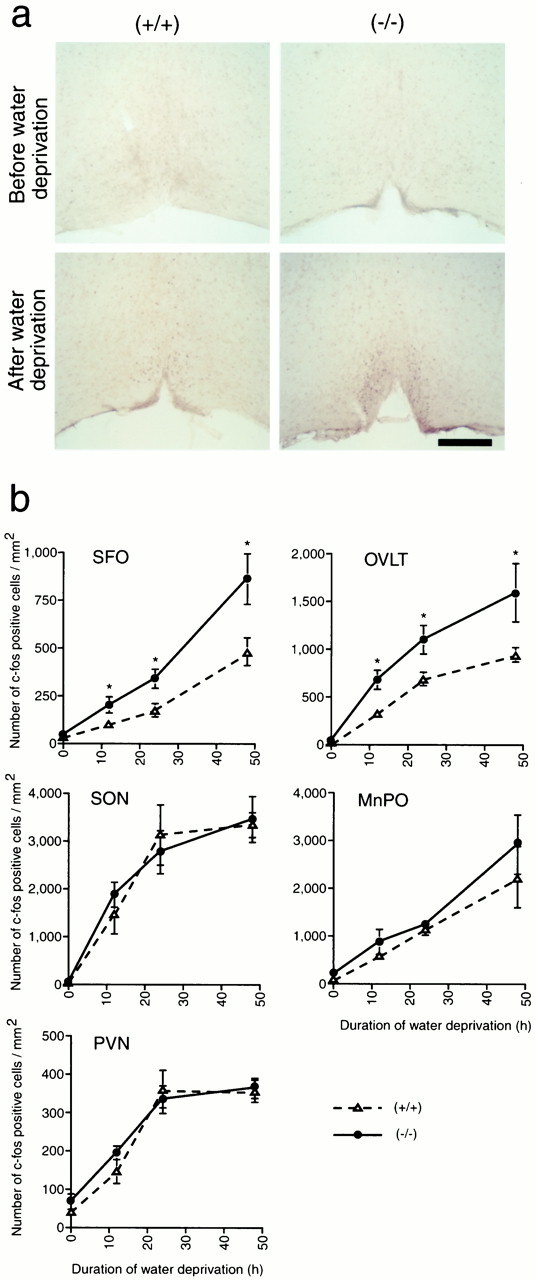
Abnormal increases of Fos-immunopositive nuclei were selectively observed in the SFO and OVLT of the null mutants under conditions of thirst. Wild-type or null mutant mice were dehydrated for 0, 12, 24, and 48 hr, and then fixed. The fixed brains were cut coronally into 50-μm-thick sections and then stained with anti-Fos polyclonal antibody. a, Typical examples of tissue sections containing the OVLT derived from euhydrated or 24 hr dehydrated wild-type (+/+) and null mutant (−/−) mice. Scale bar, 200 μm. b, Mean numbers of Fos-immunopositive cells per square millimeter in the SFO, OVLT, SON, PVN, and MnPO during water deprivation were plotted. Vertical bars indicate SE. *ttest analyses revealed a significant difference (p < 0.05) betweenmNav2−/−and mNav2+/+mice.
The taste reception ofmNav2−/−mice is not impaired
We next examined the effect of mNav2-channel deficiency on mouse behavior, focusing on water and salt intakes. For the behavioral study, the mutant mice were backcrossed with C57BL/6J for four generations to verify that the behavior was identical between the F1 and the N4 mice and was not influenced by the 129/Sv genetic background of the ES cells. Because Schwann cell clusters that are located and associated with nerve fibers beneath the taste buds were intensely positive for lacZ (data not shown), we carefully examined whether there exist abnormalities in the taste reception inmNav2−/−mice.
The taste preferences of the homozygous mutant, heterozygous mutant, and wild-type littermates were first examined by a 48 hr two-bottle preference test against pure water. Under the condition satiated with water and salt, the concentration sensitivity to a series of NaCl solutions was comparable among the three groups of mice (Fig.5a). All the groups showed maximum preference to 0.1 m NaCl and evasiveness to 0.3 m NaCl or more. Preference ratios to sweet (0.5 m sucrose), sour (0.01m HCl), and bitter [0.02 mquinine (Q)-HCl] tastants were not different among the groups (Fig. 5b).
To further verify the normality in taste responses in the null mutants, electrophysiological analysis was performed on a taste-afferent nerve tract, the chorda tympani nerve, which is known to be the nerve fiber responsible for tasting NaCl (Fig. 6). The neurophysiological responses to (in m): 0.1 NaCl, 0.1 KCl, 0.1 CH3COONa, 0.5 sucrose, 0.01 HCl, and 0.02 quinine-HCl were of similar intensity between the null-mutant and wild-type mice. Responses to 0.1 m NaCl and 0.1m CH3COONa were decreased to the same degree by amiloride application in both groups of mice, indicating that amiloride-sensitive channels in taste buds in the mutant mice function normally. Similar results were observed in the null-mutant and wild-type mice under the acute salt appetite condition (data not shown). These findings, together with the normal behavioral response to various tastants under water- and salt-sufficient conditions (Fig. 5), indicate that the taste reception of the null mutants is not impaired.
Fig. 6.

Normal responses to various tastant stimuli in the chorda tympani nerve of the null mutants. a, Sample recording of the integrated chorda tympani responses to (in m): 0.1 ammonium chloride (NH4Cl), 0.1 sodium chloride (NaCl), 0.1 NaCl with 0.1 mm amiloride, 0.1 KCl, 0.1 sodium acetate (AcNa), 0.1 AcNa with 0.1 mm amiloride, 0.5 sucrose, 0.01 hydrochloric acid (HCl), and 0.02 quinine hydrochloride (Q-HCl) in the wild-type and mNav2 null mutant mice. b, Mean magnitude of responses to various taste stimuli. The values are expressed relative to the magnitude of the response to 0.1 m NH4Cl.n = 4 (+/+) and 5 (−/−).
Abnormal intake of hypertonic saline under thirst and salt appetite conditions
In the dehydrated condition, animals take in a large quantity of water and avoid hypertonic saline to recover from the hypertonic state. We then examined the preference to hypertonic saline (0.3 mNaCl) of the three genotypes before and after 24 hr dehydration. In contrast to the wild-type and heterozygous mutant mice, which showed markedly decreased preference ratios to hyper-salt solution after dehydration, the null mutants showed no change in the preference ratio (Fig. 7a). Total water intake (water plus 0.3 m NaCl) did not differ among the groups both before and after 24 hr dehydration. The total water intake of all the groups showed a more than fourfold increase after dehydration (Fig. 7b). The electrolyte concentrations in the serum before and after dehydration were normal in both wild-type and homozygous-mutant mice (n = 6): before dehydration: 153.6 ± 0.6 and 153.0 ± 1.2 mm for Na+, respectively; 4.6 ± 0.1 and 4.7 ± 0.1 mm for K+; 118.5 ± 0.6 and 118.3 ± 0.9 mm for Cl−; after dehydration: 151.6 ± 0.8 and 150 ± 0.3 for Na+; 6.5 ± 0.2 and 6.7 ± 0.2 for K+; 116.0 ± 1.0 and 116.4 ± 0.8 for Cl−. This suggests that the null mutants immediately excreted excessive amounts of sodium into urine, and thus the renal function of null mutants works normally.
Furthermore, motivated salt appetite was induced by intraperitoneal injection of a diuretic drug, furosemide, and a sodium-free diet. The ingested volumes of pure water and 0.3 m NaCl are shown in cumulative values for every 2 hr (Fig.8). Under the control condition, in which isotonic saline was injected in place of a furosemide solution, the ingested volumes of water and 0.3 m NaCl were comparable among the three groups (two graphs at the top). Under the acute salt appetite condition induced by furosemide injection intraperitoneally with a sodium-depleted diet, however, the null mutants showed an approximately twofold increase in the ingestion of 0.3 mNaCl compared with the wild-type and heterozygous mutant mice (middle). This abnormal ingestion of hypertonic saline stopped when a sodium-containing conventional food was provided (bottom).
DISCUSSION
In this study, we applied a gene knock-out strategy to know physiological roles of subfamily 2 NaChs (Nav2), because all the attempts to functionally express Nav2 have not been successful. Here, we found that mouse NaG/SCL11 gene corresponds to Nav2.3. This finding suggests that Nav2 members identified to date from different species are species orthologs. The null mutant animals (mNav2−/−) were healthy, fertile, and apparently normal. Thus, the phenotype is quite distinct from those of subfamily 1 NaCh-deficient mice, for instance SCN2A and SCN8A, which were reported to die postnatally because of neuronal defects in electrical activities (Burgess et al., 1995; Planells-Cases et al., 2000). Here, detailed expression ofNav2 in the CNS was revealed, for the first time, from the lacZ expression analysis of the gene-targeted mice. Furthermore, interestingly,Nav2-deficient mice showed abnormal salt-intake behavior under water- and salt-depleted conditions without affecting the taste reception. This is probably attributable to a failure to detect circulating sodium concentration in the CNS.
To maintain water and mineral homeostasis, the role of central osmosensitive and sodium-sensitive neurons in water- and sodium-intake behaviors is critical (Denton et al., 1996; Bourque and Oliet, 1997). Osmo- and sodium-regulatory responses are thought to be controlled by a group of specialized neurons that sense changes in the external osmotic pressure and sodium concentration and elicit appropriate electrical signals. Changes in systemic osmolarity and sodium concentration are known to be detected mainly by the central CVOs, i.e., MnPO, OVLT, SFO, and SON. Neurons in the CVOs are located outside the blood–brain barrier, where the brain can monitor various substance levels in the plasma and CSF, either directly or indirectly.
The OVLT, SFO, MnPO, SON, and PVN are known to be the sites wherec-fos expression is upregulated in animals under thirst and acute salt-appetite conditions (Rawland, 1998). We found thatmNav2-null mutants showed a similar increase of c-fos expression in these areas except for SFO and OVLT, where the increase was approximately twofold that of the wild type (Fig. 3). Furthermore, the null mutants showed abnormal behavior in ingestion of hypertonic saline under thirst and acute salt-appetite conditions (Figs. 7, 8). Here, it was evident that this abnormality is caused by a defect in the central control because sensing of various tastes in the peripheral organ was normal including salt taste (Figs.5, 6). Under both conditions, the abnormality was observed in salt intake, but not in total water intake, indicating that these two behaviors are controlled separately in the CNS and the mNav2 channel is essential for the regulation of salt intake behavior.
The cephalic receptors were first hypothesized to be osmoreceptors in the light of Verney's elegant demonstration of anti-diuretic hormone (Verney, 1947). The magnocellular neurosecretory cells in the SON appear to exhibit intrinsic osmosensitivity (Leng, 1980). The increase in firing rate observed in the vasopression-secreting cells after direct application of hypertonic saline is thought to be related to a reduction in cell volume (Oliet and Bourque, 1993). However, mNav2 was not expressed in SON, and therefore magnocellular neurons are negative for mNav2 (Fig. 3). Glial cells in the SON and PVN have a striking morphological appearance with long astrocytic processes radiating between the magnocellular neurons. The glial-specific distribution of the water channels aquapolin-4 (AQP-4) in SON (Nielsen et al., 1997) implies that it may be glial cells, rather than neurons, that exhibit intrinsic osmosensitivity. Thus, glial cells might function as the primary “vesicular osmometer”, conveying information on the state of water homeostasis to the neural elements by transcellular water movement. Of note is that glial cells were negative formNav2 expression (Fig. 3).
This may not be true for the SFO, where the distribution of AQP-4 is restricted to the basolateral membrane of ependymal cells lining the ventricles (Nielsen et al., 1997). Here, transmembrane water movement might be a prerequisite driven by an ionic gradient established with CSF, and thus osmosensitive neurons in the SFO may respond to the ionic composition of the CSF. This mechanism may not be confined to the SFO, but may be a common feature of the other CVOs, including OVLT. On the contrary, neurons in the OVLT and MnPO have also been shown to be intrinsically osmosensitive, and the integrity of the neural connection linking the SON, OVLT, and MnPO is necessary for SON neurons to respond to osmotic stimulation (Honda et al., 1990). In vivoanalyses (Gutman et al., 1988; Honda et al., 1990) and electrophysiological experiments using slices or explants have revealed that a majority of neurons within the SFO (Sibbald et al., 1988) and OVLT (Nissen et al., 1993) are actually excited or inhibited on exposure to hypertonic or hypotonic NaCl solutions. Noteworthy is that the saline solutions are more effective in activating cells of the OVLT than the mannitol-containing equiosmolar solutions (Vivas et al., 1990).
Here, it should be recalled that water intake behavior was normal in the mutants (Figs. 7, 8). The mutants appear to be able to somehow detect hyperosmolarity because they showed increase in the total water intake under thirst conditions. The SFO and OVLT were selectively hyperactive in the CVOs in the mutant mice under thirst conditions (Fig. 4). Activities of the SFO and OVLT are known to affect both water and salt intake behavior (Andersson, 1978; Johnson et al., 1999). Sensory information on the systemic body-fluid condition is integrated in the higher central area, which controls separately salt and water intake behavior. This implies that osmosensitive and sodium-sensitive neurons make up different populations in these areas. Lesion experiments have shown that the central nucleus of the amygdala selectively regulates salt appetite, but not water-drinking behavior, induced by angiotensin II (ANG II) or sodium depletion (Galaverna et al., 1992, 1993; Zardetto-Smith et al., 1994). The expression of mNav2 in amygdala was restricted to the basolateral nucleus and did not overlap with the central nucleus (Fig.3f), suggesting that mNav2 channel deficiency in the amygdala would not directly affect salt intake behavior. Because there exists direct efferents from the lamina terminalis, particularly from the SFO, projecting to the central nucleus of the amygdala (Johnson et al., 1999), it is possible that the hyperactivated neurons, with strong c-fos expression of the SFO and OVLT in null mutants under conditions of thirst, selectively regulate the activity of the central nucleus. We envisage that these are probably sodium-sensitive neurons, and mNav2 channel is involved in the sensing mechanism for the extracellular sodium concentration. In addition, the mNav2 channels expressed in the ependymal cells might also contribute to the sodium-absorption activity from the extracellular space. As described above, a water channel, AQP-4, is reported to be involved in the transportation of water through ependymal cells in the central ventricular systems (Wells, 1998). It is thus possible to envisage that the deficiency of mNav2 channels in ependymal cells also altered the extracellular sodium concentration surrounding the central sensory neurons.
A similar situation was observed in the alveolar epithelium: an intensive expression of mNav2 was observed in alveolar type II (ATII) cells of the pulmonary alveolus (our unpublished data). These cells actively absorb sodium ion (Matalon and O'Brodovich, 1999) and water (Verkman, 1998) and regulate alveolar surface fluid conditions. Water moves mainly across alveolar type I (ATI) cells and airway epithelium into the capillaries through water channels. The sodium ion influx through nonselective cation channels and sodium ion-selective channels on the apical membrane of ATII cells seemed to be the driving force. It is thus possible to assume that the mNav2 channel is one of the molecular entitities of these cation currents detected in the ATII cells.
In gene-targeting studies, the background genotypes should be considered (Gerlai, 1996). A recent electrophysiological study showed that there exists a prominent difference between mouse strains in the presence of the amiloride-sensitive component of the salt-sensing system (Ninomiya et al., 1989). However, our electrophysiological study demonstrated the existence of the amiloride-sensitive component in the null mutants generated by backcrossing four times onto a pure C57BL/6J background (Fig. 5). As for the amiloride-sensitive channel, the presence of the α-, β-, and γ-subunits of the epithelial sodium channel (Garty and Palmer, 1997) mRNAs in similar amounts as the wild type was verified by RT-PCR analysis using tongue tissue of the null mutants (data not shown). Some investigators reported that there exist differences in voluntary NaCl intake between mouse strains (Ninomiya et al., 1989; Bachmanov et al., 1998). However, as far as we examined, the behavior of the null mutants of different stages in the genetic background (F1 and N4) was identical.
Body-fluid homeostasis is controlled also by the endocrine system. ANG II is a powerful stimulus of thirst and sodium appetite (Fitzsimons, 1997). The three CVOs, SFO, OVLT, and area postrema, are rich in ANG II receptors (Lenkei et al., 1997), where themNav2 expression is also positive. However, an increase in induction of the circulating ANG II or transportation to sensitive neurons through tanycytes (Fitzsimons, 1997) seemed not to occur in the null mutant for the following reasons: (1) the action of ANG II on the sensory CVOs helps to increase the intake of both water and salt (Buggy and Fisher, 1974). However, themNav2 null mutants showed an increase only in the intake of salt solution when thirsty, and the total fluid intake was normal. (2) When measured before and after the thirst experiment, systolic blood pressure was comparable between the null mutant and wild-type mice (our unpublished data). The adrenal mineralocorticoids, represented by aldosterone, are also known to induce salt intake (Denton, 1984). To recover the depleted sodium, they enhance reabsorption of sodium in the kidney and the motivation to consume salt. However, the levels of plasma aldosterone just after 24 hr water deprivation were comparable between the null mutant and wild-type mice (our unpublished data), and receptive loci of aldosterone (Birmingham et al., 1984) do not overlap with mNav2expression in the CNS. Therefore, the involvement of circulating aldosterone in the abnormal salt intake of the null mutants can be excluded.
In summary, our results suggest that the central mNav2 channel is involved in controlling the activities of neurons in the sensory CVOs responsible for the regulation of salt-intake behavior. TheNav2-deficient mice showed abnormal NaCl intake behavior, as if the water-depleted signal was confused with the sodium-depleted signal, and the salt-intake signal was hyperactive in moments of need. This is not surprising because the central sensing and integration mechanisms for osmotic pressure and the sodium level in cerebral arterial plasma and CSF are closely interrelated as described above, although the details are still not known. Sensory neurons in the SFO and OVLT, which showed marked c-fosexpression in themNav2−/−mouse, seem to positively regulate the salt-intake behavior, however, it is not clear at present whether they directly or indirectly project to the amygdala. In addition, it is also necessary to examine whether these neurons correspond to the cells expressingmNav2 in these organs. Because the functional reconstruction of this channel in vitro has not been successful, the channel properties and characteristics have remained obscure. Electrophysiological studies onmNav2-positive cells (tissues) comparing the mNav2-null mutant with the wild-type would be a promising approach to this issue.
Footnotes
This work was supported by grants-in-aid from Japan Science and Technology Corporation (CREST), the Ministry of Education, Science, Sports and Culture of Japan, and the Human Frontier Science Program. We are very grateful to Dr. Yoheved Berwald-Netter for providing rat NaG probes, Drs. Andras Nagy and Hisato Kondoh for R1 cells, Dr. Michael Tumkun for anti-mNav2.3 antibody, Dr. Masahiko Mori for a 129 mouse genomic library, Drs. Takeshi Yagi, Masaru Okabe, Masazumi Takahashi, Toshiyuki Takai, and Goro Katsuura for technical advice, Ms. Masae Mizoguchi and Mie Yasuda for technical assistance, and Ms. Akiko Kodama for secretarial assistance.
Correspondence should be addressed to Masaharu Noda, Division of Molecular Neurobiology, National Institute for Basic Biology, 38 Nishigonaka, Myodaiji-cho, Okazaki 444–8585, Japan. E-mail:madon@nibb.ac.jp.
REFERENCES
- 1.Akopian AN, Souslova V, Sivilotti L, Wood JN. Structure and distribution of a broadly expressed atypical sodium channel. FEBS Lett. 1997;400:183–187. doi: 10.1016/s0014-5793(96)01389-0. [DOI] [PubMed] [Google Scholar]
- 2.Andersson B. Regulation of water intake. Physiol Rev. 1978;58:582–603. doi: 10.1152/physrev.1978.58.3.582. [DOI] [PubMed] [Google Scholar]
- 3.Bachmanov AA, Tordoff MG, Beauchamp GK. Voluntary sodium chloride consumption by mice: differences among five inbred strains. Behav Genet. 1998;28:117–124. doi: 10.1023/a:1021471924143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birmingham MK, Sar M, Stumpf WE. Localization of aldosterone and corticosterone in the central nervous system, assessed by quantitative autoradiography. Neurochem Res. 1984;9:333–350. doi: 10.1007/BF00963982. [DOI] [PubMed] [Google Scholar]
- 5.Bourque CW, Oliet SH. Osmoreceptors in the central nervous system. Annu Rev Physiol. 1997;59:601–619. doi: 10.1146/annurev.physiol.59.1.601. [DOI] [PubMed] [Google Scholar]
- 6.Buggy J, Fisher AE. Evidence for a dual central role for angiotensin in water and sodium intake. Nature. 1974;250:733–735. doi: 10.1038/250733a0. [DOI] [PubMed] [Google Scholar]
- 7.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease”. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 8.Catterall WA. Structure and function of voltage-gated ion channels. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 9.Chae HE, Heideman PD. Water-deprived white-footed mice express c-fos on a day/night cycle graded according to the duration of deprivation. Brain Res. 1998;791:1–10. doi: 10.1016/s0006-8993(97)01509-6. [DOI] [PubMed] [Google Scholar]
- 10.Denton DA. The hunger for salt: an anthropological, physiological and medical analysis. Springer-Verlag; New York: 1984. [Google Scholar]
- 11.Denton DA, McKinley MJ, Weisinger RS. Hypothalamic integration of body fluid regulation. Proc Natl Acad Sci USA. 1996;93:7397–7404. doi: 10.1073/pnas.93.14.7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felipe A, Knittle TJ, Doyle KL, Tamkun MM. Primary structure and differential expression during development and pregnancy of a novel voltage-gated sodium channel in the mouse. J Biol Chem. 1994;269:30125–30131. [PubMed] [Google Scholar]
- 13.Felts PA, Black JA, Dib-Hajj SD, Waxman SG. NaG: a sodium channel-like mRNA shared by Schwann cells and other neural crest derivatives. Glia. 1997;21:269–276. doi: 10.1002/(sici)1098-1136(199711)21:3<269::aid-glia2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson AV, Bains JS. Electrophysiology of the circumventricular organs. Front Neuroendocrinol. 1996;17:440–475. doi: 10.1006/frne.1996.0012. [DOI] [PubMed] [Google Scholar]
- 15.Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1997;78:583–686. doi: 10.1152/physrev.1998.78.3.583. [DOI] [PubMed] [Google Scholar]
- 16.Galaverna O, De Luca LAJ, Schulkin J, Yao SZ, Epstein AN. Dificits in NaCl ingestion after damage to the central nucleus of the amygdala in the rat. Brain Res Bull. 1992;28:89–98. doi: 10.1016/0361-9230(92)90234-o. [DOI] [PubMed] [Google Scholar]
- 17.Galaverna OG, Seeley RJ, Berridge KC, Grill HJ, Epstein AN, Schulkin J. Lesions of the central nucleus of the amygdala. I. Effects on taste reactivity, taste aversion learning and sodium appetite. Behav Brain Res. 1993;59:11–17. doi: 10.1016/0166-4328(93)90146-h. [DOI] [PubMed] [Google Scholar]
- 18.Garty H, Palmer LG. Epitherial sodium channels: Function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 19.Gautron S, Dos Santos G, Pinto-Henrique D, Koulakoff A, Gros F, Berwald-Netter Y. The glial voltage-gated sodium channel: cell- and tissue-specific mRNA expression. Proc Natl Acad Sci USA. 1992;89:7272–7276. doi: 10.1073/pnas.89.15.7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George ALJ, Knittle TJ, Tamkun MM. Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc Natl Acad Sci USA. 1992;89:4893–4897. doi: 10.1073/pnas.89.11.4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation of the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 22.Goldin AL. Diversity of mammalian voltage-gated sodium channels. In: Rudy B, Seeburg P, editors. Molecular and diversity of ion channels and receptors. New York Academy of Sciences; New York: 1999. pp. 38–50. [DOI] [PubMed] [Google Scholar]
- 23.Gutman MB, Ciriello J, Mogeson GJ. Effects of plasma angiotensin and hypernatoremia on subfornical neurons. Am J Physiol. 1988;254:R746–R754. doi: 10.1152/ajpregu.1988.254.5.R746. [DOI] [PubMed] [Google Scholar]
- 24.Hatton GI. Pituicytes, glia and control of terminal secretion. J Exp Biol. 1988;139:67–79. doi: 10.1242/jeb.139.1.67. [DOI] [PubMed] [Google Scholar]
- 25.Honda K, Negoro H, Dyball REJ, Higuchi T, Takano S. The osmoreceptor complex in the rat: evidence for interactions between the supraoptic and other diencephalic nuclei. J Physiol (Lond) 1990;431:225–241. doi: 10.1113/jphysiol.1990.sp018328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson AK, Gross PM. Sensory circumventricular organs and brain homeostatic pathways. FASEB J. 1993;7:678–686. doi: 10.1096/fasebj.7.8.8500693. [DOI] [PubMed] [Google Scholar]
- 27.Johnson AK, de Olmos J, Pastsuskovas CV, Zardetto-Smith AM, Vivas L. The extended amygdala and salt appetite. Ann NY Acad Sci. 1999;877:258–280. doi: 10.1111/j.1749-6632.1999.tb09272.x. [DOI] [PubMed] [Google Scholar]
- 28.Kayano T, Noda M, Flockerzi V, Takahashi H, Numa S. Primary structure of rat brain sodium channel III deduced from the cDNA sequence. FEBS Lett. 1988;228:187–194. doi: 10.1016/0014-5793(88)80614-8. [DOI] [PubMed] [Google Scholar]
- 29.Knittle TJ, Doyle KL, Tamkun MM. Immunolocalization of the mNav2.3 Na+ channel in mouse heart: upregulation in myometrium during pregnancy. Am J Physiol. 1996;270:C688–C696. doi: 10.1152/ajpcell.1996.270.2.C688. [DOI] [PubMed] [Google Scholar]
- 30.Leng G. Rat supraoptic neurones: the effects of locally applied hypertonic saline. J Physiol (Lond) 1980;304:405–414. doi: 10.1113/jphysiol.1980.sp013332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenkei Z, Palkovits M, Corvol P, Llorens-Cortes C. Expression of angiotensin type-1 (AT1) and type-2 (AT2) receptor mRNAs in the adult rat brain: a functional neuroanatomical review. Front Neuroendocrinol. 1997;18:383–439. doi: 10.1006/frne.1997.0155. [DOI] [PubMed] [Google Scholar]
- 32.Matalon S, O'Brodovich H. Sodium channels in aveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 33.Morien A, Garrard L, Rowland NE. Expression of Fos immunoreactivity in rat brain during dehydration: effect of duration and timing of water deprivation. Brain Res. 1999;816:1–7. doi: 10.1016/s0006-8993(98)00828-2. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen S, Nagelhus EA, Amirty-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: High resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ninomiya Y, Sako N, Funakoshi M. Strain differences in amiloride inhibition of NaCl responses in mice, Mus musculus. J Comp Physiol [A] 1989;166:1–5. doi: 10.1007/BF00190204. [DOI] [PubMed] [Google Scholar]
- 36.Nissen R, Bourque CW, Renaud LP. Membrane properties of organum vasculosum lamina terminalis neurons recorded in vitro. Am J Physiol. 1993;264:R811–R815. doi: 10.1152/ajpregu.1993.264.4.R811. [DOI] [PubMed] [Google Scholar]
- 37.Noda M. Structure and function of sodium channels. Ann NY Acad Sci. 1993;707:20–37. doi: 10.1111/j.1749-6632.1993.tb38039.x. [DOI] [PubMed] [Google Scholar]
- 38.Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Kurasaki M, Takahashi H, Numa S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature. 1986;320:188–192. doi: 10.1038/320188a0. [DOI] [PubMed] [Google Scholar]
- 39.Oliet SHR, Bourque CW. Mechanosensitive channels transduce osmosensitivity in supraoptic neurons. Nature. 1993;364:341–343. doi: 10.1038/364341a0. [DOI] [PubMed] [Google Scholar]
- 40.Pilgrim C. Transport function of the hypothalamic tanycyte ependyma: how good is the evidence? Neuroscience. 1978;3:277–283. doi: 10.1016/0306-4522(78)90076-3. [DOI] [PubMed] [Google Scholar]
- 41.Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, Masliah E, Montal M. Neuronal death and perinatal lethality in voltage-gated sodium channel α(II)-deficient mice. Biophys J. 2000;78:2878–2891. doi: 10.1016/S0006-3495(00)76829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rawland NE. Brain mechanisms of mammalian fluid homeostasis: insights from use of immediate early gene mapping. Neurosci Biobehav Rev. 1998;23:49–63. doi: 10.1016/s0149-7634(97)00068-7. [DOI] [PubMed] [Google Scholar]
- 43.Ritchie JM. Voltage-gated ion channels in Schwann cells and glia. Trends Neurosci. 1992;15:345–351. doi: 10.1016/0166-2236(92)90052-a. [DOI] [PubMed] [Google Scholar]
- 44.Shintani T, Watanabe E, Maeda N, Noda M. Neurons as well as astrocytes express proteoglycan-type protein tyrosine phosphataseζ/RRTPβ: analysis of mice in which the PTPζ/RRTPβ gene was replaced with the lacZ gene. Neurosci Lett. 1998;247:135–138. doi: 10.1016/s0304-3940(98)00295-x. [DOI] [PubMed] [Google Scholar]
- 45.Sibbald JR, Hubbard JE, Sirrett NE. Resposes from osmosensitive neurons of the rat subfornical organ in vitro. Brain Res. 1988;461:205–214. doi: 10.1016/0006-8993(88)90251-x. [DOI] [PubMed] [Google Scholar]
- 46.Sontheimer H. Voltage-dependent ion channels in glial cells. Glia. 1994;11:156–172. doi: 10.1002/glia.440110210. [DOI] [PubMed] [Google Scholar]
- 47.Sontheimer H, Black JA, Waxman SG. Voltage-gated sodium channels in glia: Properties and possible functions. Trends Neurosci. 1996;19:325–332. doi: 10.1016/0166-2236(96)10039-4. [DOI] [PubMed] [Google Scholar]
- 48.Ueta Y, Yamashita H, Kawata M, Koizumi K. Water deprivation induces regional expression of c-fos protein in the brain of inbred polydipsic mice. Brain Res. 1995;677:221–228. doi: 10.1016/0006-8993(95)00142-d. [DOI] [PubMed] [Google Scholar]
- 49.Verkman AS. Role of aquaporin water channels in kidney and lung. Am J Med Sci. 1998;316:310–320. doi: 10.1097/00000441-199811000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Verney EB. The antidiuretic hormone and the factors which determine its release. Proc R Soc Lond B Biol Sci. 1947;135B:25–106. [PubMed] [Google Scholar]
- 51.Vivas L, Chiaraviglio E, Carrer HF. Rat organum vasculosum laminae terminalis in vitro: responses to changes in sodium concentration. Brain Res. 1990;519:294–300. doi: 10.1016/0006-8993(90)90091-o. [DOI] [PubMed] [Google Scholar]
- 52.Wells T. Vesicular osmometers, vasopression secretion and aquaporin-4: a new mechanism for osmoreception? Mol Cell Endocrinol. 1998;136:103–107. doi: 10.1016/s0303-7207(97)00219-0. [DOI] [PubMed] [Google Scholar]
- 53.Yagi T, Nada S, Watanabe N, Tamemoto H, Kohmura N, Ikawa Y, Aizawa S. A novel negative selection for homologous recombinants using diphtheria toxin A fragment gene. Anal Biochem. 1993;214:77–86. doi: 10.1006/abio.1993.1459. [DOI] [PubMed] [Google Scholar]
- 54.Zardetto-Smith AM, Beltz TG, Johnson AK. Role of the central nucleus of the amygdala and bed nucleus of the stria terminalis in experimentally-induced salt appetite. Brain Res. 1994;645:123–134. doi: 10.1016/0006-8993(94)91645-4. [DOI] [PubMed] [Google Scholar]