

Abstract
Neural activity producing a transient increase in intracellular Ca2+ concentration can induce long-term potentiation (LTP) at visual cortical inhibitory synapses similar to those seen at various excitatory synapses. Here we report that low-frequency neural activity is required to maintain LTP at these inhibitory synapses. Inhibitory responses of layer 5 cells evoked by layer 4 stimulation were studied in developing rat visual cortical slices under a pharmacological blockade of excitatory synaptic transmission using intracellular and whole-cell recording methods. Although LTP induced by high-frequency stimulation (HFS) persisted while test stimulation was applied at 0.1 Hz, it was not maintained in approximately two-thirds of cells after test stimulation was stopped for 30 min. In the rest of the cells, LTP seemed to be maintained by spontaneous presynaptic spikes, because presynaptic inhibitory cells discharged spontaneously in our experimental condition and because LTP was totally abolished by a temporary application of Na+ channel blockers. Experiments applying various Ca2+ channel blockers and Ca2+ chelators after HFS demonstrated that LTP maintenance was mediated by presynaptic Ca2+ entries through multiple types of high-threshold Ca2+channels, which activated Ca2+-dependent reactions different from those triggering transmitter release. The Ca2+ entries associated with action potentials seemed to be regulated by presynaptic K+ channels, presumably large-conductance Ca2+-activated K+ channels, because the application of blockers for these channels facilitated LTP maintenance. In addition, noradrenaline facilitated the maintenance of LTP. These findings demonstrate a new mechanism by which neural activity regulates the continuation and termination of LTP at visual cortical inhibitory synapses.
Keywords: long-term potentiation, maintenance, inhibitory synaptic transmission, Ca2+ channels, Ca2+-activated K+ channels, visual cortex
Activity-dependent long-term modification of synaptic transmission is thought to underlie information storage and learning (Brown et al., 1990; Bliss and Collingridge, 1993; Hawkins et al., 1993). Induction and maintenance phases can be distinguished in this modification process. During the induction phase, neural activity producing a transient increase in intracellular Ca2+ concentration ([Ca2+]i) initiates a series of biochemical reactions that lead to enduring modification of synaptic strength (Lynch et al., 1983; Malenka et al., 1988). For example, at excitatory synapses in hippocampal CA1 pyramidal cells, high-frequency stimulation (HFS) of presynaptic fibers can produce postsynaptic Ca2+ entry through NMDA receptors, leading to long-term potentiation (LTP) (Gustafsson and Wigstrom, 1988; Brown et al., 1990; Madison et al., 1991; Bliss and Collingridge, 1993). During the maintenance phase, modified synaptic strength is preserved by mechanisms independent of neural activity, which have still not been clarified sufficiently. It was proposed that NMDA receptor-mediated postsynaptic [Ca2+]i increase produces autophosphorylation of Ca2+/calmodulin-dependent protein kinase II (CaM kinase II), converting the molecule into a Ca2+-independent active form, which maintains LTP by persistent phosphorylation of its substrates (Lisman, 1994).
LTP is also produced by HFS of presynaptic fibers in GABA-mediated inhibitory synaptic transmission of rat visual cortex (Komatsu and Iwakiri, 1993). This LTP is input-specific and associative and requires postsynaptic [Ca2+]i increase for induction (Komatsu, 1994, 1996), as is the case with NMDA receptor-dependent LTP. However, the induction of LTP lacks dependence on postsynaptic membrane potentials, and postsynaptic [Ca2+]i increase originates from internal Ca2+ stores rather than from an extracellular source (Komatsu, 1994, 1996). The activation of GABAB receptors, which is also required for induction, seems to contribute to postsynaptic [Ca2+]i increase by facilitating monoamine receptor-mediated inositol trisphosphate formation, which produces Ca2+ release from the internal stores (Komatsu, 1996). This LTP occurs far more frequently in developing than in mature animals (Komatsu, 1994), suggesting that it is a cellular mechanism underlying experience-dependent development of visual responsiveness in cortical cells.
Although LTP at visual cortical inhibitory synapses resembles the NMDA-receptor dependent LTP in basic properties, such as input specificity, associativity, and involvement of postsynaptic [Ca2+]i increases in induction, the present study demonstrates that the former is a new form of LTP, different from the latter in that the maintenance of increased synaptic strength requires a low-frequency firing of presynaptic cells. This maintenance is mediated by action potential-associated presynaptic [Ca2+]i increases, which activate Ca2+-dependent reactions different from those triggering transmitter release.
MATERIALS AND METHODS
Sprague Dawley rats at postnatal ages of 20–29 d were deeply anesthetized with isoflurane before the whole brain was removed from the skull and immersed in an ice-cold oxygenated (95% O2 and 5% CO2) Krebs'–Ringer's solution containing (in mm): 124 NaCl, 5 KCl, 1.3 MgSO4, 4 CaCl2, 1.2 KH2PO4, 26 NaHCO3, and 10 glucose. Then, coronal slices of primary visual cortex (400-μm-thick) were prepared using a Microslicer (DTK-1000; Dosaka, Kyoto, Japan) and kept in a recovery chamber perfused with Krebs'–Ringer's solution at 33°C. During recording experiments, the medium, maintained at 33°C, contained 100 μmdl-2-amino-5-phosphonovaleric acid (APV), an NMDA receptor antagonist, and 40 μm6,7-dinitroquinoxaline-2,3-dione (DNQX), a non-NMDA receptor antagonist. For some experiments, the concentration of CaCl2 was altered without changing other solutes.
Two pairs of bipolar stimulating electrodes (Fig.1A, s1,s2) made of tungsten wires (diameter, 100 μm; interpolar distance, 200 μm) were placed in layer 4, separated from each other by ∼0.5 mm. Layers 2–4 was surgically cut between s1 ands2 to ensure that separate groups of presynaptic fibers were activated. Test stimulation was applied alternately to s1and s2 at intervals of 5 sec. As a conditioning stimulation, HFS (50 Hz, 1 sec) was applied to one of the electrodes 10 times at intervals of 10 sec. The intensity of the test stimulation and HFS was adjusted to 1.5–2 and 5 times the threshold intensity to evoke IPSPs, respectively.
Fig. 1.

Temporary cessation of test stimulation disturbs LTP maintenance. A, Experimental arrangement of stimulating (s1 and s2) and recording electrodes (r), with the leftfigures and a dashed line indicating cortical layers and surgical cuts, respectively. B, C, LTP was maintained in cells in which test stimulation (0.l Hz) was continued after HFS. B, Top(a) and middle(b) traces show superimposed average (n = 6) responses in a cell before and after HFS for conditioned (left) and unconditioned (right) pathways, respectively. Bottom traces (a, b) are thetop and middle traces superimposed. Recorded time of the traces is indicated in C. Resting membrane potential was −54 and −55 mV before and 100 min after HFS, respectively. Input resistance (42 MΩ) was unchanged before and 100 min after HFS. C, The initial falling slope of IPSPs (percent of the mean baseline) plotted against the time after HFS for 12 cells. Squares and triangles(mean ± SEM) represent responses for conditioned and unconditioned pathways, respectively. No significant difference (p > 0.4; n = 12) was found in either resting membrane potential or input resistance before (−55 ± 2 mV, 44 ± 4 MΩ) and 100 min after HFS (−56 ± 3 mV, 44 ± 5 MΩ), which was also the case for other experimental groups of cells to which HFS was applied.D, E, Similar to B andC, but test stimulation for the conditioned pathway (s1) was stopped for the period indicated by thehorizontal bar in E. D,Left and right superimposedtraces represent responses in two cells that did not and did maintain LTP after cessation of test stimulation, respectively.E, Squares and circlesrepresent responses for conditioned pathways that did (n = 3) and did not maintain LTP (n = 5), respectively, and trianglesindicate unconditioned pathways (n = 8).
Intracellular recording was conducted with microelectrode pipettes containing 2 m K-methylsulfate (50–100 MΩ). The electrode was mounted on a three-dimensional oil-driven micromanipulator (MO-103; Narishige, Tokyo, Japan). For analysis, we selected cells with a stable resting membrane potential hyperpolarized more than −50 mV. When the resting membrane potential was deeper than −60 mV and consequently the amplitude of IPSPs evoked by test stimulation was too small, the membrane potential was depolarized by current injection through the recording electrode to increase the IPSP amplitude. Input resistance was monitored throughout the experiments by injecting 0.05–0.1 nA hyperpolarizing current pulses. A bridge circuit was used to record the membrane potential while current injection was made through the recording electrode (Axoclamp 2A; Axon Instruments, Foster City, CA). In experiments recording spontaneous IPSCs, whole-cell recording was made from pyramidal cells using infrared differential contrast optics (BX50WI; Olympus Optical, Tokyo, Japan). Patch pipettes (5–6 MΩ) were filled with a solution containing (in mm): 126 d-gluconate, 140 CsOH, 8 CsCl, 2 NaCl, 0.2 EGTA, 10 HEPES, 3 MgATP, and 0.5 Na2GTP, pH 7.2 with CsOH. We selected cells with a high seal resistance (>1 GΩ) and a series resistance <25 MΩ (15–21 MΩ) for analysis. Cells were voltage clamped at 0 mV, and series resistance was not compensated. The laminar location of the stimulating and recording electrodes was identified on histological sections stained with cresyl violet after the recordings, as described previously (Komatsu, 1994). Data are expressed as mean ± SEM, and either Student'st test or Welch's test was applied.
The compounds used were obtained from the following sources: APV, DNQX, and CHS50911 from Tocris Cookson (Bristol, UK); iberiotoxin, tetrodotoxin (TTX), bicuculline methiodide, noradrenaline, and serotonin from Sigma (St. Louis, MO); 4-aminopyridine, charybdotoxin, ω-conotoxin GVIA, and nifedipine from Research Biochemicals (Natick, MA); ω-agatoxin IVA from Peptide Institute (Osaka, Japan); EGTA-AM from Calbiochem (La Jolla, CA); saxitoxin (STX) from Alexis (San Diego, CA); and isoflurane from Abbott Laboratories (North Chicago, IL).
RESULTS
Intracellular recording studies were conducted in visual cortical slices of developing rats at ages of postnatal 20–29 d, when LTP of IPSP occurs frequently (Komatsu, 1994) and visual responses are easily modified by visual experience (Fagiolini et al., 1994). IPSPs evoked by layer 4 stimulation were recorded from layer 5 neurons under a blockade of excitatory synaptic transmission using high doses of the NMDA receptor antagonist APV (100 μm) and the non-NMDA receptor antagonist DNQX (40 μm). In all the cases in which the LTP was analyzed, sharp electrodes were used to record IPSPs, because stable recording was possible for longer periods compared with patch electrodes. One of two stimulating electrodes placed in layer 4 was used to test the effect of HFS, and the other served as a control (Fig. 1A). Synaptic strength was assessed by test stimulation at a low frequency (0.1 Hz), which is in a range commonly used in studies of LTP at excitatory synapses. Experiments were conducted in the solution containing a high concentration (4 mm) of Ca2+(Komatsu, 1994, 1996) in which strong HFS used in this study always (12 of 12) produced LTP lasting >120 min as long as the recording remained stable (Fig. 1B,C).
LTP maintenance requires presynaptic action potentials but not postsynaptic responses
To test whether neural activity is necessary to maintain LTP, test stimulation of the conditioned pathway was stopped for 30 min after HFS. Figure 1, D and E, illustrates cases in which stimulation cessation was started 30 min after HFS. Before stimulation cessation, the responses for the conditioned pathway were significantly larger (p < 0.005) than the baseline level for all of the tested cells. After test stimulation was resumed, potentiated responses returned almost to the baseline level (p > 0.2) in approximately two-thirds of the cells (circles), whereas almost the same level of potentiation, which was significantly larger (p< 0.01) than the baseline, was maintained for the rest of cells (squares). The same effect was produced by cessation started either soon, or 30 or 120 min after HFS as shown in Figure2, which shows the magnitude of LTP before and after stimulation cessation (filled squares). No such LTP abolition was found when test stimulation was continued (open circles). Once LTP was abolished, no recovery was found, although test stimulation was continued for 30–75 min. To rule out the possibility that cessation of the stimulation reduces test responses irrespective of LTP, test stimulation was stopped for a separate group of cells to which HFS was not applied. No change was found in the responses after the stimulation cessation (99 ± 3% of the values before cessation; p > 0.3; n = 6).
Fig. 2.

Summary of effects of test stimulation cessation on LTP. A, Magnitude of LTP at 50–60 min after HFS in cells for which test stimulation was not (open circles) and was stopped from 0 to 30 min after HFS (filled squares). B, Magnitude of LTP (filled squares) before (20–30 min after HFS;left) and after (80–90 min after HFS;right) test stimulation cessation (30–60 min after HFS) and those (open circles) for corresponding values when test stimulation was not stopped. C, Similar toB, but test stimulation was stopped from 120 to 150 min after HFS.
We tested the possibility that spontaneous presynaptic spikes contributed to LTP, which persisted even after stimulation cessation, because inhibitory neurons fire spontaneously in rat somatosensory cortical slices even under a pharmacological blockade of ionotropic glutamate receptors (Salin and Prince, 1996). We first confirmed that inhibitory cells innervating layer 5 cells also discharged spontaneously in our experimental conditions. For this purpose, we tested whether spontaneous IPSCs were sensitive to a pharmacological blockade of action potentials using the whole-cell recording method, which is more effective in the detection of small responses than the method using sharp electrodes. Bath application of the Na+ channel blocker TTX (1 μm) significantly (p < 0.05) reduced the mean frequency of spontaneous IPSCs from 10.1 ± 2.4 to 3.4 ± 0.6 Hz and their mean amplitude from 21.9 ± 1.7 to 14.0 ± 1.2 pA in layer 5 cells (n = 6), as shown in Figure 3, indicating that these cells received inhibitory inputs, mediated by spontaneous presynaptic spikes, at a frequency of ∼7 Hz (3.2–14.6 Hz) on average. Therefore, these spontaneous spikes may have contributed to maintaining LTP during the cessation of test stimulation. This supposition was supported by experiments in which action potentials were temporarily abolished after HFS (Fig.4A,B). We used another sodium channel blocker, STX, instead of TTX to block spikes, because it is washed out more rapidly than TTX (Narahashi, 1974). Test responses disappeared temporarily and returned to the baseline level for both conditioned and unconditioned pathways after washout at the time when LTP was still demonstrated in the control experiment (Fig. 1C). Similarly, a temporary application of STX also abolished the LTP, which had persisted even after the cessation of test stimulation (Fig. 4C,D). These results indicate that presynaptic action potentials occurring at a low frequency are necessary to maintain LTP.
Fig. 3.

Inhibitory presynaptic cells discharge spontaneously. A–C, Spontaneous IPSCs recorded from a layer 5 cell, which was voltage clamped at 0 mV. Tracesin A, B, and C show spontaneous postsynaptic responses in the control solution containing both non-NMDA and NMDA receptor antagonists (DNQX and APV), after adding TTX (1 μm), a sodium channel blocker, and after further addition of bicuculline methiodide (20 μm), a GABAA receptor antagonist, respectively. D, Frequency histogram of peak amplitudes of IPSCs, recorded from the cell shown in A and B in the control solution (broken line) and in the presence of TTX (solid line). The total number of events was 1290 (control) and 341 (TTX).
Fig. 4.

Blockade of presynaptic action potentials, but not of GABA receptors, abolishes LTP. A, B, Effects of STX (0.1 μm), a sodium channel blocker, on LTP maintenance (n = 6). After LTP induction, STX was applied for the period indicated by the horizontal barin B. Recorded time of traces inA is indicated in B.Squares and triangles represent responses for conditioned and unconditioned pathways, respectively.C, D, Similar to A andB, but STX was applied to cells (n = 3) in which LTP was maintained even after test stimulation for the conditioned pathway (S1 off) was stopped for 30 min. E, F, Similar toA and B, but selective antagonists for GABAA receptors (bicuculline methiodide, 20 μm) and GABAB receptors (SCH 50911, 50 μm) were simultaneously applied for the period indicated by the horizontal bar in F(n = 6). Filled triangles inF indicate responses of unconditioned pathways for STX application, which were obtained from the experiments shown inB.
The effect of presynaptic action potentials on LTP maintenance can be mediated by postsynaptic responses. To test this possibility, we applied high doses of the selective GABAAreceptor antagonist bicuculline methiodide and the selective GABAB receptor antagonist CHS 50911 after HFS (Fig. 4E,F). IPSPs were completely abolished by these compounds. After washout, the responses of conditioned and unconditioned pathways returned to the respective levels before the drug application. A comparison of unconditioned pathway responses for GABA receptor antagonists (open triangles) and STX (filled triangles) in Figure4F demonstrated that the time course of IPSP blockade by the two treatments was almost the same. These results suggest that the maintenance of LTP requires neither postsynaptic responses nor activation of either GABAA or GABAB receptors.
LTP maintenance is mediated by presynaptic Ca2+entry through multiple types of high-threshold Ca2+channels
The above results prompted us to test the involvement of presynaptic Ca2+ channels in LTP maintenance. Accordingly, we applied Ca2+channel blockers after HFS. LTP always disappeared without any changes in unconditioned pathway responses after application of 20 μm nifedipine (Fig.5A), a selective L-type Ca2+ channel blocker (Fox et al., 1987;Aosaki and Kasai, 1989). The same effects were produced by low doses of ω-agatoxin IVA (30–50 nm), blocking selectively P-type Ca2+ channels at this range of doses (Fig. 5B) (Mintz et al., 1992; Sather et al., 1993). At a high dose (1 μm), which also blocks Q-type Ca2+ channels (Mintz et al., 1992;Sather et al., 1993; Wheeler et al., 1994), both conditioned and unconditioned pathway responses disappeared (Fig. 5C). Application of a high dose of ω-conotoxin GVIA (1 μm), a selective N-type Ca2+ channel blocker (Aosaki and Kasai, 1989; Plummer et al., 1989), also abolished LTP and reduced slightly both conditioned (84 ± 2% of the baseline at 70–80 min after HFS; n = 5) and unconditioned pathway responses (80 ± 2%). Although the reduction was significant for both pathways (p < 0.02), no significant difference (p > 0.1) was found in the amount of reduction between the two pathways (Fig. 5D). Application of 50 μm Ni2+, which rather selectively blocks T- and R-type Ca2+ channels (Fox et al., 1987; Narahashi et al., 1987; Zhang et al., 1993), affected neither LTP nor baseline responses (Fig. 5E). These results indicate that transmitter release is mediated mostly by Q-type and partially by N-type Ca2+ channels, whereas Ca2+ entries through P-, N-, and L-type Ca2+ channels are all necessary for LTP maintenance. We could not test the involvement of Q-type Ca2+ channels in maintenance because no specific blockers are available.
Fig. 5.

Effects of Ca2+channel blockers and chelators on LTP maintenance. A, Nifedipine (20 μm) was applied for the period indicated by the horizontal bar (n = 5).B–F, Similar to A, but 30–50 nm (B) or 1 μmω-agatoxin IVA (C), 1 μmω-conotoxin GVIA (D), 50 μmNi2+ (E), or 2 μm EGTA-AM (F) was applied instead. The number of cells was five for each of A–E and eight for F. Squares andtriangles represent responses for conditioned and unconditioned pathways, respectively.
It is likely that presynaptic, but not postsynaptic, high-threshold Ca2+ channels contribute to LTP maintenance, because test stimulation produced only small hyperpolarizing responses in postsynaptic cells and because LTP was maintained under voltage clamp at hyperpolarized (−90 mV) or very depolarized (between +20 and +40 mV) membrane potentials (Komatsu, 1994) and even under no postsynaptic responses (Fig.4E,F). This suggests that LTP maintenance requires presynaptic [Ca2+]i elevation. We tested this possibility with EGTA-AM, which is membrane-permeable and converted into EGTA, a slow Ca2+chelator, in the cytoplasm. Bath application of the compound (2 μm) after HFS abolished LTP without any effects on the unconditioned pathway (Fig. 5F). LTP persisted during the whole-cell recording with patch electrodes containing 1 mm EGTA (Komatsu, 1994, 1996). These results are consistent with the idea that increases in presynaptic [Ca2+]i are necessary for LTP maintenance.
Temporary reduction in presynaptic Ca2+ entry abolishes LTP
Temporarily applied Ca2+ channel blockers and chelators might remain in tissues after washout, because this procedure is not easily accomplished. If so, abolition of LTP can be attributable to continuous, but not temporary, reduction in presynaptic [Ca2+]i increases. We, thus, tested the effect of temporary reduction in Ca2+ entry on LTP maintenance by changing extracellular Ca2+ concentrations ([Ca2+]o). For this purpose, we first examined the dependence of LTP on [Ca2+]o. LTP (>15% increase from the baseline at 40–50 min after HFS) occurred rarely at a normal value (2.4 mm) of [Ca2+]o, more frequently with the increase of [Ca2+]o, and always at 3.6–4.8 mm (Fig.6A). When [Ca2+]o was temporarily reduced from a high to normal value, LTP was abolished and never recovered after restoration of high [Ca2+]o for 40–90 min (Fig. 6B). This suggests that, once LTP is abolished by a reduction in Ca2+ entry, it could not be reestablished, even by restoring the previous amount of Ca2+ entry. In consideration of this result and the experiment using EGTA-AM, it is extremely unlikely that the complete abolition of LTP by any of the three Ca2+ channel blockers (Fig.5A,B,D) merely resulted from the blockade of Ca2+ channels recruited to transmitter releases after HFS, although such recruitment did take place. High [Ca2+]o was not required for LTP induction, because HFS, applied under normal [Ca2+]o, produced LTP if high [Ca2+]o was resumed immediately after HFS (Fig. 6C).
Fig. 6.

Temporary reduction in [Ca2+]o abolishes LTP.A, Incidence of LTP (>15% increase from the baseline at 40–50 min after HFS) plotted against [Ca2+]o with figures indicating the number of tested cells. B, C, [Ca2+]o was changed between 4 and 2.4 mm, indicated in the middle. The number of cells was six (B) and five (C). Squares andtriangles represent responses for conditioned and unconditioned pathways, respectively.
Presynaptic K+ channels regulate the maintenance of LTP
The above results suggest that the amount of Ca2+ entry was insufficient to maintain LTP at normal [Ca2+]o in our experimental conditions (Fig.7A). Using various K+ channel blockers, we tested the possibility that open K+ channels disturb LTP maintenance, because they reduce Ca2+entry associated with action potentials. In the presence of 60 nm charybdotoxin or 100 nmiberiotoxin, LTP occurred frequently (7 of 9 for charybdotoxin, and 5 of 5 for iberiotoxin) even at normal [Ca2+]o, although the magnitude was significantly smaller (p < 0.02) than that at high [Ca2+]o (compare Fig. 7B with Fig. 1C). These toxins are known to block large-conductance Ca2+-activated K+ (BK) channels (Miller et al., 1985;Galvez et al., 1990). However, they could block some other K+ channels (Lewis and Cahalan, 1988;MacKinnon et al., 1988; Schweitz et al., 1989; Galvez et al., 1990). The same effect was produced by 1 mmCs+, which blocks various K+ channels including BK channels (Hille, 1992), without affecting baseline responses (Fig. 7C). In contrast, 4-aminopyridine (10–50 μm), which blocks various K+ channels but not BK channels (Hermann and Hartung, 1982; Hille, 1992), was ineffective for LTP production, although it increased baseline responses to 154 ± 23% (n = 7) of the value before drug application (Fig.7D). This increase may be attributable to enhanced transmitter release, as shown for various synapses (Thesleff, 1980). If LTP is expressed presynaptically, the ineffectiveness of this blocker for LTP production could be attributable to an occlusion of LTP by the enhanced transmitter release before HFS. However, this possibility was not supported by similar experiments conducted at high [Ca2+]o. Application of the blocker increased test responses to 158 ± 6% of the control value, but HFS still produced LTP (186 ± 10% at 30–40 min after HFS compared with the value just before HFS;n = 3). Cs+ was used to confirm that blockade of these K+ channels is involved in the maintenance of LTP, because it was far more quickly washed in and out than those toxins. LTP occurred when Cs+ was added to the solution after, but not during, HFS (Fig. 8). Thus, it is thought that closure of BK or pharmacologically related K+ channels facilitates the maintenance of LTP.
Fig. 7.

Effects of K+ channel blockers on LTP. A, Effect of HFS on IPSPs at normal (2.4 mm) [Ca2+]o(n = 8). B, Effect of charybdotoxin (open symbols; n = 9) and iberiotoxin (filled symbols;n = 5) on LTP at normal [Ca2+]o. Both toxins were present all the time. C, Effect of 1 mmCs+ on LTP at normal [Ca2+]o (n = 8). Blocker application time is indicated by the horizontal bar. D, Similar to C, but 10–50 μm 4-aminopyridine was applied (n = 7).
Fig. 8.
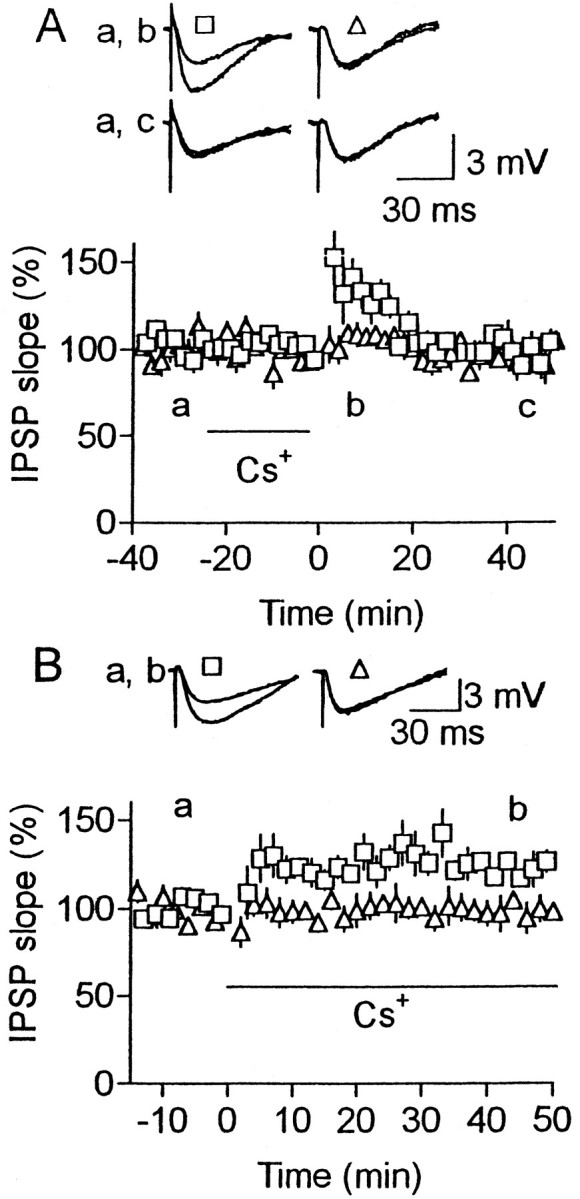
Cs+ facilitates the maintenance of LTP. Similar to Figure 7C, but Cs+(1 mm) was applied to the bath during (A) or after HFS (B). Number of cells was five (A) and six (B).
Noradrenaline facilitates the maintenance of LTP
The inability to maintain LTP at normal [Ca2+]o might result from the absence of neuromodulatory inputs from other parts of the brain to the cells. We tested the involvement of noradrenaline and serotonin. In the presence of noradrenaline (5 μm), LTP was consistently (8 of 8) produced under normal [Ca2+]o (Fig.9), although the magnitude was significantly smaller (p < 0.01) than that under high [Ca2+]o(Fig. 1C). On the other hand, no LTP was produced (95 ± 4% of the baseline; n = 5) in the presence of high doses of serotonin (100 μm). These results suggest that noradrenaline controls the maintenance of LTP by modulating the presynaptic Ca2+ and/or K+ channels.
Fig. 9.

Noradrenaline enables the maintenance of LTP under normal [Ca2+]o. Effects of HFS on IPSPs in the presence of noradrenaline (5 μm) under normal [Ca2+]o (2.4 mm). Noradrenaline was present all the time. Number of cells was eight.
DISCUSSION
This study demonstrated that low-frequency spike activity of presynaptic neurons is required to maintain LTP at visual cortical inhibitory synapses. The effect of action potentials is mediated by presynaptic Ca2+ entry through multiple types of high-threshold Ca2+ channels, which activates Ca2+-dependent reactions different from those triggering transmitter release. Our results also suggest that presynaptic K+ channels regulate the Ca2+ entry involved in maintenance.
Our results suggest that triggering presynaptic spikes at a frequency of 0.1 Hz is almost sufficient for the maintenance of LTP. However, one cannot exclude the possibility that this maintenance could be observed using a lower presynaptic spike frequency. Although the time course of the [Ca2+]iincrease associated with action potentials at axon terminals of these inhibitory cells has not been clarified, it has been demonstrated that Ca2+ transients at hippocampal excitatory nerve terminals return to the baseline level within a few to several seconds, a period shorter than the stimulus intervals used in this study (Regehr et al., 1994; Wu and Saggau, 1994). Thus, it is likely that intermittent occurrences of Ca2+transients or a continuously elevated low level of [Ca2+]i is sufficient for the activation of Ca2+-dependent processes responsible for LTP maintenance.
The inhibitory synaptic transmission studied in this experiment was mediated by ω-agatoxin IVA- and ω-conotoxin GVIA-sensitive Ca2+ channels, as was the case in other central excitatory and inhibitory synapses (Dunlap et al., 1995;Reuter, 1996). Because dihydropyridines did not affect transmitter release at central synapses, it has been suggested that L-type Ca2+ channels are almost absent at the presynaptic terminals of the CNS, although they contribute to hormone secretion from neuroendocrine cells (Dunlap et al., 1995;Reuter, 1996). The results of the present study provided evidence for the presence of L-type Ca2+ channels at central nerve terminals, which was also reported recently for cultured hippocampal cells in which they were involved in post-tetanic potentiation but not basal responses (Jensen et al., 1999). It has been suggested that multiple types of Ca2+channels, which are involved in transmitter release, coexist at a single nerve terminal (Dunlap et al., 1995). Our results showed that the blockade of any of the multiple types of Ca2+ channels involved in LTP maintenance abolished LTP totally in all of the tested cells, suggesting that these channels also coexist at a nerve terminal. If only a subset of these channels are present at individual terminals, the blockade of one type of these channels may produce a partial, but not total, blockade of LTP. The question of why such multiple channels are involved in LTP maintenance remains unclarified.
The observation that charybdotoxin and iberiotoxin enabled LTP production at normal [Ca2+]o suggests that BK channels are involved in the regulation of LTP maintenance. Although charybdotoxin blocks K+ channels other than BK channels (Lewis and Cahalan, 1988; MacKinnon et al., 1988; Schweitz et al., 1989; Galvez et al., 1990), iberiotoxin seems to block BK channels more specifically. However, we cannot still deny the possibility that the latter toxin blocks some other K+ channels. Therefore, we conclude at present that BK or pharmacologically related K+ channels are involved in the regulation of LTP maintenance. Neuronal BK channels are usually opened by simultaneous Ca2+ increase and depolarization in association with action potentials, contributing to repolarization of action potentials (Lancaster and Nicoll, 1987; Storm, 1987). Therefore, activation of these channels may reduce the Ca2+ entry associated with action potentials, contributing to the abolition of LTP.
At squid giant synapses, presynaptic injections of EGTA blocked both HFS-induced short-term potentiation and slow Ca2+ signals, whereas they had no effect on basal transmitter release triggered by fast Ca2+ signals (Swandulla et al., 1991). Thus, the experiment using the membrane-permeable Ca2+ chelator EGTA-AM suggests that presynaptic slow Ca2+ signals activate reactions that maintain LTP. Although this Ca2+-dependent process is unresolved yet, Ca2+-dependent protein kinases and phosphatases are possible candidates for molecules mediating these Ca2+ signals. These reactions could act on some transmitter releasing processes modified by HFS, because the presynaptic localization of maintenance mechanisms strongly suggests the presynaptic expression of LTP. The BK or related K+ channels may be located apart from transmitter release sites and control Ca2+entry relevant to LTP maintenance but not triggering transmitter release, because Cs+ facilitated LTP maintenance but not baseline responses, whereas 4-aminopyridine facilitated baseline responses but not LTP maintenance. This supposition is compatible with the observation that P- and L-type Ca2+ channels contributed to LTP maintenance but not the baseline responses. In addition, Ca2+ imaging of presynaptic axons has suggested that high-threshold Ca2+channels are also located at sites other than transmitter release sites, although their density is less than those at release sites (Mackenzie et al., 1996).
Although the maintenance of LTP depends on neural activity and Ca2+ signals at visual cortical inhibitory synapses, it has been reported that LTP at hippocampal excitatory synapses persists after the blockade of presynaptic spikes or Ca2+ channels (Malgaroli and Tsien, 1992;Manabe et al., 1992; Cormier et al., 1993; Castillo et al., 1994;Wheeler et al., 1994). A mechanism has been proposed for NMDA receptor-dependent LTP, which is suggested to be expressed, at least partly, by phosphorylation of AMPA receptors by CaM kinase II (Lisman, 1994; Pettit et al., 1994; Barria et al., 1997). The phosphorylated state could be maintained by Ca2+-dependent autophosphorylation of the kinase, switching the molecule into an active state even at low [Ca2+]i (Saitoh and Schwartz, 1985; Miller and Kennedy, 1986).
In regard to synaptic plasticity as a basis of learning and memory, the reversibility of modified synaptic strength could be important. At CA1 excitatory synapses, HFS (10–100 Hz)-induced LTP is reversed by low-frequency (1–5 Hz) stimulation continued for 10–15 min (Staubli and Lynch, 1990; Fujii et al., 1991). This type of LTP reversal has been reported for other excitatory synapses (Kirkwood et al., 1993;Chen et al., 1996; Tzounopoulos et al., 1998). In contrast, at visual cortical inhibitory synapses, LTP is induced by stimulation of a wide range (2–50 Hz) of frequencies (Komatsu, 1994), and its reversal is attained by reducing the frequency of stimulation to a level lower than that for test stimulation. This difference suggests that the two forms of LTP operate very differently and play different functional roles.
LTP at these inhibitory synapses was consistently produced in the presence of noradrenaline at normal [Ca2+]o, suggesting that the activity of noradrenergic cells, which were shown to be involved in visual cortical plasticity (Kasamatsu and Pettigrew, 1976; Bear and Singer, 1986), regulates the maintenance mechanism by modulating presynaptic K+ and/or Ca2+ channels. Because this synaptic plasticity could underlie the experience-dependent refinement of visual responsiveness proceeding gradually during a critical period in early life (Komatsu, 1994), a single episode of neural activity evoked by visual inputs may produce LTP, which is reversible depending on the following neural activity. The modified visual responsiveness consequent to the LTP may not persist unless the strengthened inhibitory synapses are activated at least at a low frequency. Orientation selectivity is improved to almost adult level in the middle of the critical period (Frégnac and Imbert, 1984), and deprivation of light in that period, even for a few days, degrades the selectivity (Freeman et al., 1981). This observation supports the idea that neural activity plays a crucial role to maintain visual responsiveness refined depending on experience. If LTP at the inhibitory synapses indeed contributes to developmental plasticity of visual responsiveness, a temporary pharmacological blockade of spike activity in cortical inhibitory cells during the critical period may degrade the selectivity of visual responses, which is experimentally testable. Repetitive use of the modified synapse during the critical period might lead eventually to a more persistent form independent of activity, requiring RNA and protein synthesis, as is the case in the late phase of hippocampal NMDA receptor-dependent LTP (Frey et al., 1988; Nguyen et al., 1994).
Footnotes
This work was supported by Grants-in Aid for Scientific Research Projects 07279104, 08458271, and 12053228 from the Japanese Ministry of Education, Science, Sports, and Culture.
Correspondence should be addressed to Dr. Yukio Komatsu, Department of Visual Neuroscience, Research Institute of Environmental Medicine, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464–8601, Japan. E-mail: komatsu@riem.nagoya-u.ac.jp.
REFERENCES
- 1.Aosaki T, Kasai H. Characterization of two kinds of high-voltage-activated Ca-channel currents in chick sensory neurons. Differential sensitivity to dihydropyridines and ω-conotoxin GIVA. Pflügers Arch. 1989;414:150–156. doi: 10.1007/BF00580957. [DOI] [PubMed] [Google Scholar]
- 2.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 3.Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- 4.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 5.Brown TH, Kairiss EW, Keenan CL. Hebbian synapses: biophysical mechanisms and algorithms. Annu Rev Neurosci. 1990;13:475–511. doi: 10.1146/annurev.ne.13.030190.002355. [DOI] [PubMed] [Google Scholar]
- 6.Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 7.Chen WR, Lee S, Kato K, Spencer DD, Shepherd GM, Williamson A. Long-term modifications of synaptic efficacy in the human inferior and middle temporal cortex. Proc Natl Acad Sci USA. 1996;93:8011–8015. doi: 10.1073/pnas.93.15.8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cormier RJ, Mauk MD, Kelly PT. Glutamate iontophoresis induces long-term potentiation in the absence of evoked presynaptic activity. Neuron. 1993;10:907–919. doi: 10.1016/0896-6273(93)90206-7. [DOI] [PubMed] [Google Scholar]
- 9.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 10.Fagiolini M, Pizzorusso T, Berardi N, Domenici L, Maffei L. Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 1994;34:709–720. doi: 10.1016/0042-6989(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 11.Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurons. J Physiol (Lond) 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman RD, Mallach R, Hartley S. Responsivity of normal kitten striate cortex deteriorates after brief binocular deprivation. J Neurophysiol. 1981;45:1074–1084. doi: 10.1152/jn.1981.45.6.1074. [DOI] [PubMed] [Google Scholar]
- 13.Frégnac Y, Imbert M. Development of neuronal selectivity in primary visual cortex of cat. Physiol Rev. 1984;64:325–434. doi: 10.1152/physrev.1984.64.1.325. [DOI] [PubMed] [Google Scholar]
- 14.Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- 15.Fujii S, Saito K, Miyakawa H, Ito K, Kato H. Reversal of long-term potentiation (depotentiation) induced by tetanus stimulation of the input to CA1 neurons of guinea pig hippocampal slices. Brain Res. 1991;555:112–122. doi: 10.1016/0006-8993(91)90867-u. [DOI] [PubMed] [Google Scholar]
- 16.Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channels from venom of the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- 17.Gustafsson B, Wigstrom H. Physiological mechanisms underlying long-term potentiation. Trends Neurosci. 1988;11:156–162. doi: 10.1016/0166-2236(88)90142-7. [DOI] [PubMed] [Google Scholar]
- 18.Hawkins RD, Kandel ER, Siegelbaum SA. Learning to modulate transmitter release. Annu Rev Neurosci. 1993;16:625–665. doi: 10.1146/annurev.ne.16.030193.003205. [DOI] [PubMed] [Google Scholar]
- 19.Hermann A, Hartung K. Properties of a Ca2+ activated K+ conductance in Helix neurones investigated by intracellular Ca2+ ionophoresis. Pflügers Arch. 1982;393:248–253. doi: 10.1007/BF00584078. [DOI] [PubMed] [Google Scholar]
- 20.Hille B. Ionic channels of excitable membranes. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 21.Jensen K, Jensen MS, Lambert JDC. Role of presynaptic L-type Ca2+ channels in GABAergic synaptic transmission in cultured hippocampal neurons. J Neurophysiol. 1999;81:1225–1230. doi: 10.1152/jn.1999.81.3.1225. [DOI] [PubMed] [Google Scholar]
- 22.Kasamatsu T, Pettigrew JD. Depletion of brain catecholamine: failure of ocular dominance shift after monocular occlusion in kitten. Science. 1976;194:206–209. doi: 10.1126/science.959850. [DOI] [PubMed] [Google Scholar]
- 23.Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF. Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science. 1993;260:1518–1521. doi: 10.1126/science.8502997. [DOI] [PubMed] [Google Scholar]
- 24.Komatsu Y. Age-dependent long-term potentiation of inhibitory synaptic transmission in rat visual cortex. J Neurosci. 1994;14:6488–6499. doi: 10.1523/JNEUROSCI.14-11-06488.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komatsu Y. GABAB receptors, monoamine receptors, and postsynaptic inositol trisphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci. 1996;16:6342–6352. doi: 10.1523/JNEUROSCI.16-20-06342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komatsu Y, Iwakiri M. Long-term modification of inhibitory synaptic transmission in developing visual cortex. NeuroReport. 1993;4:907–910. doi: 10.1097/00001756-199307000-00017. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol (Lond) 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis RS, Cahalan MD. Subset-specific expression of potassium channels in developing murine T lymphocytes. Science. 1988;239:771–775. doi: 10.1126/science.2448877. [DOI] [PubMed] [Google Scholar]
- 29.Lisman J. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 30.Lynch G, Larson J, Kelso S, Barrionuevo G, Schotler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 31.Mackenzie PJ, Umemiya M, Murphy TH. Ca2+ imaging of CNS axons in culture indicates reliable coupling between single action potentials and distal functional release sites. Neuron. 1996;16:783–795. doi: 10.1016/s0896-6273(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 32.MacKinnon R, Reinhart PH, White MM. Charybdotoxin block of shaker K+ channels suggests that different types of K+ channels share common structural features. Neuron. 1988;1:997–1001. doi: 10.1016/0896-6273(88)90156-0. [DOI] [PubMed] [Google Scholar]
- 33.Madison DV, Malenka RC, Nicoll RA. Mechanisms underlying long-term potentiation of synaptic transmission. Annu Rev Neurosci. 1991;14:379–397. doi: 10.1146/annurev.ne.14.030191.002115. [DOI] [PubMed] [Google Scholar]
- 34.Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- 35.Malgaroli A, Tsien RW. Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature. 1992;357:134–139. doi: 10.1038/357134a0. [DOI] [PubMed] [Google Scholar]
- 36.Manabe T, Renner P, Nicoll RA. Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature. 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- 37.Miller C, Moczydlowski E, Latorre R, Phillips M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature. 1985;313:316–318. doi: 10.1038/313316a0. [DOI] [PubMed] [Google Scholar]
- 38.Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 39.Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- 40.Narahashi T. Chemicals as tools in the study of excitable membranes. Physiol Rev. 1974;54:813–889. doi: 10.1152/physrev.1974.54.4.813. [DOI] [PubMed] [Google Scholar]
- 41.Narahashi T, Tsunoo A, Yoshii M. Characterization of two types of calcium channels in mouse neuroblastoma cells. J Physiol (Lond) 1987;383:231–249. doi: 10.1113/jphysiol.1987.sp016406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 43.Pettit DL, Perlman S, Malinow R. Potentiated transmission and prevention of further LTP by increased CAMKII activity in postsynaptic hippocampal slice neurons. Science. 1994;266:1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- 44.Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- 45.Regehr WG, Delaney K, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reuter H. Diversity and function of presynaptic calcium channels in the brain. Curr Opin Neurobiol. 1996;6:331–337. doi: 10.1016/s0959-4388(96)80116-4. [DOI] [PubMed] [Google Scholar]
- 47.Saitoh T, Schwartz JH. Phosphorylation-dependent subcellular translocation of a Ca2+/calmodulin-dependent protein kinase produces an autonomous enzyme in Aplysia neurons. J Cell Biol. 1985;100:835–842. doi: 10.1083/jcb.100.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salin PA, Prince DA. Spontaneous GABAA receptor-mediated inhibitory currents in adult rat somatosensory cortex. J Neurophysiol. 1996;75:1573–1588. doi: 10.1152/jn.1996.75.4.1573. [DOI] [PubMed] [Google Scholar]
- 49.Sather WA, Tanabe T, Zhang J-F, Mori Y, Adams ME, Tsien RW. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel α1 subunits. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- 50.Schweitz H, Stansfeld CE, Bidard J-N, Fagni L, Maes P, Lazdunski M. Charybdotoxin blocks dendrotoxin-sensitive voltage-activated K+ channels. FEBS Lett. 1989;250:519–522. doi: 10.1016/0014-5793(89)80788-4. [DOI] [PubMed] [Google Scholar]
- 51.Staubli U, Lynch G. Stable depression of potentiated synaptic responses in the hippocampus with 1–5 Hz stimulation. Brain Res. 1990;513:113–118. doi: 10.1016/0006-8993(90)91096-y. [DOI] [PubMed] [Google Scholar]
- 52.Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol (Lond) 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swandulla D, Hans M, Zipser K, Augustine GJ. Role of residual calcium in synaptic depression and posttetanic potentiation: fast and slow calcium signaling in nerve terminals. Neuron. 1991;7:915–926. doi: 10.1016/0896-6273(91)90337-y. [DOI] [PubMed] [Google Scholar]
- 54.Thesleff S. Aminopyridines and synaptic transmission. Neuroscience. 1980;5:1413–1419. doi: 10.1016/0306-4522(80)90002-0. [DOI] [PubMed] [Google Scholar]
- 55.Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- 56.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 57.Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J-F, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL, Tsien RW. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]