

Abstract
Reactive oxygen species, including superoxide, generally are considered neurotoxic molecules whose effects can be alleviated by antioxidants. Different from this view, we show that scavenging of superoxide with an antioxidant enzyme is associated with deficits in hippocampal long-term potentiation (LTP), a putative neural substrate of memory, and hippocampal-mediated memory function. Using transgenic mice that overexpress extracellular superoxide dismutase (EC-SOD), a superoxide scavenger, we found that LTP was impaired in hippocampal area CA1 despite normal LTP in area CA3. The LTP impairment in area CA1 could be reversed by inhibition of EC-SOD. In addition, we found that EC-SOD transgenic mice exhibited impaired long-term memory of fear conditioning to contextual cues despite exhibiting normal short-term memory of the conditioning experience. These findings strongly suggest that superoxide, rather than being considered exclusively a neurotoxic molecule, should also be considered a signaling molecule necessary for normal neuronal function.
Keywords: EC-SOD, superoxide, LTP, associative memory, contextual fear conditioning, hydrogen peroxide
Long-term potentiation (LTP) in the hippocampus is an activity-dependent increase in synaptic strength that has been hypothesized to underlie mammalian learning and memory (Bliss and Collingridge, 1993). The most intensely studied form of LTP is induced by high-frequency stimulation (HFS) of the Schaffer collateral–commissural input to the pyramidal neurons of area CA1. The induction of LTP in area CA1 is dependent on the activation of NMDA receptors (Collingridge et al., 1983) followed by an influx of Ca2+ into the postsynaptic cell (Lynch et al., 1983; Malenka et al., 1988). The influx of Ca2+ results in the activation of a number of Ca2+-dependent enzymes, some of which produce small signaling molecules, including cAMP, nitric oxide-cGMP, and arachidonic acid (for review, see Roberson et al., 1996).
An additional class of signaling molecules likely to be produced in response to LTP-inducing HFS are reactive oxygen species (ROS), particularly superoxide. Consistent with this possibility, it was shown that superoxide is produced in response to NMDA receptor activation in hippocampal area CA1 (Bindokas et al., 1996). In addition, several studies showed that removal of superoxide alters hippocampal LTP in area CA1. For example, cell-permeable superoxide scavengers of superoxide were shown to block LTP (Klann, 1998), and cell-impermeable superoxide scavengers were shown to strongly attenuate LTP (Klann et al., 1998). Finally, hippocampal slices from transgenic mice that overexpress cytoplasmic superoxide dismutase (SOD-1) were found to exhibit deficient LTP (Gahtan et al., 1998). These data suggest that ROS, such as superoxide, are critical components of the signal transduction machinery necessary for LTP in hippocampal area CA1.
On the basis of a large body of evidence that links hippocampal LTP and certain types of memory function (Martin et al., 2000), the finding that cell-impermeable scavengers strongly attenuate LTP suggests the possibility that extracellular superoxide may be important not only for LTP but also for hippocampal-dependent memory function. To test this hypothesis, we performed studies with transgenic mice that overexpress extracellular superoxide dismutase (EC-SOD). Herein we report that EC-SOD transgenic mice exhibit a reversible impairment of LTP in hippocampal area CA1. Furthermore, we report that EC-SOD transgenic mice exhibit impaired long-term but not short-term memory in contextual fear conditioning, a hippocampal-dependent memory task. Taken together, our findings suggest that extracellular superoxide is critical for both hippocampal LTP and hippocampal-dependent memory function.
MATERIALS AND METHODS
EC-SOD transgenic mice. EC-SOD transgenic mice were generated as previously described (Oury et al., 1992). Briefly, purified EC-SOD DNA was injected into the pronuclei of fertilized eggs isolated from mice [(C57BL/6 × C3H)F1 × (C57BL/6 × C3H)F1]. Mouse eggs surviving microinjection then were implanted into the oviducts of pseudopregnant foster mothers (CD1) following standard procedures. Mice carrying the human EC-SOD transgene were identified by Southern blot analysis of tail DNA probed with the entire human EC-SOD cDNA. Transgenic founders were bred with (C57BL/6 × C3H)F1 (14–16 back-crosses) to produce offspring for further studies. Heterozygote mice expressing human EC-SOD were compared with wild-type mice from the same litter for each experiment.
Nissl staining. Brains from wild-type and EC-SOD transgenic mice were sectioned as described previously (Oury et al., 1999). Sections were collected sequentially in four bins of cryoprotectant and stored at −20°C until processing. One bin of tissue from each brain was processed for staining with cresyl violet. Sections were analyzed with a Zeiss (Thornwood, NY) Axioplan photomicroscope equipped with differential interference optics. Images of immunoreactive cells were digitized with a DAGE (Michigan City, IN) video camera (MTI 3CCD) and an image analysis system (Simple 32; C-Imaging Systems).
Reverse transcription-PCR. Hippocampi were dissected from the brains of EC-SOD transgenic mice and nontransgenic littermates, and RNA was isolated using acid phenol-chloroform extractions as previously described (Chomczynski and Sacchi, 1987). Message for human EC-SOD or mouse EC-SOD was detected by reverse transcription (RT)-PCR using a Geneamp rtTh PCR kit (Perkin-Elmer, Norwalk, CT) and primers specific for either mouse EC-SOD (forward, 5′-CCCATGCTCTCCGCCTCTAGAA-3′; reverse, 5′-AAAGTCATTGCCTTGGCGCATG-3′) or human EC-SOD (forward, 5′-AGACACCTTCCACTCTGAGG-3′; reverse, 5′-GTTTCGGTACAAATGGAGGC-3′). PCR products were detected after agarose gel electrophoresis by ethidium bromide staining.
Immunocytochemical staining. Wild-type and EC-SOD transgenic mice were killed by lethal injection with Nembutol. The brain was perfused and fixed by injecting 20 ml of 4% buffered formalin through the left ventricle of the heart. Hippocampal sections then were processed for standard paraffin embedding. Five-micrometer-thick sections were cut onto poly-l-lysine-coated slides for immunochemical labeling using a rabbit polyclonal antibody specific to human EC-SOD with minor variations to a previously described protocol (Oury et al., 1996). Briefly, the primary antibody was diluted into a wild-type mouse brain homogenate at 1:300 and allowed to incubate overnight at 4°C. The solution was centrifuged to remove particulates and applied to tissue sections that had been treated with 0.1% pepsin in 0.01N HCl for 10 min for antigen retrieval. After washing, the primary antibody was detected using biotinylated goat anti-rabbit IgG and the Vectastain ABC reagent (Vector Laboratories, Burlingame, CA) followed by development with diaminobenzidine. As a control, serial sections were labeled with nonimmune rabbit IgG. To further control for potential nonspecific staining, sections from wild-type mice lacking human EC-SOD were labeled with this antibody against human EC-SOD to ensure that the labeling present in the EC-SOD transgenic mice was specific for human EC-SOD.
Western blots. Equivalent amounts of hippocampal protein from wild-type and EC-SOD transgenic mice were resolved with 10% SDS-PAGE, blotted electrophoretically to Immobilon membranes (Millipore, Bedford, MA), and incubated in Tris-buffered saline with Tween 20 (50 mm Tris-HCl, pH 7.5–8.0, 150 mmNaCl, and 0.1% Tween 20) containing 3% bovine serum albumin. Blots were incubated with either an antibody specific for human EC-SOD or an antibody specifc for mouse EC-SOD, followed by horseradish peroxidase-linked secondary antibody, and developed using enhanced chemiluminescence (Amersham Pharmacia Biotech, Arlington Heights, IL). Densitometric analysis of immunoreactivity was conducted using NIH Image software.
SOD activity. EC-SOD activity and total SOD activity (cytoplasmic SOD and mitochondrial SOD) remaining after extraction of EC-SOD were measured by inhibition of cytochrome c reduction at pH 10 as described previously (Crapo et al., 1978).
Preparation of hippocampal slices. Hippocampi from male heterozygote EC-SOD transgenic mice and male wild-type mice 3–6 months old (∼25–35 gm) were removed, and 400 μm slices were prepared with a McIlwain tissue chopper. The slices were perfused for 1–2 hr with a standard saline solution (in mm: 124 NaCl, 4.4 KCl, 26 NaHCO3, 10 d-glucose, 2 CaCl2, and 2 MgCl2, gassed with 95% O2 and 5% CO2, pH 7.4) in an interface tissue slice chamber at 30–32°C.
Induction of paired-pulse facilitation and post-tetanic potentiation. For assessment of either paired-pulse facilitation (PPF) or post-tetanic potentiation (PTP), a pair of bipolar stimulating electrodes were placed into the st. radiatum of area CA1 to stimulate the Schaffer collateral–commissural fibers, and a recording electrode was placed into the st. radiatum of area CA1. PPF is a presynaptic facilitation revealed when two stimuli are presented in rapid succession; the response to the second stimulus is enhanced depending on the interstimulus interval. The initial slope of the extracellular field EPSP (fEPSP) for each stimulus was measured over a range of interstimulus intervals (25–400 msec). Facilitation was measured by examining the ratio of the fEPSP slope to stimulus 2/fEPSP slope to stimulus 1. PTP was analyzed by measuring the fEPSP slope every second for the first 15 sec after HFS. To avoid the potentially confounding effects of LTP on measurements of PTP, the NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (APV; 100 μm) was included in the perfusing solution.
Induction of LTP. For induction of LTP in area CA1, a pair of bipolar stimulating electrodes was placed into the st. radiatum of area CA1 to stimulate the Schaffer collateral–commissural fibers, and a recording electrode was placed into the st. radiatum of area CA1. Responses to electrical stimulation in area CA1 were monitored for at least 20 min before the delivery of LTP-inducing HFS. In most experiments, test stimuli (50 μsec duration) were given at a current (30–100 μA) that produced 50% of the maximum initial slope fEPSP. Responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz). One train of LTP-inducing HFS consisted of 100 pulses delivered at 100 Hz using a current (60–100 μA) that elicited the maximum fEPSP. Multiple trains of HFS were delivered with an intertrain interval of 20 sec. Responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz) for 60 min after the final train of HFS. Post-HFS responses were elicited using the same test stimulation intensity as that used before HFS. LTP was defined as a ≥20% increase in the initial slope of the fEPSP compared with pre-HFS control levels (within-slice comparison).
For induction of mossy fiber LTP in area CA3, mouse hippocampal slices were prepared as described previously for rat hippocampal slices (Urban and Barrionuevo, 1996). Slices were transferred to an incubation chamber containing a standard saline solution containing (in mm): 125 NaCl, 2 KCl, 26 NaHCO3, 10 dextrose, 1 CaCl2, and 6 MgCl2, gassed with 95% O2and 5% CO2, pH 7.4, at room temperature. After incubation, slices were transferred to a recording chamber and submerged in the same standard saline solution described above, with the exception that the divalent ion concentrations were (in mm): 2.5 CaCl2 and 1 MgCl2, and the solution contained 100 μm APV to block NMDA receptors. The temperature of the recording chamber was 30–32°C. A bipolar stimulating electrode was placed into the granule cell layer of the dentate gyrus to stimulate the mossy fibers; a recording electrode was placed into the st. radiatum of area CA3. Mossy fiber LTP was induced with three trains of HFS. Each train consisted of 100 pulses delivered at 100 Hz with an intertrain interval of 10 sec (Urban and Barrionuevo, 1996). Responses to test stimuli were measured every 2.5 min as an average of four individual traces (0.1 Hz) for 60 min after the final train of HFS. Post-HFS responses were elicited using the same test stimulation intensity as that used before HFS. LTP was defined as a ≥20% increase in the initial amplitude of the fEPSP compared with pre-HFS control levels (within-slice comparison).
For all of the LTP experiments, n was the number of mice used in each experimental condition.
Whole-cell recordings. Slices were placed in a submersion recording chamber and perfused with the following external solution (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 10 glucose, and 0.01 bicuculline, gassed with 95% O2 and 5% CO2, pH 7.4, at 30–32°C. Whole-cell recordings were obtained from CA1 pyramidal neurons identified visually by differential interference contrast and infrared video microscopy (Stuart et al., 1993). Gigaohm seals (>2 GΩ) were obtained with patch pipettes (3–7 MΩ) pulled from borosilicate glass and filled with the following internal solution (in mm): 122.5 Cs gluconate, 11.5 CsCl, 8 NaCl, 10 HEPES, 0.5 EGTA, 4 ATP, 0.3 GTP, and 14 phosphocreatine, pH 7.2–7.4, 290–300 mOsm. Recordings were made with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA) operating in voltage-clamp mode. Series resistance (12–20 MΩ) was not compensated, and recordings were discarded for analysis if the series resistance changed >20%. EPSCs were evoked at 0.1 Hz by stimulation of the Schaffer collateral–commissural pathway using bipolar stimulation electrodes placed in the st. radiatum 100–200 μm from the st. pyramidale. Stimulation parameters were adjusted to elicit EPSCs with a peak amplitude of 50–150 pA when recorded at −70 mV. The AMPA and NMDA components of the EPSC were measured as the mean current in 10 msec time windows placed at the current peak (AMPA) or 50–80 msec after the peak (NMDA). Current–voltage plots were made for each neuron after currents were normalized relative to the AMPA component (measured at −60 or +60 mV, respectively).
Contextual fear conditioning: unsignaled shocks. To test the animals' ability to associate neutral contextual stimuli with an aversive stimulus (foot shock) in the absence of competing discrete stimuli paired with shock, the animals were placed into a standard mouse conditioning chamber (13 × 10.5 × 13 cm) equipped with a house light (28 V), a loudspeaker, and a floor consisting of 19 equally spaced metal rods (2.8 mm diameter). The fear-conditioning chambers were housed in sound-attenuating cubicles (56 × 50 × 41 cm) equipped with a background noise-generating fan to overshadow extraneous sounds. The onset of the cubicle fan and the illumination of the house light signaled the onset of the training session. A 2 sec scrambled foot shock (0.75 mA) was presented 120 and 240 sec after session onset. The session ended 30 sec after the second shock, as indicated by the extinguishment of the house light and the cubicle fan. The animals were removed immediately and returned to their home cages. To test fear conditioning to the contextual cues, the animals were returned to the training context 24 hr after training for a 5 min test session. As previously, session onset and offset were indicated by the turning on and off, respectively, of the house light and the cubicle fan. No shocks were presented during the test session. The occurrence of freezing (no movement other than respiratory movement), an indicator of fear, was measured every 10 sec during training, except the two 10 sec bins during which the shocks occurred, and every 10 sec throughout testing. A summary of this and the next protocol is provided in Table1.
Table 1.
Contextual fear conditioning protocols for assessing associative memory in wild-type and EC-SOD transgenic mice
| Contextual fear conditioning | Training | Testing |
|---|---|---|
| Unsignaled shocks | Context A | Context A |
| 2 × 2 sec shock, | No shocks, | |
| 4.5 min trial | 5 min trial | |
| Signaled shocks | Context A | Context A |
| 2 × 30 sec tone followed by 2 sec shock | No shocks or tones | |
| 4.5 min trial | Either 2 min (immediate test) or 5 min trial (delayed test) | |
| Context B | ||
| No shocks, 3 min tone in second trial half | ||
| 6 min trial |
Contextual fear conditioning: signaled shocks. The training session was identical to the one described above, except that a 30 sec tone (80 dB, 2 kHz) preceded and terminated with the onset of each of the two shocks. Conditioning to the training context was tested either 3 min or 24 hr after training with either a 2 min (immediate test) or a 5 min (delayed test) test session as described above. To test fear conditioning to the cue, the animals were returned to their home cages for either 24 hr (immediate context test) or 2 hr (delayed context test) after testing conditioning to the context. The test chamber was modified with respect to tactile, spatial, visual, and olfactory properties to create a novel test environment. The animals were placed into the modified chamber for a 6 min test session, with the tone present continuously during the second 3 min of that session. The occurrence of freezing was measured during training and test sessions, as described above.
Open-field behavior. To test the animals' locomotor function and exploratory behavior in a novel context, the animals were placed for 10 min in an open-field chamber (28 × 28 × 40 cm) whose floor was divided into 16 equal-sized square fields, and the frequency of the following behaviors was recorded: (1) entries into each of the 16 fields, (2) rearing, (3) nose poking into small holes (1 cm diameter) equally spaced 2 cm above the floor along two opposite walls of the chamber, and (4) freezing bouts, an indicator of fear.
Pain threshold. To test the animals' sensitivity to pain, we placed the animals individually on a surface (25.5 × 25.5 cm) surrounded by Plexiglas walls and heated to 52 ± 0.1°C and recorded the latency to (1) forepaw lick, (2) hindpaw lick, (3) hindpaw shake, and (4) jumping. The animals were removed from the surface immediately after the jump or after 5 min had elapsed, whichever came first.
RESULTS
Anatomical and biochemical characterization of EC-SOD transgenic mice
The mutant mice used in these studies overexpress human EC-SOD. These transgenic animals appeared to be healthy and exhibited no obvious neurological abnormalities. Adult brains of the mice were fixed and examined for histological abnormalities. The appearance and size of the brains from EC-SOD transgenic mice were indistinguishable from those of wild-type mice from the same litter. We examined the gross anatomy of various brain regions with Nissl staining and observed no obvious abnormalities in EC-SOD transgenic mice compared with wild-type mice, including the hippocampal formation (Fig.1A). Thus, at a gross neuroanatomical level, EC-SOD transgenic mice appeared to be identical to their wild-type littermates.
Fig. 1.

Anatomical and biochemical comparison of wild-type and EC-SOD transgenic mice. A, Nissl stains of sagittal sections through the hippocampus of wild-type (left) and EC-SOD transgenic (right) mice. B, RT-PCR of mouse hippocampal RNA. MW, Molecular weight marker; lane 1, RT-PCR kit positive control;lane 2, wild-type mouse hippocampal RNA; lane 3, wild-type mouse hippocampal RNA with RNase; lane 4, EC-SOD trangenic mouse hippocampal RNA; lane 5, EC-SOD transgenic mouse hippocampal RNA with Rnase.C, Western blot of hippocampal homogenates from wild-type (WT) and EC-SOD transgenic (TG) mice. The blots were probed with antibodies specific for either human EC-SOD (top) or mouse EC-SOD (bottom). D, Immunocytochemistry of either wild-type (left) or EC-SOD transgenic (right) mice showing diffuse expression of human EC-SOD in the hippocampi of EC-SOD transgenic mice. Scale bars, 200 μm.E, EC-SOD activity measured in hippocampal homogenates from wild-type and EC-SOD transgenic mice. Error bars indicate SEM for four determinations. *Statistical significance with a paired Student'st test (p < 0.05).
We proceeded to analyze EC-SOD transgenic mice at the biochemical level. RT-PCR analysis revealed that human EC-SOD mRNA was highly expressed in the hippocampus, with no effect on endogenous levels of mouse EC-SOD mRNA (Fig. 1B). Similarly, Western blot analysis revealed that EC-SOD transgenic mice overexpressed human EC-SOD protein in the hippocampus without alteration of endogenous levels of EC-SOD protein (Fig. 1C). Using immunohistochemical techniques, we observed that human EC-SOD was expressed diffusely throughout the hippocampi of transgenic mice (Fig.1D). In enzymatic assays we found that EC-SOD transgenic mice had ∼10-fold more hippocampal EC-SOD activity than wild-type mice did (Fig. 1E). Taken together, these results demonstrate that the EC-SOD transgenic mice overexpress an enzymatically competent EC-SOD in the hippocampus.
Impaired LTP in hippocampal area CA1 in EC-SOD transgenic mice
We previously showed that cell-impermeable superoxide scavengers strongly attenuate LTP in area CA1 of rat hippocampal slices (Klann et al., 1998). Because of the overexpression of EC-SOD in the hippocampus, we hypothesized that LTP would be impaired in EC-SOD transgenic mice. In the first set of experiments, we induced LTP in area CA1 of hippocampal slices from wild-type and EC-SOD transgenic mice with one train of HFS (100 pulses at 100 Hz). This induction paradigm produced LTP in slices from wild-type mice (fEPSP slope = 145 ± 7% of control; n = 9; Fig.2A,B). In contrast, the same induction protocol failed to produce LTP in slices from EC-SOD transgenic mice (fEPSP slope = 100 ± 3% of control;n = 9; Fig. 2A,B). We also performed experiments in area CA1 using three trains of HFS. With this induction paradigm we observed robust potentiation in slices from wild-type mice (fEPSP slope = 165 ± 5% of control; n = 10; Fig. 2C) and strongly attenuated potentiation in slices from EC-SOD transgenic mice (118 ± 5% of control; n = 10; Fig. 2C). Taken together, these results are consistent with the idea that extracellular superoxide is necessary for the full expression of LTP in hippocampal area CA1.
Fig. 2.

Impaired LTP in hippocampal area CA1 of EC-SOD transgenic mice. A, Stable baseline responses were recorded in hippocampal area CA1 of slices from either wild-type (WT) or EC-SOD transgenic (EC-SOD TG) mice for 20 min. LTP-inducing HFS consisted of one train of HFS (100 pulses at 100 Hz). Error bars indicate SEM for 10 determinations. B, Representative fEPSPs recorded in area CA1 before and 60 min after delivery of HFS to slices from either WT or EC-SOD TG mice. Calibration: 2 mV, 3 msec. C,Similar to A, except LTP-inducing HFS consisted of three trains of HFS (100 Hz) delivered 20 sec apart. Error bars indicate SEM for 10 determinations. D, Stable baseline responses were recorded in area CA3 of slices from either WT or EC-SOD TG mice for 10 min. LTP-inducing HFS consisted of three trains of HFS delivered 10 sec apart. Error bars indicate SEM for five determinations.
To determine whether extracellular superoxide also was necessary for LTP hippocampal area CA3, we induced LTP at the mossy fiber→CA3 pyramidal cell synapse in slices from wild-type and EC-SOD transgenic mice with three trains of HFS. In contrast to the forms of LTP examined in Figure 2, A and C, mossy fiber LTP is NMDA receptor-independent (Harris et al., 1984). As illustrated in Figure 2D, we observed no difference in mossy fiber LTP between slices from wild-type mice (fEPSP amplitude = 153 ± 15% of control; n = 5) and EC-SOD transgenic mice (fEPSP amplitude = 165 ± 19% of control; n = 5). These data suggest that extracellular superoxide is not necessary for mossy fiber LTP in area CA3, and that superoxide production in the hippocampus is likely to be coupled to NMDA receptor activation (Bindokas et al., 1996).
EC-SOD transgenic mice exhibit normal synaptic transmission, post-tetanic potentiation, and paired pulse facilitation
We performed several experiments to determine whether the impairment of LTP in hippocampal area CA1 of EC-SOD transgenic mice was attributable to alterations in either synaptic transmission or NMDA receptor function. First, we tested the synaptic input–output relation in area CA1 by using a range of stimulus intensities (10–70 μA) to elicit synaptic responses and comparing either the fiber volley amplitude versus the stimulus intensity (Fig.3A) or the fEPSP slope versus the fiber volley amplitude (Fig. 3B). We observed no significant differences between wild-type and EC-SOD transgenic mice with either comparison (Fig. 3A,B). Using whole-cell recordings we similarly detected no differences between wild-type and EC-SOD transgenic mice in the amplitude and time course of synaptic currents at various voltages (data not shown). Furthermore, the current–voltage relationships for the non-NMDA component (Fig.3C) as well as the NMDA component (Fig. 3D) of the synaptic currents were indistinguishable between wild-type and EC-SOD transgenic mice (Fig. 3C,D). Finally, there were no significant differences in high-frequency synaptic transmission between slices from wild-type and EC-SOD transgenic mice, measured as either total depolarization during HFS [integrating the entire response to HFS: wild-type, 106 ± 8% of control; n = 4; EC-SOD transgenic (TG), 103 ± 5% of control; n = 4] or the steady-state depolarization produced during HFS (averaged over the last 50 msec of the HFS: wild-type, 99 ± 7% of control;n = 4; EC-SOD TG, 104 ± 6% of control;n = 4). Overall, these results indicate that the LTP deficit in EC-SOD transgenic mice cannot be attributed to alterations in either synaptic transmission or NMDA receptor function.
Fig. 3.

Baseline synaptic transmission and synaptic currents are normal in EC-SOD transgenic mice. A, Plot of fiber volley amplitude across stimulation intensities. There was no significant difference between wild-type (WT) and EC-SOD transgenic (EC-SOD TG) mice. Error bars indicate SEM for seven determinations. B, Plot of fEPSP slope across fiber volley amplitudes. There was no significant difference between WT and EC-SOD TG mice. Error bars indicate SEM for seven determinations. C, Averaged amplitudes of AMPA receptor-mediated responses. D, Averaged amplitudes of NMDA receptor-mediated responses. Error bars in C andD indicate SEM for five determinations. The AMPA-mediated component was measured 5 msec after onset of the EPSC; the NMDA component was determined from the averaged response 50–100 msec after the stimulus.
We examined two short-lasting forms of presynaptic plasticity to determine whether the blockade of LTP in EC-SOD transgenic mice was attributable to abnormal presynaptic function. We first examined PTP in hippocampal slices from wild-type and EC-SOD transgenic mice. For that purpose, we analyzed the enhancement of synaptic responses during the first 15 sec after HFS (Zucker, 1989; Chapman et al., 1995). To avoid potential confounding effects of LTP on PTP measurements, we performed the experiments in the presence of APV (50 μm), an NMDA receptor antagonist. As shown in Figure4A, PTP in area CA1 was indistinguishable between hippocampal slices from wild-type and EC-SOD transgenic mice. Next, we examined PPF in hippocampal slices from wild-type and EC-SOD transgenic mice. PPF is a presynaptic facilitation revealed by the enhanced magnitude of the response evoked by the second pulse of a pair of pulses delivered at short intervals (McNaughton, 1982; Hess et al., 1987; Muller and Lynch, 1989). We observed no significant difference in PPF between slices taken from wild-type and EC-SOD transgenic mice (Fig. 4B). Taken together, these data suggest that the blockade of LTP in EC-SOD transgenic mice cannot be attributed to abnormal presynaptic function.
Fig. 4.

Post-tetanic potentiation and paired-pulse facilitation are normal in EC-SOD transgenic mice. A,Post-tetanic potentiation in wild-type (WT) and EC-SOD transgenic (EC-SOD TG) mice. The graph includes 2 responses measured before HFS, 15 responses measured every second immediately after HFS, and 2 additional responses measured 5 min after HFS. Error bars indicate SEM for seven determinations.B, Paired pulse stimulation in WT and EC-SOD TG mice. The graph represents responses to paired pulses in which the fEPSP slope of the response to the second stimulus is expressed as a percentage of the fEPSP slope of the response to the first stimulus versus the interpulse interval of paired pulses. Error bars indicate SEM for five determinations.
Blockade of LTP in EC-SOD transgenic mice is reversible
EC-SOD is a Cu- and Zn-dependent enzyme (Marklund et al., 1982). Therefore, we determined whether the copper chelator diethyldithiocarbamate (DDCA) could reverse the blockade of LTP observed in hippocampal area CA1 of EC-SOD transgenic mice. For that purpose, we delivered one train of LTP-inducing HFS to slices from either wild-type or EC-SOD transgenic mice in either the presence or absence of 5 mm DDCA. We observed that DDCA reversed the blockade of LTP in slices from EC-SOD transgenic mice (−DDCA, fEPSP slope = 99 ± 7% of control; n = 6; +DDCA, fEPSP slope = 145 ± 7% of control; n = 6; Fig. 5A) but had no statistically significant effect on LTP in slices from wild-type mice (−DDCA, fEPSP slope = 156 ± 6% of control;n = 6; +DDCA, fEPSP slope = 165 ± 8% of control; n = 6; Fig. 5A). These results show that the blockade of LTP in EC-SOD transgenic mice can be reversed by inhibition of EC-SOD. In addition, these results suggest that the underlying signaling cascades responsible for the induction and expression of LTP remain intact in the hippocampi of EC-SOD transgenic mice.
Fig. 5.
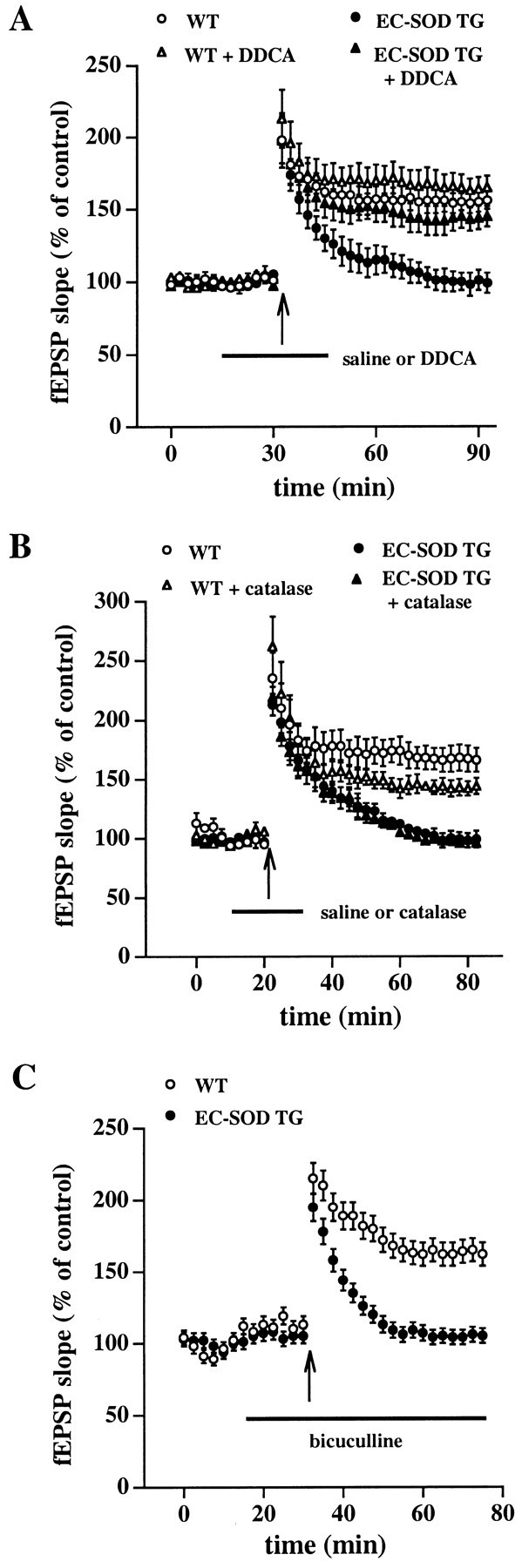
Impaired LTP in EC-SOD transgenic mice is rescued by DDCA but not by catalase or bicuculline. A, Rescue of impaired LTP by DDCA. Responses recorded from slices from either wild-type (WT) or EC-SOD transgenic (EC-SOD TG) mice given LTP-inducing HFS in the presence of 5 mm DDCA were compared with responses recorded from control slices from the same animal (in an adjacent recording chamber) given LTP-inducing HFS in the absence of the compound. Error bars indicate SEM for six determinations. B, Impaired LTP is not reversed by catalase. Responses recorded from slices given LTP-inducing HFS in the presence of catalase (260 U/ml) were compared with responses recorded from control slices from the same animal (in an adjacent recording chamber) given LTP-inducing HFS in the absence of the compound. Error bars indicate SEM for six determinations.C, Slices from either WT or EC-SOD TG mice were given LTP-inducing HFS in the presence of 10 μm bicuculline. Error bars indicate SEM for four determinations.
EC-SOD converts superoxide to hydrogen peroxide and oxygen (Halliwell, 1992). Because hydrogen peroxide has been shown to inhibit LTP (Pellmar et al., 1991; Auerbach and Segal, 1997), it is possible that the blockade of LTP in EC-SOD transgenic mice was caused by altered hydrogen peroxide metabolism. To examine the potential involvement of altered hydrogen peroxide metabolism in the impairment of LTP in EC-SOD transgenic mice, slices from wild-type and EC-SOD transgenic mice were given LTP-inducing stimulation in either the presence or absence of catalase (260 U/ml), an enzyme that scavenges hydrogen peroxide (Halliwell, 1992). Catalase had no effect on the blockade of LTP in slices from EC-SOD transgenic mice (−catalase, fEPSP slope = 99 ± 7% of control; n = 6; +catalase, fEPSP slope = 96 ± 5% of control; n = 6; Fig.5B). Interestingly, in experiments with slices from wild-type mice, we observed that catalase caused a small but statistically significant decrease in the magnitude of LTP (−catalase, fEPSP slope = 166 ± 10% of control; n = 6; +catalase, fEPSP slope = 144 ± 7% of control;n = 6; Fig. 5B). These results indicate that blockade of LTP in EC-SOD transgenic mice is not caused by overproduction of hydrogen peroxide. Furthermore, these results indicate that production of hydrogen peroxide may be necessary for the full expression of LTP in area CA1 of wild-type mice.
Mice that overexpress cytoplasmic SOD (SOD-1) exhibit impaired LTP that has been attributed to upregulation of GABAergic neurotransmission (Levkovitz et al., 1999). To determine whether a similar mechanism was involved in the blockade of LTP in EC-SOD transgenic mice, we delivered one train of LTP-inducing HFS to slices from wild-type and EC-SOD transgenic mice in the presence of bicuculline (10 μm), a selective GABAA receptor antagonist. Bicuculline had no effect on the blockade of LTP in slices from EC-SOD transgenic mice (fEPSP slope = 105 ± 3% of control; n= 4; Fig. 5C) or on LTP in slices from wild-type mice (fEPSP slope = 162 ± 8% of control; n = 4; Fig.5C). These results indicate that the lack of LTP in EC-SOD transgenic mice cannot be attributed to upregulation of GABAergic neurotransmission.
EC-SOD transgenic mice exhibit deficient hippocampal-dependent associative memory
Under the assumption that hippocampal LTP underlies hippocampal-dependent memory, our observations of compromised LTP in area CA1 of hippocampal slices from EC-SOD transgenic mice lead to the prediction that hippocampal-dependent memory function also is compromised in these mice. To test this prediction, we used two versions of the contextual fear-conditioning paradigm (Kim and Fanselow, 1992; Phillips and LeDoux, 1992). In the first paradigm, which involved conditioning with unsignaled shocks (Table1), wild-type and EC-SOD transgenic mice received two 2 sec foot shocks during a 5 min training session (2 min intershock interval) and were tested 24 hr later for conditioning to the training context. Figure6A shows that whereas both groups of animals exhibited comparably low levels of freezing during training, EC-SOD transgenic mice exhibited significantly less freezing (19 ± 6% freezing; n = 11) than did wild-type mice (42 ± 7% freezing; n = 11) during testing. In the second paradigm, which involved conditioning with signaled shocks, wild-type and EC-SOD transgenic mice similarly received two 2 sec foot shocks; however, the shocks were preceded by a 30 sec tone. Animals trained in this paradigm were tested for conditioning to the training context either 3 min after training, to assess short-term memory of the conditioning experience, or, similar to the first paradigm, 24 hr after training, to assess long-term memory of the conditioning experience. Figure 6B shows that wild-type and EC-SOD transgenic mice exhibited comparably low levels of freezing during training irrespective of delay to testing. When tested for conditioning to the context 3 min after training, both wild-type and EC-SOD transgenic mice exhibited elevated levels of freezing, with the freezing level being comparable between the two groups (wild-type mice, 33 ± 10% freezing; n = 6; EC-SOD transgenic mice, 29 ± 13% freezing; n = 8; Fig.6B). In contrast, when tested for conditioning to the context 24 hr after training, EC-SOD transgenic mice exhibited significantly lower levels of freezing to the context than did wild-type mice (wild-type mice, 58 ± 9% freezing;n = 8; EC-SOD transgenic mice, 17 ± 6% freezing;n = 8), similar to the pattern observed with the first paradigm. Collectively, these data indicate that the initial acquisition and short-term memory of contextual fear conditioning is normal in EC-SOD transgenic mice, but that consolidation of contextual fear conditioning over a long retention interval is significantly impaired in EC-SOD transgenic mice.
Fig. 6.
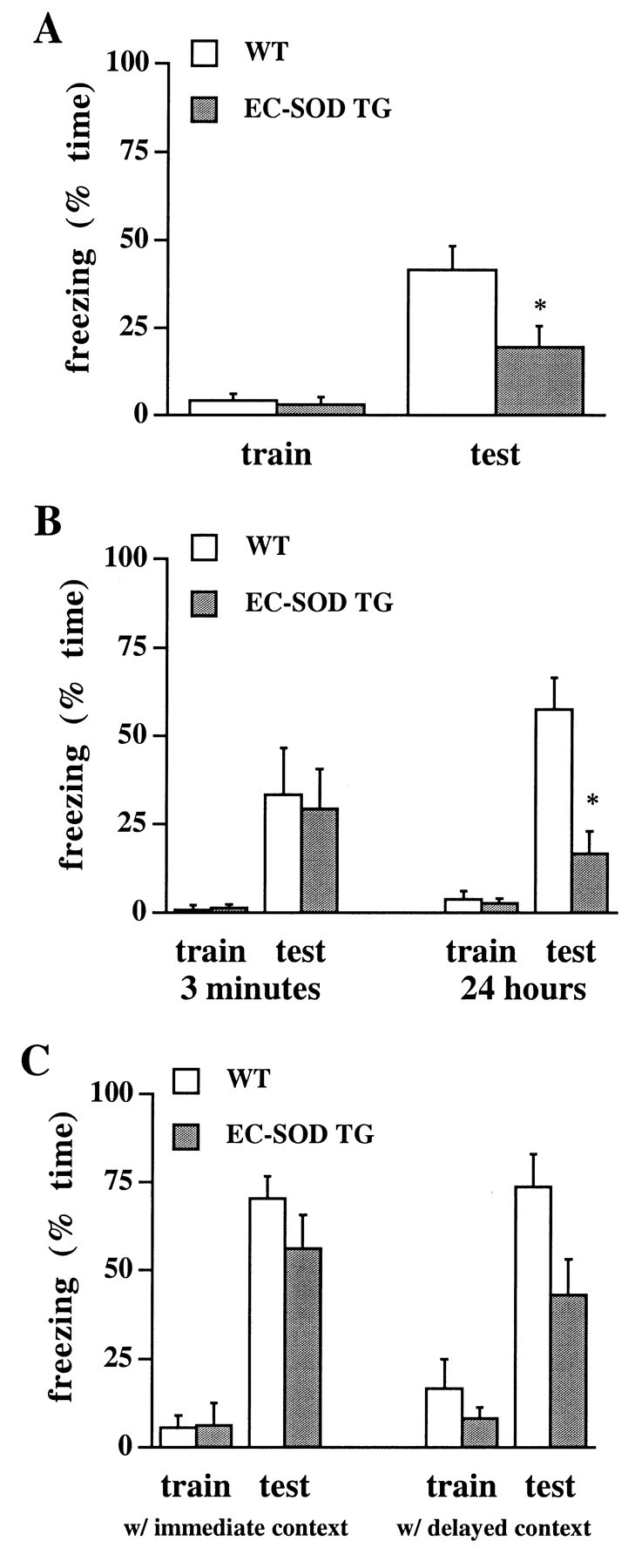
Impaired associative memory in EC-SOD transgenic mice. A, Group data of the percentage of time engaged in freezing to the training context during training (train) and testing (test) for wild-type mice (WT; open bars) and EC-SOD transgenic mice (EC-SOD TG; filled bars). Animals were conditioned with unsignaled shocks and tested 24 hr after training. Error bars indicate SEM for 11 determinations per group.B, Group data of freezing behavior as shown above, except that animals were conditioned with signaled shocks and tested either 3 min or 24 hr after training. Error bars indicate SEM for eight determinations per group, except for the group of wild-type animals tested 3 min after training, for which the number of determinations was six. C, Group data of freezing behavior to the tone during training (train) and testing (test) for wild-type mice (open bars) and EC-SOD transgenic mice (filled bars). All animals were tested for conditioning to the tone 26 hr after training. Data on the left show results for animals that received a context test trial 3 min after training (with immediate context test); data on the right show results for animals that received a context test trial 24 hr after training (with delayed context test). The trend toward stronger conditioning to the tone by wild-type mice compared with EC-SOD transgenic mice failed to reach statistical significance regardless of whether animals received the immediate, 2 min context test or the delayed, 5 min context test. The trend, however, raises the possibility that conditioning to the tone may also have been affected by the overexpression of EC-SOD, albeit to a considerably lesser extent than conditioning to the context. Error bars indicate SEM for eight determinations per group, except for the group of wild-type animals with the immediate context test, for which the number of determinations was six. *Statistically significant difference (p < 0.01) between wild-type and EC-SOD transgenic mice determined with Tukey's HSD method for post hoc comparisons after a repeated measures ANOVA with one within factor (trial) and one between factor (group).
The lack of a short-term memory deficit by EC-SOD mice suggests that the long-term memory deficit cannot be attributed to a difference between the groups in exploratory behavior or sensorimotor function. Nevertheless, to rule out such an alternative explanation of the findings, we performed two additional behavior analyses. First, we observed exploratory behavior of wild-type and EC-SOD transgenic mice in an open-field chamber for 10 min, recording the frequency of entries into equal-sized square fields drawn onto the floor of the chamber, rearing, nose poking into holes in the chamber walls, and freezing. We found no systematic differences between the two groups of mice with respect to any of the measures, including the percentage of entries into the center fields of the chamber (Table2). Second, we tested the pain threshold of wild-type and EC-SOD mice by placing the animals on a heated platform. We observed no differences in the latency to lick the forepaw, lick the hindpaw, shake the hindpaw, or jump off the hot platform (Table 2). Taken together, these observations render it unlikely that the long-term memory deficit in EC-SOD transgenic mice can be explained in terms of altered sensorimotor function.
Table 2.
Open-field behavior and pain threshold are indistinguishable between wild-type and EC-SOD transgenic mice
| Frequency (mean ± SEM) of various behaviors in the open-field test | ||||
|---|---|---|---|---|
| Group | Field entries | Rearing | Nose poking | Freezing |
| Wild-type (n = 7) | 238 ± 32 | 84 ± 9 | 32 ± 4 | 0 ± 0 |
| EC-SOD TG (n = 6) | 218 ± 31 | 64 ± 8 | 41 ± 8 | 0 ± 0 |
| Latency (in sec; mean ± SEM) to various behaviors in the pain threshold test | ||||
| Group | Forepaw lick | Hindpaw lick | Hindpaw shake | Jump |
| Wild-type (n = 10) | 22 ± 3 | 62 ± 18 | 35 ± 5 | 190 ± 27 |
| EC-SOD TG (n = 8) | 18 ± 3 | 48 ± 7 | 36 ± 5 | 220 ± 21 |
This conclusion was strengthened by our tests of conditioning to the tone in animals trained in the fear-conditioning paradigm with signaled shocks (Table 1). To assess fear conditioning to the tone, a hippocampal-independent conditioning paradigm (Kim and Fanselow, 1992;Phillips and LeDoux, 1992), we modified the conditioning chamber with respect to spatial, tactile, olfactory, and visual properties to render the test context novel. Animals were tested in the novel context 26 hr after training, with the tone being presented during the second 3 min of the 6 min test. Analysis of freezing during the first 3 min of the test revealed that none of the mice generalized from the training to the novel test context. Figure 6C depicts fear conditioning to the tone for mice that received a context test trial 3 min after training (with immediate context) and animals that received a context test trial 24 hr after training (with delayed context). Irrespective of the context test protocol, wild-type and EC-SOD transgenic mice showed comparably low levels of freezing to the tone during training and significantly enhanced levels of freezing to the tone during testing (with immediate context: wild-type mice, 70 ± 6% freezing;n = 6; EC-SOD transgenic mice, 56 ± 9% freezing;n = 8; with delayed context: wild-type mice, 74 ± 9% freezing; n = 8; EC-SOD transgenic mice, 43 ± 10% freezing; n = 8). These data show that EC-SOD transgenic mice can consolidate associative representations over a long retention interval provided that the associations involve simple stimulus relations whose mnemonic processing is relatively independent of the integrity of the hippocampus.
DISCUSSION
A growing body of evidence suggests that ROS, such as superoxide, are necessary to induce the full expression of LTP in hippocampal area CA1. For example, cell-permeable superoxide scavengers have been shown to prevent the induction of LTP (Klann, 1998), and transgenic mice that overexpress the superoxide-scavenging enzyme SOD-1, a cytoplasmic SOD, exhibit impaired LTP (Gahtan et al., 1998). In addition, cell-impermeable scavengers of superoxide have been shown to strongly attenuate LTP (Klann et al., 1998). Similarly, we found that, depending on the LTP induction paradigm, LTP was either blocked or strongly attenuated in area CA1 of hippocampal slices from EC-SOD transgenic mice (Fig. 2A,B). These findings are consistent with the idea that superoxide is a critical molecule in the biochemical milieu necessary for LTP in hippocampal area CA1.
In contrast to LTP, synaptic transmission appeared to be normal in EC-SOD transgenic mice (Fig. 3). In addition, we observed no deficit in either PTP or PPF in EC-SOD transgenic mice (Fig. 4). These data are important because they indicate that the overexpression of EC-SOD in the transgenic mice interferes with neither presynaptic nor postsynaptic function. The blockade of LTP in these mice, therefore, is unlikely to be attributable to nonspecific effects of EC-SOD. Furthermore, we were able to reverse the LTP deficit by inhibiting EC-SOD (Fig. 5A), which suggests that there are no fundamental differences in the biochemical machinery in area CA1 between wild-type and EC-SOD transgenic mice.
Interestingly, we found that mossy fiber LTP in EC-SOD transgenic mice was indistinguishable from mossy fiber LTP in wild-type mice (Fig.2C), which suggests that superoxide does not play a critical role in this form of synaptic plasticity. The induction of mossy fiber LTP is NMDA receptor-independent; instead, its induction is dependent on the activation of voltage-gated Ca2+channels (Jaffe and Johnston, 1990; Castillo et al., 1994). In rat hippocampal slices, glutamate receptor (AMPA, kainate, and NMDA) activation has been shown to result in the production of superoxide, whereas membrane depolarization that activates voltage-gated Ca2+ channels did not (Bindokas et al., 1996). Thus, superoxide production in the hippocampus is likely to be more closely coupled to NMDA receptor activation than to the activation of voltage-gated Ca2+channels. It will be of interest to determine whether other NMDA receptor-dependent forms of LTP in the hippocampus, such as at the perforant path→dentate granule cell synapse, are also dependent on production of superoxide.
The enzymatic action of EC-SOD is to convert superoxide to hydrogen peroxide and oxygen (Halliwell, 1992), and long incubations of hippocampal slices with hydrogen peroxide have been shown to prevent the full expression of LTP (Pellmar et al., 1991; Auerbach and Segal, 1997). Furthermore, mice that overexpress SOD-1 have impaired LTP that can be reversed partially by the hydrogen peroxide scavenger catalase (Gahtan et al., 1998). In contrast to these studies, catalase did not reverse the impaired LTP observed in EC-SOD transgenic mice (Fig.5B). Moreover, hydrogen peroxide may be necessaryfor the full expression of LTP, because we found that catalase slightly attenuated LTP in slices from wild-type mice (Fig. 5B), a finding consistent with previous observations (Katsuki et al., 1998). One way to reconcile these apparently contradictory findings is to hypothesize that hydrogen peroxide is necessary for the full expression of LTP, and that prolonged exposure to exogenous hydrogen peroxide, in addition to endogenous hydrogen peroxide, is deleterious to the full expression of LTP. This hypothesis remains to be tested.
Our findings are consistent with the hypothesis that superoxide is produced in response to LTP-inducing stimulation. If this hypothesis is correct, then several questions arise. One question is the cellular action of superoxide. It is possible that superoxide acts as a signaling molecule. Superoxide can activate several protein kinases in the hippocampus, including protein kinase C (PKC; Knapp and Klann, 2000) and extracellular signal-regulated kinase 2 (Kanterewicz et al., 1998), that are necessary for LTP (Malinow et al., 1989; English and Sweatt, 1997). In addition, superoxide scavengers have been shown to block LTP-associated increases in PKC activity (Klann et al., 1998). Alternatively, superoxide can inactivate the calcium/calmodulin-dependent phosphatase calcineurin (Wang et al., 1996). Such an inactivation has been shown to occur with electrical stimulation of cultured hippocampal neurons (Bito et al., 1996) and might serve to enhance the phosphorylation of critical proteins involved in LTP. It also is possible that superoxide interacts with nitric oxide. The role of nitric oxide in LTP is controversial (Holscher, 1997); however, there is evidence that nitric oxide is produced via NMDA receptor activation after LTP-inducing stimulation (Chetkovich et al., 1993). Superoxide and nitric oxide are known to interact rapidly to form peroxynitrite, a very reactive oxidant that can also nitrate target proteins (Torreilles et al., 1999). It remains to be determined whether peroxynitrite either oxidizes or nitrates proteins after LTP-inducing stimulation.
Another question raised by our findings concerns the source of superoxide production. Superoxide can be a product of the actions of lipoxygenase on arachidonic acid (Kukreja et al., 1986), and lipoxygenase inhibitors have been shown to block LTP (Lynch et al., 1989). Although, as mentioned above, the role of nitric oxide synthase in LTP is controversial (Holscher, 1997), this enzyme has been shown to produce superoxide (Pou et al., 1999). Finally, NMDA receptor activation in hippocampal slices has been shown to increase production of superoxide via the mitochondrial electron transport chain (Bindokas et al., 1996). It will be interesting to determine whether LTP-inducing stimulation produces superoxide via any of the aforementioned mechanisms.
Our studies suggest that superoxide is necessary not only for LTP but also for associative memory. We found that EC-SOD transgenic mice exhibited a pronounced deficit in contextual fear conditioning with both unsignaled and signaled shocks when there was a delay of 24 hr between training and testing (Fig. 6A,B). This learning deficit appears to be caused by impairment in long-term memory, i.e., information consolidation rather than information acquisition, because EC-SOD transgenic mice exhibited similar levels of contextual fear conditioning as did wild-type mice when the delay between training and testing was short (Fig. 6B). The long-term memory deficit appears to be relatively specific to hippocampal-mediated learning, because EC-SOD transgenic mice did not exhibit a significant deficit in fear conditioning to the cue despite a 26 hr delay between training and testing. However, there was a trend for EC-SOD transgenic mice to freeze less to the tone than wild-type mice (Fig. 6C), which in light of the role of the amygdala in mediating this type of learning (Phillips and LeDoux, 1992; Maren et al., 1996) suggests the intriguing possibility that superoxide is involved in synaptic plasticity in the amygdala (Rogan and LeDoux, 1995).
In addition to the role that superoxide plays in the type of associative memory documented herein, superoxide appears to be necessary for other types of learning. For example, EC-SOD transgenic mice were shown to exhibit deficits in the acquisition of the eight-arm radial maze task (Levin et al., 1998), a hippocampal-dependent spatial memory task. In addition, transgenic mice that overexpress SOD-1 were found to be deficient in their ability to acquire the spatial version of the Morris water maze task (Gahtan et al., 1998). This finding is particularly intriguing, because in Down's syndrome, the phenotypic manifestation of trisomy 21, the activity of SOD-1 is elevated. The learning deficit observed in SOD-1 transgenic mice has been attributed to a defect in ROS metabolism (Gahtan et al., 1998); however, given the role of superoxide as a signaling molecule, it is possible that removal of superoxide by either SOD-1 or EC-SOD interferes with signaling cascades critical for learning.
Our studies demonstrate that superoxide is necessary for hippocampal synaptic plasticity and consolidation of associative memories. Although traditionally superoxide has been considered a neurotoxic molecule, our data suggest that improper scavenging of superoxide could be involved in the pathogenesis of a wide variety of neurodegenerative diseases of learning and memory, including Alzheimer's disease (Maulthaup et al., 1997). Whereas previous studies have suggested that scavenging of superoxide should limit neuronal degeneration, a growing body of evidence suggests that we consider superoxide and similar molecules as part of the biochemical signaling necessary for normal neuronal function (Lander, 1997; Klann and Thiels, 1999).
Footnotes
This work was supported by National Institutes of Health Grants NS36180 (E.T.), NS24288 (G.B.), and NS34007 (E.K.). We thank Lisa M. Schaefer and Kimberly D. Palangio for technical assistance, Dr. Lauren T. Knapp for helpful comments on this manuscript, Dr. J. Patrick Card for help with Nissl and immunocytochemical staining of sections, and Dr. Jerry W. Rudy for invaluable advice concerning the behavioral experiments.
Correspondence should be addressed to Dr. Eric Klann, Department of Neuroscience, University of Pittsburgh, 446 Crawford Hall, Pittsburgh, PA 15260. E-mail: eklann+@pitt.edu.
Dr. Urban's present address: Max-Planck-Institut für Medizinische Forschung, Abteilung Zellphysiologie, 29 Jahnstrasse, D-69120 Heidelberg, Germany.
REFERENCES
- 1.Auerbach JM, Segal M. Peroxide modulation of slow onset potentiation in rat hippocampus. J Neurosci. 1997;17:8695–8701. doi: 10.1523/JNEUROSCI.17-22-08695.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hyrdoethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 4.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 5.Castillo PE, Weisskopf MG, Nicoll RA. The role of calcium channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 6.Chapman PF, Frenguelli BG, Smith A, Chen C-M, Silva AJ. The α-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron. 1995;14:591–597. doi: 10.1016/0896-6273(95)90315-1. [DOI] [PubMed] [Google Scholar]
- 7.Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- 8.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 9.Collingridge GL, Kehl SJ, McClennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol (Lond) 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crapo JD, McCord JM, Fridovich I. Preparation and assay of superoxide dismutases. Methods Enzymol. 1978;53:382–393. doi: 10.1016/s0076-6879(78)53044-9. [DOI] [PubMed] [Google Scholar]
- 11.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 12.Gahtan E, Auerbach JM, Groner Y, Segal M. Reversible impairment of long-term potentiation in transgenic Cu/Zn-SOD mice. Eur J Neurosci. 1998;10:538–544. doi: 10.1046/j.1460-9568.1998.00058.x. [DOI] [PubMed] [Google Scholar]
- 13.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 14.Harris EW, Ganong AH, Cotman CW. Long-term potentiation in the hippocampus involves activation of N-methyl-d-aspartate receptors. Brain Res. 1984;323:132–137. doi: 10.1016/0006-8993(84)90275-0. [DOI] [PubMed] [Google Scholar]
- 15.Hess G, Kuhnt U, Voronin LL. Quantal analysis of paired-pulse facilitation in guinea pig hippocampal slices. Neurosci Lett. 1987;77:187–192. doi: 10.1016/0304-3940(87)90584-2. [DOI] [PubMed] [Google Scholar]
- 16.Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- 17.Jaffe D, Johnston D. Induction of long-term potentiation at hippocampal mossy fiber synapses follows a Hebbian rule. J Neurophysiol. 1990;64:948–960. doi: 10.1152/jn.1990.64.3.948. [DOI] [PubMed] [Google Scholar]
- 18.Kanterewicz BI, Knapp LT, Klann E. Stimulation of p42 and p44 mitogen-activated protein kinases by reactive oxygen species and nitric oxide in hippocampus. J Neurochem. 1998;70:1009–1016. doi: 10.1046/j.1471-4159.1998.70031009.x. [DOI] [PubMed] [Google Scholar]
- 19.Katsuki H, Noguchi K, Matsuki N. Modulation of synaptic plasticity by hydrogen peroxide in the hippocampus. Soc Neurosci Abstr. 1998;24:1070. [Google Scholar]
- 20.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 21.Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- 22.Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- 23.Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- 24.Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- 25.Kukreja RC, Kontos HA, Hess ML, Ellis EF. PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res. 1986;59:612–619. doi: 10.1161/01.res.59.6.612. [DOI] [PubMed] [Google Scholar]
- 26.Lander H. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- 27.Levin ED, Brady TC, Hochrein EC, Oury TD, Jonsson LM, Marklund SL, Crapo JD. Molecular manipulations of extracellular superoxide dismutase: functional importance for learning. Behav Genet. 1998;28:381–390. doi: 10.1023/a:1021673703129. [DOI] [PubMed] [Google Scholar]
- 28.Levkovitz Y, Avigonne E, Groner Y, Segal M. Upregulation of GABA neurotransmission suppresses hippocampal excitability and prevents long-term potentiation in transgenic superoxide dismutase-overexpressing mice. J Neurosci. 1999;19:10977–10984. doi: 10.1523/JNEUROSCI.19-24-10977.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynch G, Larson K, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 30.Lynch MA, Errington ML, Bliss TVP. Nordihydroguariaretic acid blocks the synaptic component of long-term potentiation and the associated increases in release of glutamate and arachidonate: an in vivo study in the dentate gyrus of the rat. Neuroscience. 1989;30:693–701. doi: 10.1016/0306-4522(89)90162-0. [DOI] [PubMed] [Google Scholar]
- 31.Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- 32.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC and CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 33.Maren S, Aharonov G, Stote DL, Fanselow MS. N-Methyl-d-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 34.Marklund SL, Holme E, Hellner L. Superoxide dismutase in extracellular fluids. Clin Chim Acta. 1982;126:41–51. doi: 10.1016/0009-8981(82)90360-6. [DOI] [PubMed] [Google Scholar]
- 35.Martin SJ, Grimwood JD, Morris RGM. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 36.Maulthaup G, Ruppert T, Schlicksupp A, Hesse L, Beher D, Masters CL, Beyreuther K. Reactive oxygen species and Alzheimer's disease. Biochem Pharmacol. 1997;54:533–539. doi: 10.1016/s0006-2952(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 37.McNaughton BL. Long-term synaptic enhancement and short-term potentiation in rat fascia dentata act through different mechanisms. J Physiol (Lond) 1982;324:249–262. doi: 10.1113/jphysiol.1982.sp014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller D, Lynch G. Evidence that changes in presynaptic calcium currents are not responsible for long-term potentiation in hippocampus. Brain Res. 1989;479:290–299. doi: 10.1016/0006-8993(89)91631-4. [DOI] [PubMed] [Google Scholar]
- 39.Oury TD, Ho Y-S, Piantadosi CA, Crapo JD. Extracellular superoxide dismutase, nitric oxide, and central nervous system toxicity. Proc Natl Acad Sci USA. 1992;89:9715–9719. doi: 10.1073/pnas.89.20.9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase in vessels and airways of humans and baboons. Free Radic Biol Med. 1996;20:957–965. doi: 10.1016/0891-5849(95)02222-8. [DOI] [PubMed] [Google Scholar]
- 41.Oury TD, Card JP, Klann E. Localization of extracellular superoxide dismutase in adult mouse brain. Brain Res. 1999;850:96–103. doi: 10.1016/s0006-8993(99)02103-4. [DOI] [PubMed] [Google Scholar]
- 42.Pellmar TC, Hollinden GE, Sarvey JM. Free radicals accelerate the decay of long-term potentiation in field CA1 of guinea-pig hippocampus. Neuroscience. 1991;44:353–359. doi: 10.1016/0306-4522(91)90060-2. [DOI] [PubMed] [Google Scholar]
- 43.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 44.Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J Biol Chem. 1999;274:9573–9580. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- 45.Roberson ED, English JD, Sweatt JD. A biochemist's view of long-term potentiation. Learn Mem. 1996;3:1–24. doi: 10.1101/lm.3.1.1. [DOI] [PubMed] [Google Scholar]
- 46.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 47.Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 48.Torreilles F, Salman-Tabcheh S, Guerin M, Torreilles J. Neurodegenerative disorders: the role of peroxynitrite. Brain Res Rev. 1999;30:153–163. doi: 10.1016/s0165-0173(99)00014-4. [DOI] [PubMed] [Google Scholar]
- 49.Urban NN, Barrionuevo G. Induction of Hebbian and non-Hebbian mossy fiber long-term potentiation by distinct patterns of high-frequency stimulation. J Neurosci. 1996;16:4293–4299. doi: 10.1523/JNEUROSCI.16-13-04293.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Culotta VC, Klee CB. Superoxide dismutase protects calcineurin from inactivation. Nature. 1996;383:434–437. doi: 10.1038/383434a0. [DOI] [PubMed] [Google Scholar]
- 51.Zucker RS. Short-term plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]