

Abstract
The inhibition of presynaptic calcium channels via G-protein-dependent second messenger pathways is a key mechanism of transmitter release modulation. We used the calyx-type nerve terminal of the chick ciliary ganglion to examine which G-proteins are involved in the voltage-sensitive inhibition of presynaptic N-type calcium channels. Adenosine caused a prominent inhibition of the calcium current that was totally blocked by pretreatment with pertussis toxin (PTX), consistent with an exclusive involvement of Go/Gi in the G-protein pathway. Immunocytochemistry was used to localize these G-protein types to the nerve terminal and its transmitter release face. We used two approaches to test for modulation by other G-protein types. First, we treated the terminals with ligands for a variety of G-protein-linked neurotransmitter receptor types that have been associated with different G-protein families. Although small inhibitory effects were observed, these could all be eliminated by PTX, indicating that in this terminal the Gi family is the sole transmitter-induced G-protein inhibitory pathway. Second, we examined the kinetics of calcium channel inhibition by uncaging the nonselective and irreversible G-protein activator GTPγS, bypassing the receptors. A large fraction of the rapid GTPγS-induced inhibition persisted, consistent with a Go/Gi-independent pathway. Immunocytochemistry identified Gq, G11, G12, and G13 as potential PTX-insensitive second messengers at this terminal. Thus, our results suggest that whereas neurotransmitter-mediated calcium channel inhibition is mainly, and possibly exclusively, via Go/Gi, other rapid PTX-insensitive G-protein pathways exist that may involve novel, and perhaps transmitter-independent, activating mechanisms.
Keywords: nerve terminal, G-protein, G-protein type, calcium channel, presynaptic, calcium channel modulation, calcium channel inhibition, transmitter release, synaptic strength, chick, calyx synapse, chick ciliary ganglion
The inhibition of presynaptic calcium channels via trimeric G-protein second messenger pathways is a key mechanism whereby transmitter release, and hence, synaptic strength can be modulated (Hille, 1994). However, relatively little is known about which specific G-proteins are involved in this pathway in intact nerve terminals. We have used the large presynaptic nerve terminal of the chick ciliary ganglion to examine the diversity of G-protein types involved in calcium channel regulation.
The chick calyx nerve terminal preparation (Stanley and Goping, 1991) has several key advantages for an analysis of G-protein action. First, it is sufficiently large to allow direct recording of whole-cell calcium currents. Second, the channels are almost exclusively N-type (Stanley, 1991; Yawo and Momiyama, 1993) and located in the presynaptic region (Stanley, 1993; Haydon et al., 1994). Third, the large size allows protein components to be localized to the surface membrane by immunocytochemistry and, by costaining for vesicle clusters, to the transmitter release site regions (Stanley and Mirotznik, 1997).
The N-type calcium channel is known to be sensitive to transmitter-mediated, G-protein-dependent, inhibition (for review, see Dolphin, 1998; Ikeda and Dunlap, 1999). A major element of this inhibition is via a voltage-sensitive mechanism (Bean, 1989) that can be relieved, and hence assayed, by a strong preceding depolarizing pulse (Grassi and Lux, 1989; Elmslie, 1990).
There is considerable diversity in G-protein pathways leading to N-type calcium channel inhibition. A wide range of metabotropic receptor types may be involved, and a number of different G-proteins are capable of acting as second messengers, indicating a high level of heterogeneity. The pertussis toxin (PTX)-sensitive G-proteins Goand Gi are the most commonly identified types in both primary neurons (Diversé-Pierluissi and Dunlap, 1993; Hille, 1994; Filippov et al., 1998; Park and Dunlap, 1998) and cell lines (Toth et al., 1996; Morikawa et al., 1998). However, PTX-insensitive G-proteins can also modulate these channels. Thus, Gs can modulate N-type channels in rat sympathetic neurons (Zhu and Ikeda, 1994), G13does so in the NG 108–15 cell line (Wilk-Blaszczak et al., 1994), and Gz will substitute for PTX-sensitive G-proteins when overexpressed in superior cervical ganglia neurons (Jeong and Ikeda, 1998).
The presynaptic calcium channels in the chick calyx are inhibited via a G-protein-dependent pathway. Adenosine, a potent modulator of transmitter release at a variety of synapses, inhibits N-type calcium channels (Yawo and Chuhma, 1993). G-protein-dependent, voltage-sensitive inhibition of the calcium channels has been shown directly using the nonhydrolyzable (and irreversible) GTP analog GTPγS (Stanley and Mirotznik, 1997).
The goal of this study was to determine which G-protein type or types modulate presynaptic N-type calcium channels at the chick calyx synapse. Our results indicate that extracellular transmitters, including adenosine, all appear to act via Go/Gi. However, we also demonstrate that other classes of G-protein are present, and we present evidence that one or more of these can also inhibit presynaptic calcium channel activity.
MATERIALS AND METHODS
Electrophysiology
Calyx nerve terminal preparation. Ciliary ganglia were removed from embryonic day 15 (E15) chicks and were enzymatically dissociated in minimal Eagle's medium (MEM), as previously described (Stanley and Goping, 1991; Haydon et al., 1994). The dissociated preparation was transferred to a coverslip recording chamber containing the external solution, and calyx nerve terminals were identified visually under high-power magnification with oil-immersion lenses (40–60×; 1.35–1.4 numerical aperture). The cells were washed three times in MEM before use.
Patch-clamp recording. Currents were recorded using the whole-cell variant of the patch-clamp technique. Patch electrodes (1.5 mm outside diameter, thin wall microcapillary glass; World Precision Instruments), were fire-polished and had resistances in the range of 3.5–4.5 MΩ when filled with the internal solution described below. Currents were amplified, and cell membrane capacitance and series resistance were electronically compensated (Axopatch 200A; Axon Instruments, Foster City, CA). Voltage protocol generation and data acquisition were performed using pClamp 7.0 software. Current traces were generally low-pass filtered at 5 kHz, and leak currents were subtracted with a standard P/6 protocol, using a positive polarity leak subtraction pulse. All recordings were performed at room temperature.
The external (bath) medium was (in mm): NaCl 160, CaCl2 5, MgCl2 1,d-glucose 5, 4-aminopyridine 2, tetrodotoxin 0.001, HEPES-Na 10, and the patch electrode internal solution was: Cs-gluconate 120, CsCl 10, EGTA-Cs 10, MgCl2 1, HEPES-Cs 10, tetraethylamonium-Cl 20, MgATP 1, with GTP 0.1 (except where specified). GTPγS (0.1 mm) was included in the internal solution as described. PTX treatment was performed by incubating the preparation at 20°C (8% CO2) in MEM overnight with or without 0.5–10 μg/ml PTX (Research Biochemicals, Natick, MA). Overnight incubation reduced the number of available calyx terminals for recording, consistent with the degeneration of the nerve stump after nerve section (Stanley and Drachman, 1980), and greatly increased the difficulty of these experiments. For flash photolysis, release of intracellular GTPγS,S-DMNPE-caged GTPγS (400 μm; Molecular Probes, Eugene, OR) was added to the internal solution, and a single 200 msec flash of unfiltered light from a mercury bulb (Uniblitz shutter; 40×; 1.35 NA quartz objective) was used to liberate the free nucleotide.
Voltage protocol and data analysis. A double trial protocol was used to measure voltage-sensitive G-protein inhibition. The cells were held at −80 mV. In the first trial a single 80 msec test pulse to 0 mV activated a calcium current. In the second trial, delivered 5 sec later, the test pulse was preceded by a 60 msec depolarization to +80 mV, a conditioning pulse that maximally relieves prepulse-sensitive G-protein inhibition in this nerve terminal (Stanley and Mirotznik, 1997). Data were acquired at 5–10 sec intervals. Current recruitment was measured as the maximal difference between the amplitude of the current induced by the test pulse with and without the conditioning pulse, at ∼10 msec after the onset of the test pulse. Data were analyzed using ClampFit 6.0 (pClamp suite) and are presented as mean ± SEM. Student's t test was used to determine statistical significance.
Drug treatments. Drugs were dissolved in external solution and were applied either by addition to the bath or by pulse-triggered pressure ejection (Medical Systems) from a puff pipette (∼5 μm diameter) at a distance of ∼10 μm. The chamber was not perfused because only one calyx was treated in each dish, and the recordings were typically of short duration and with few individual drug applications. The following G-protein receptor agonists were used: substance P, bradykinin, somatostatin, neuropeptide Y (Peninsula), BRL52537 (Tocris), VIP, serotonin, ATP (Sigma, St. Louis, MO), adenosine, and noradrenalin (Research Biochemicals). The drugs were diluted in external solution and were applied by a pneumatic pressure ejection from a closely positioned micropipette or by addition to the bath.
Immunocytochemistry
Antibody characterization. Primary G-protein antibodies (Table 1) were characterized by standard Western blot (10% SDS gel) techniques. After electrophoresis, the proteins were transferred onto nitrocellulose membrane and probed with the indicated G-protein antibodies. Western blots were visualized with enhanced chemiluminescence.
Table 1.
Antibodies used in this study
| Antibody | Source | Immunostain dilution | Western blot dilution |
|---|---|---|---|
| Poly anti-Gαo/i | J. K. Northup | 1:200 | 1:1000 |
| Poly anti-Gαil−3 | Santa Cruz (sc262) | 1:100 | 1:1000 |
| Poly anti-Gαo+i3 | DuPont (NEI-803) | 1:100 | 1:1000 |
| Poly anti-Gαi3 | Calbiochem (371729) | 1:100 | 1:1000 |
| Mono anti-Gαo (Ab-1) | Lab Vision | 1:200 | 1:1000 |
| Mono anti-Gαo (Ab-2) | Lab Vision | 1:200 | 1:1000 |
| Mono anti-Gαi1 | Lab Vision | 1:100 | 1:1000 |
| Mono anti-Gαi2 | Lab Vision | 1:100 | 1:1000 |
| Poly anti-Gαq/11 | DuPont | 1:100 | 1:1000 |
| Poly anti-Gαq/11 | Calbiochem | 1:100 | 1:1000 |
| Poly anti-Gαq/11 | Santa Cruz | 1:100 | 1:1000 |
| Poly anti-Gα12,13 | Gutkind | 1:100 | 1:1000 |
| Poly anti-Gαs | DuPont | 1:100 | 1:1000 |
| Poly anti-Gαz | Calbiochem | 1:100 | 1:500 |
| Mono SV2 | DSHB | 1:0–1:1 | |
| Poly SV2A | StressGen | 1:200 | |
| Mono neurofilament | DSHB | 1:1 | |
| Mono tubulin α | Lab Vision | 1:100 | |
| Mono tubulin β | Lab Vision | 1:100 |
G-protein antibodies used in this study are listed in Table 1. All primary antibodies were first tested for cross-reactivity against chick G-proteins by Western blot against chick brain protein (10 μg/lane; Fig. 1A). We only relied on antibodies that gave bands restricted to the appropriate molecular weight for the Gα subunit (∼40 kDa).
Fig. 1.

Characterization of G-protein antibodies.A, All antibodies gave an appropriate ∼40 kDa band for chick brain protein (10 μg/lane). Lane 1, Monoclonal anti-Go (Ab-1); 2, monoclonal anti-Go (Ab-2); 3, polyclonal anti-Go (J. K. Northup); 4, polyclonal anti-Gi3 (Calbiochem); 5, polyclonal anti-Gi1–3 (Santa Cruz Biotechnology); 6, polyclonal anti-Go+i3 (DuPont); 7, polyclonal anti-Gq/11 (DuPont); 8, polyclonal anti-Gq/11 (Santa Cruz Biotechnology); 9, polyclonal anti-Gq/11 (Calbiochem); 10, polyclonal anti-G12,13 (Gutkind); 11, polyclonal anti-Gs (DuPont); 12, polyclonal anti-Gz (Calbiochem). B, Blot of antibodies against the recombinant Gα subunits: Go, Gs, Gq, G11, Gi1, Gi2, Gi3, and G13, as well as a G-protein mixture that includes Gz.
The specificity of each antibody for its particular G-protein subtype was tested against the following individual recombinant Gα subunits (Calbiochem, La Jolla, CA): Go, Gs, Gq, G11, Gi1, Gi2, and Gi3 (Fig.1B, panels 1–12). Anti-Gαz was tested against all of these plus a mix of G-proteins (Calbiochem) reported to contain Gαi1–3, Gαo, Gαs, Gαz, Gαβs, and Gαγ (Fig.1B, panel 12). The G12,13 antibody was tested against the above single recombinant G-proteins plus recombinant G13 (Calbiochem) (Fig. 1B, panel 10). Recombinant Gα12 was unavailable.
On the whole, the antibodies distinguished far better between G-protein families (Go/Gi, Gs, Gq/11, and G12/13) than between members of the same family (Fig. 1B, panels 1–12). We made considerable effort to differentiate between specific PTX-sensitive members of the Go/Gi family (Go, Gi1, Gi2, and Gi3). Monoclonal antibodies against Gαo (Fig. 1B, lanes 1,2) and Gαi (data not shown) were particularly selective, but of these only the anti-Gαo antibodies were of use for immunocytochemistry. Commercially available polyclonal anti-G-protein antibodies for particular members of the Go/Gi group typically exhibited little specificity and cross-reacted with all members (Fig.1B, panels 4–6). Although we were unable to obtain recombinant Gαz to test the anti-Gαz antibody, our evidence suggests that this antibody was specific because a band of the appropriate molecular weight was observed against the Gαz-containing mixture of G-proteins, whereas none was observed with any of the other recombinant G-proteins alone (Fig. 1B, panel 12).
Immunostaining of calyx nerve terminals. Ganglia from E15 chicks were dissociated and plated on coverslips, as described above. The preparation was fixed in 2% paraformaldehyde for 45 min and then permeabilized in 0.5% polyoxyethylene-20-cetyl ether with 0.5% paraformaldehyde for 10 min. Cells were stained by exposure to primary antibodies overnight. To identify the nerve terminal and the transmitter release zones, all preparations were double-labeled with the appropriate complimentary monoclonal or polyclonal antibody against the synaptic vesicle protein SV2 (Table 1). FITC and LRSC-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were applied at 1:50 dilution for 1 hr. Definitive localization of staining to the transmitter release face or the external, Schwann cell face of the nerve terminal was only possible in the dissociated preparations when the calyces remained attached to the postsynaptic ciliary neuron.
Immunostaining was also performed on ciliary ganglia slices prepared by cryostat section, without previous treatment with dissociation enzymes. Ganglia were fixed in 4% paraformaldehyde in 15% picric acid for 1–1½ hr, infiltrated with 15% sucrose for 1 hr and 30% sucrose overnight, and were then sliced on a cryostat into 12 μm sections. Staining was performed as above and, except where noted, the staining patterns between dissociated cells and cryostat cells were consistent.
Dissociated cells and cryostat slices were visualized under fluorescent illumination on a Zeiss Axiophot with a 63 or 100×, 1.4 NA lens. Images were acquired and analyzed using a Scanalytics Cellscan deconvolution system as described (Juhaszova et al., 2000). This system uses Exhaustive Photon Reassignment to yield confocal-like images of slices through the sample. At least 30 calyces were examined for each stain combination.
RESULTS
Detection of G-protein-dependent inhibition of presynaptic calcium channels
The object of this study was to determine which G-protein families are involved in the modulation of calcium currents in the presynaptic terminal of the chick ciliary ganglion. G-protein-dependent inhibition was monitored as the percentage of current increase after a strong depolarizing prepulse (Fig. 2) and is termed here the “prepulse recruitment.” There was no evidence of calcium channel inhibition in the absence of intracellular GTP (Fig. 2,top left panel).
Fig. 2.

Modulation of chick ciliary ganglion presynaptic calcium current by neurotransmitter. The degree of current recruitment was determined using a double-trial protocol (see Materials and Methods) before, during, and after a puff of transmitter onto the terminal. Each panel shows before (left column) and during (right column) transmitter application in a single calyx nerve terminal. GTP (0.1 mm) was included in the internal solution in all experiments except in panel 1.Panel 1, Adenosine (10 μm) in the absence of internal GTP. Panel 2, Adenosine (10 μm) treatment. Panel 3, Treatment with a mix of substance P (0.5 μm), bradykinin (1 μm), neuropeptide Y (0.1 μm), and BRL52537 (1 μm). Panel 4, Treatment with a mix of noradrenaline (100 μm) and somatostatin (10 μm). −pp, Without prepulse; +pp, with prepulse.
Adenosine modulates presynaptic calcium channels via a PTX-sensitive G-protein pathway
Transmitter-induced inhibition of calcium channels can involve many different G-protein species, the most common of which appear to be Go and Gi. Adenosine, which inhibits N-type calcium channels in many neuronal cells, is believed to act solely via Go/Gi, and this agent inhibits calcium influx at the calyx nerve terminal (Yawo and Chuhma, 1993). Thus, our first objective was to demonstrate that this inhibition involved a characteristic voltage-sensitive inhibition mechanism. We then tested if the adenosine inhibition pathway was via Go/Gi by blocking these G-proteins with the selective toxin PTX.
A step voltage depolarization of the calyx nerve terminal triggered a calcium current with characteristic properties of rapid activation with little inactivation during the current pulse, terminated by a rapid, monotonic tail current on return to the resting potential (Fig. 2,left column). In the absence of drug treatment with or without intracellular GTP (Fig. 2, left column) or in the presence of adenosine but while omitting intracellular GTP (Fig. 2,top right panel), little or no prepulse recruitment was observed. However, when adenosine (10 μm) was puff-applied in the presence of intracellular GTP (Fig. 2,second panel) a brief puff application caused a significant calcium current inhibition and a prepulse recruitment of 16.1 ± 2.7%, (n = 9), consistent with voltage-sensitive inhibition via the G-protein pathway. Inhibition was maintained only during the 5 sec puff application but persisted beyond the puff with longer treatment durations that can be attributed to extracellular accumulation. Repeated applications did not show significant desensitization (Fig.3A).
Fig. 3.

Effect of PTX pretreatment on adenosine-induced calcium current inhibition. A, Time course of calcium channel inhibition with puff application of transmitter (horizontal bar) with or without overnight PTX treatment. The effects of three consecutive trials given ∼1 min apart to each group of calyces are shown. Top series,Adenosine (10 μm) treatment (control,n = 6; PTX, n = 14).Bottom series, Treatment with a cocktail of substance P (0.5 μm), bradykinin (1 μm), neuropeptide Y (0.1 μm), and BRL52537 (1 μm; control, n = 4; PTX,n = 3). B, Bath application of adenosine. Current inhibition was monitored after the addition (t = 0) of adenosine (0.2 mm) to control (open symbols, n = 6) or PTX-treated (filled symbols,n = 6) calyces. In both A andB current inhibition is monitored by the percentage of prepulse recruitment.
We next tested for Go/Giinvolvement in the adenosine pathway by pretreatment with PTX. Block with PTX typically requires hours of exposure necessitating the development of a method for long-term maintenance of the dissociated calyces. After ∼15 hr incubation (in MEM at 20°C, 8% CO2) nerve terminals survived but were less common and were more fragile. Control calyces were incubated in the same conditions in the absence of PTX. In these a prominent inward calcium current was still present, and adenosine-dependent prepulse recruitment was similar to calyces before incubation at 13.6 ± 2.4% (n = 6; p > 0.05). PTX was tested at concentrations ranging from 0.5 to 10 μg/ml but was fully effective at 1 μg/ml. Pretreatment with PTX reduced adenosine-induced prepulse recruitment to undetectable levels at 1.6 ± 2.2% (n = 14; Fig. 3A).
In an attempt to saturate the effect of adenosine, we bath-applied the transmitter at a high concentration (0.2 mm). In the absence of PTX, adenosine treatment for ∼2 min resulted in 23.5 ± 6.1% (n = 8) prepulse recruitment, which was maintained for up to 6 min without significant desensitization (Fig.3B). PTX pretreatment eliminated prepulse recruitment (−2.6 ± 3.2%; n = 6; p < 0.01; Fig. 3B). Thus, adenosine inhibits the calcium current via a Go/i pathway, in agreement with findings from other preparations.
Go/Gi are located in the presynaptic nerve terminal
We used high-resolution immunocytochemistry to determine whether Go/Gi proteins were present in the calyx. The Gi protein family comprises Go1, Go2, Gi1, Gi2, Gi3, Gz, Gt1, Gt2, and Ggust. Go, Gi and Gz have been implicated in the modulation of N-type calcium channels. With the exception of Gz, all are blocked by PTX. The synaptic vesicle marker SV2 was used to positively identify calyx nerve terminals, and the bright patches of staining for this protein mark the clusters of vesicles at the transmitter release sites.
We examined calyces that remained attached to a postsynaptic neuron and also those that had become fully detached. The former provides a view of the target protein locations at an intact synapse, whereas the latter can be used to demonstrate unambiguously that the G-protein is located in the presynaptic terminal and that we are not observing staining in the synaptic space or on the surface of the postsynaptic ciliary neuron. We also examined staining in calyces from whole fixed cryostat-sectioned ciliary ganglion (Fig.4A). With this technique we were able to reproduce the main features of our findings in the absence of enzymatic dissociation.
Fig. 4.
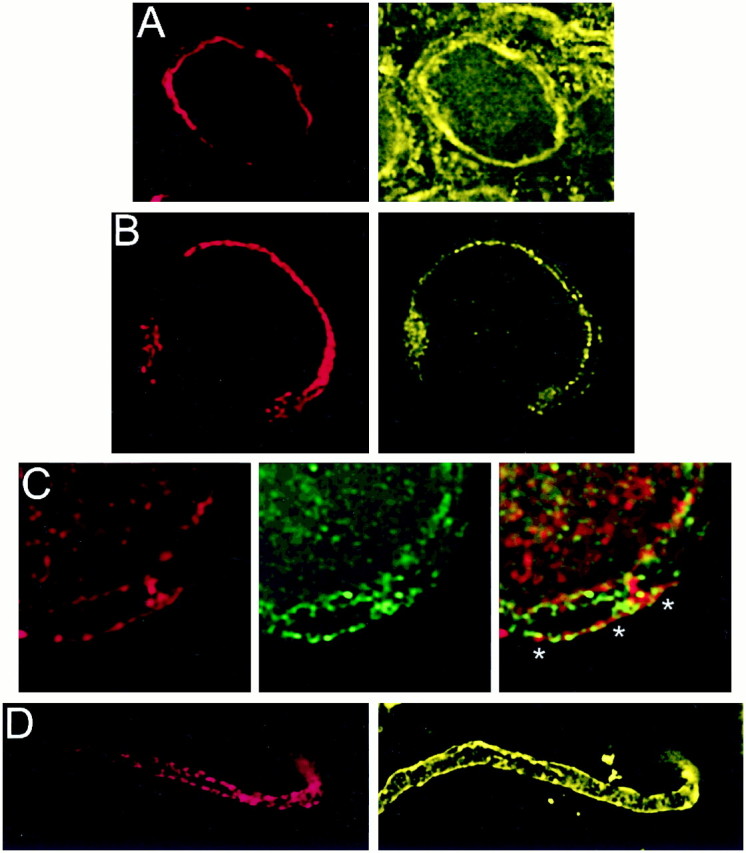
Localization of PTX-sensitive G-proteins. Gαo and Gαi localized to the membrane of the calyx presynaptic terminal. Calyx terminals are identified by SV2 (red) in the left panel of each pair except in C. A, Monoclonal antibodies (Ab-1 + Ab-2, yellow) localized Go to the membrane of calyx terminals in cryostat sections of whole ciliary ganglia. B, In dissociated preparations, monoclonal anti-Gαo (Ab-2) stained the membranes of presynaptic calyces with especially bright, patchy staining at the synaptic interface. C, Gi was localized by a multistaining approach. Go plus Gi were stained in red with polyclonal anti-Gαo (Northup;red; left panel) whereas Go was stained in green with monoclonal anti-Gαo(Ab-1 plus Ab-2; middle panel). Both labeled the calyx membrane. Superimposing the two stains (right panel) localizes Go by the costained regions (yellow). The distinct regions of red staining (e.g., asterisks) identify membrane regions with Gi but not Go. D,Fully isolated nerve terminals exhibited staining of the surface membrane with polyclonal anti-Gαo (Northup), confirming a presynaptic localization of Go/Gi.
Go was localized with two different monoclonal antibodies (Ab-1 and Ab-2). These antibodies gave essentially the same staining pattern in cryostat sections (Fig. 4A) and in dissociated ganglia (Fig. 4B). Staining was almost exclusively on the surface membrane of both the back (Schwann cell) and release-face aspects of the terminal, with particularly bright, patchy staining at the synaptic interface. We could not find an antibody suitable for selective immunostaining of chick Gi. However, a polyclonal antibody, anti-Gαo/i (Table 1), recognized Go and all three Gi α subunits (Fig. 1B, panel 3). If we assume that the monoclonal antibodies against Go localize all of this G-protein, then the distribution of Gi (or, more accurately, the Gi that is not colocalized with Go) can be deduced as the regions stained with anti-Gαo/i but not with either of the monoclonal antibodies. Thus, in Figure 4C, Go/Gi is stained in red (left panel), Go in green (middle panel), and costained regions are yellow in the superimposed images (right panel). While much of the Go/Gi and Go staining is colocalized and indistinguishable from Go alone, some distinct regions of red staining at the calyx membrane were evident (asterisks). These spots were seen near the vesicle clusters and are consistent with the presence of Gi at the transmitter release face of the calyx nerve terminal.
In fully isolated nerve terminals anti-Gαo/istaining was noted primarily on the surface membrane (Fig.4D). Staining partially corresponded with SV2 staining, indicating the presence of Go/Gi at the transmitter release sites, but with a distribution that also extended to nonterminal regions. Other polyclonal antibodies against Go/Gi gave similar results (data not shown).
Calcium channel inhibition by metabotropic neurotransmitter receptors
Whereas the inhibition of N-type calcium channels via the adenosine pathway involves primarily the PTX-sensitive members of the Gi family, other metabotropic receptor types are known to use G-protein from different families (for review, see Hille et al., 1995). As a first attempt to identify inhibitory pathways that involved other G-protein types, we screened a number of different receptor agonists for voltage-dependent inhibition of the calcium channels. Ligands were puff-applied, and we tested for the presence of prepulse-dependent calcium current recruitment.
A mix (combined to speed the screening process) of substance P (0.5 μm), neuropeptide Y (0.1 μm), bradykinin (1 μm), and BRL52537 (1 μm) resulted in a weak, but significant, current recruitment (7.0 ± 2.4%,n = 4, p < 0.05; Figs. 2, third panel, 3A). Current inhibition by this cocktail was, however, also blocked by PTX pretreatment (Fig. 3A) and, hence, was also consistent with the involvement a Go/Gi pathway. Other receptor ligands including noradrenaline (100 μm) and somatostatin (3 μm; Fig. 2, bottom panel), VIP, ATP, or serotonin (both 10 μm; data not shown) did not inhibit the calcium current. Thus, this approach failed to demonstrate the involvement of other G-protein types. In fact, our findings suggest that the PTX-sensitive Go/Gi may mediate all neurotransmitter-induced, voltage-sensitive calcium channel inhibition via G-proteins at this nerve terminal.
Calcium channel inhibition by GTPγS
We used GTPγS to test if there was any evidence for calyx calcium channel inhibition by non-Go/Gi G-proteins. GTPγS is a nonhydrolyzable analog of GTP that irreversibly activates all trimeric G-proteins and has been demonstrated to strongly inhibit the calcium channels at this nerve terminal (Stanley and Mirotznik, 1997). Our strategy was to first compare adenosine and GTPγS-dependent calcium channel inhibition and then to test for persisting GTPγS inhibitory effects after Go/Gi block with PTX.
Infusion of GTPγS (0.1 mm) into the untreated calyx terminals resulted in a robust and maintained prepulse-sensitive calcium channel inhibition (Fig.5A; Stanley and Mirotznik, 1997). The degree of current inhibition was double that observed with bath application of adenosine (GTPγS: 47.1 ± 12.6%,n = 5; adenosine: 23.5 ± 6.1%, n= 8 as above, p < 0.01), suggesting that GTPγS activates a larger pool of inhibitory trimeric G-proteins than adenosine. This conclusion was supported by the finding that, in contrast to the results with adenosine, almost half of the GTPγS-induced current inhibition persisted after PTX pretreatment of the nerve terminals (Fig. 3A; 18.2 ± 4.1%,n = 6).
Fig. 5.

Effect of PTX pretreatment on GTPγS-induced calcium channel inhibition. A, Intracellular infusion of GTPγS. Calcium currents were recorded every 10 sec after membrane rupture in each experiment, and the percentage of recruitment was averaged for 30 sec periods. Control, n = 5 experiments (open symbols); PTX, n = 6 (closed symbols). B, Flash photolysis of caged GTPγS with or without PTX pretreatment. Caged GTPγS was included in the internal solution, and ∼2 min after membrane seal rupture the caged nucleotide was released by a 200 msec flash (arrow). Immediately after, current trials were initiated (control, n = 7; PTX,n = 5). Insert, Representative current traces recorded 5–10 sec after the flash in a control and a PTX-treated terminal. −pp, Without prepulse; +pp, with prepulse. Symbols as inA.
The above results suggest, but do not prove, that the more pronounced calcium channel inhibition observed with GTPγS than adenosine is attributable to non-Go/Gipathways. Although it is known that GTPγS can still activate PTX-inhibited G-protein, published evidence suggests that its action is markedly slowed (see Discussion). We therefore compared the kinetics of GTPγS-dependent inhibition with or without previous PTX treatment using flash photolysis of caged GTPγS (Dolphin et al., 1988). No prepulse-sensitive calcium channel inhibition was detected before flash treatment (Fig. 5B). In control terminals flash photolysis resulted in significant prepulse-dependent calcium current recruitment (23.8 ± 6%, n = 7). Maximum inhibition was detected within 5 sec, the interval between trials. The effect of flash photolysis after PTX was essentially indistinguishable, with a prepulse-dependent recruitment of 25.5 ± 4% (n = 5) and an abrupt onset within the first 5 sec. An anomaly in these results was that the amplitude of inhibition with flash photolysis was significantly less than that observed when (free) GTPγS was introduced directly into the nerve terminal. We do not know the reason for this disparity but it may reflect, in part, the liberation of a lower concentration of intracellular GTPγS from the caged compound or perhaps a component that is inhibited with a much slower time constant (possibly via recruitment of PTX-inhibited Go/Gi) than could not be reliably detected within the limitations of the uncaging technique. Because of the technical difficulty of these experiments (see Materials and Methods) these possibilities were not examined further. The important point was, however, that rapid inhibition could still be detected, even after PTX treatment, in stark contrast to the findings with adenosine.
PTX-insensitive G-proteins localized at the presynaptic nerve terminal
We used immunocytochemistry to test for the presence and distribution in the calyx nerve terminal of PTX-insensitive G-proteins in the Gq, G12, and Gs families and also for Gz, a PTX-insensitive member of the Gi family.
Members of the Gq subfamily were examined with three polyclonal antibodies that cross-reacted with Gαq and Gα11 (Table 1, Fig. 1), and all three gave similar staining patterns at the calyx nerve terminal. The results obtained with the DuPont (Billerica, MA) antibody are presented. Attached calyces had staining throughout the nerve terminal that often colocalized with SV2 (Fig.6A) and was particularly bright at the synaptic interface. Fully isolated calyx nerve terminals also exhibited spotty staining that localized to both the membrane and the cytoplasm (Fig. 6B).
Fig. 6.

Localization of PTX-insensitive G-proteins: Gq/11. Gαq/11 (DuPont antibody) localized to the membrane and cytoplasm of the presynaptic terminal. Calyx terminals were identified by SV2 (red) in theleft panel. Attached (A) and isolated (B) calyces had bright staining throughout the terminal that frequently colocalized with SV2.
The G12 subfamily of G-proteins contains two members, G12 and G13, and our antibody recognized both (Fig. 1). With most attached calyces, the staining of the presynaptic terminal was comparable in intensity to the postsynaptic soma and thus difficult to distinguish. However, some somata were less brightly stained, and the calyx could then be seen to exhibit clear spotty staining throughout the terminal (Fig.7A). However, unlike Go or Gq/11, prominent staining of the synaptic interface was not observed. Fully isolated calyces stained both at the terminal region, as identified by the SV2 staining, and further up the length of the axon (Fig.7B).
Fig. 7.

Localization of PTX-insensitive G-proteins: G12,13. Gα12,13 localized to both the membrane and cytoplasm of the calyx terminal. Calyx terminals are identified by SV2 (red) in the left panel. A, Attached calyces had spotty staining throughout the membrane and the cytoplasm. B, The staining remained in isolated calyces.
The Gs family contains Golfand Gs. We only investigated Gs staining because the former is expressed solely in the olfactory system. An antibody that recognized only Gαs (Fig. 1B, panel 11) produced bright staining of the soma that rivaled that of the calyx (Fig. 8A). The staining was, if anything, reduced along the synaptic interface. This impression was supported by the observation that in fully isolated calyx nerve terminals, punctate Gs staining was negatively correlated with SV2 staining and, thus, with the presynaptic region (Fig. 8B).
Fig. 8.

Localization of PTX-insensitive G-proteins: Gs. Gαs predominantly localized to the calyx cytoplasm. Calyx terminals are identified by SV2 (red) in each panel pair. A, Attached calyces were as stained as the postsynaptic neuron with reduced staining at the interface. B, Isolated calyces showed bright GαS staining along the axon that decreased at the terminal region.
Anti-Gαz gave a novel staining pattern, not seen for any other G-protein. Staining was limited to a bright fibrous band that coursed through the cytoplasm of the calyx axon and into the terminal (Fig. 9A,B). Staining for Gz and SV2 were mutually exclusive, and no clear Gz staining was associated with the surface membrane in the nerve terminal. In the postsynaptic soma, the stained fibrous pattern formed a faint web-like pattern just beneath the membrane that came together to course out of the soma and stream down the axon (data not shown). Because this pattern of staining was strongly suggestive of the cytoskeleton, we colabeled Gz with cytoskeletal proteins. Gz did not colocalize with tubulin α or β (Fig. 9C shows tubulin α) but exhibited near perfect colocalization with the phosphorylated 200 kDa subunit of neurofilament protein (Fig. 9D). This Gz staining pattern was not attributable to cross-reaction of the antibody with chick neurofilament protein because Western blots for the antibody did not show a 200 kDa band (data not shown) or, for that matter, any band other than the G-protein itself (Fig. 1).
Fig. 9.
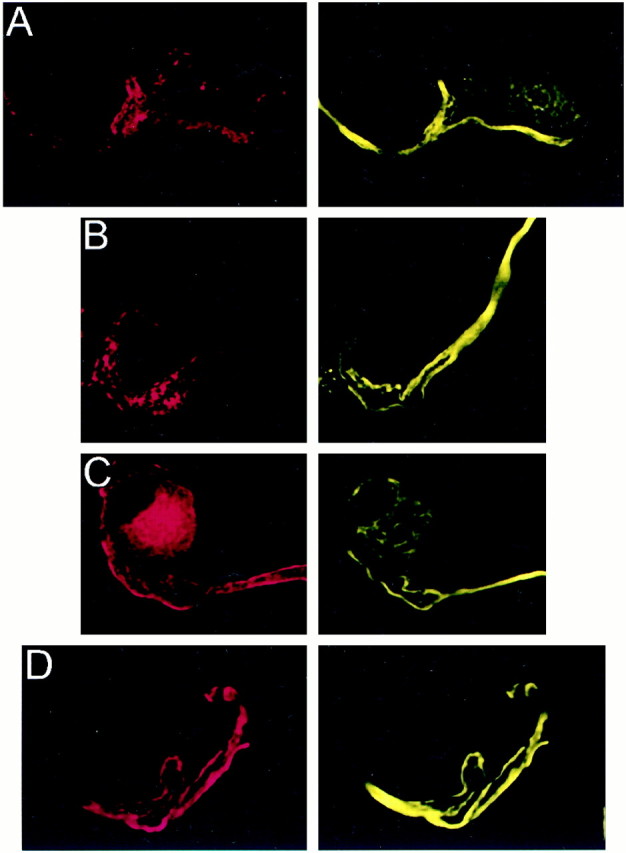
Localization of PTX-insensitive G-proteins: Gz. Attached (A) and isolated (B) calyces had bright fibrous staining for Gz that coursed through the axon and partially into the terminal and was inversely correlated with SV2 (red, left panel). This staining pattern was suggestive of colocalization with the cytoskeleton. C, Tubulin α (red, left panel) stained predominantly near the calyx membrane and, hence, did not colocalize with the predominantly intracellular staining of Gz (yellow; right panel). D, Gz showed almost perfect colocalization (yellow, right panel) with the phosphorylated 200 kDa subunit of neurofilament protein (red, left panel).
DISCUSSION
We have examined the diversity of G-proteins involved in the voltage-dependent modulation of N-type calcium channels at an identified presynaptic nerve terminal. Our main findings are first, numerous G-proteins from several families exist at this nerve terminal but that only certain types are closely associated with the transmitter release face. Second, PTX-sensitive members of the Gi family are involved in the adenosine-dependent, and possibly all, neurotransmitter-induced calcium channel inhibition. Third, we present evidence that PTX-independent G-proteins can also modulate the presynaptic calcium channels, although their role in nerve terminal function remains to be established.
The isolated chick calyx synapse preparation exhibits several methodological advantages for the study of presynaptic calcium channels and their modulation. These include excellent visualization of the whole nerve terminal and the ability to achieve an effective voltage clamp of membrane currents. Furthermore, immunofluorescent staining can be performed on cryostat sections, dissociated calyx synapses free from surrounding cells, and even fully isolated nerve terminals, ensuring unambiguous localization of staining to the nerve terminal.
Adenosine-induced calcium current inhibition was demonstrated to occur via a PTX-sensitive pathway. Go was localized to the membrane of the calyx nerve terminal (Fig. 4A,B), and evidence was presented for similar localization of Gi (Fig. 4C). Thus, we conclude that one or both of these G-protein species mediate the adenosine inhibitory pathway at the calyx presynaptic nerve terminal.
The involvement of PTX-insensitive G-proteins in calcium channel inhibition was initially tested by treating the nerve terminal with a variety of neurotransmitters that have been associated with non-Go/Gi inhibitory pathways. Ligands for noradrenaline, somatostatin, P2Y, muscarinic, VIP, serotonin, substance P, NPY, bradykinin, and κ-opiate G-protein receptors were tested. However, minimal inhibition of the calcium channels was observed, and even this was blocked by PTX. Thus, our results suggest that at this nerve terminal, neurotransmitter-dependent inhibition of presynaptic calcium channels occurs exclusively via the PTX-sensitive G-proteins Go and Gi.
A more general test for G-proteins involved in calcium channel inhibition was based on the nonselective G-protein activator GTPγS. Intracellular treatment with this agent caused a far greater calcium current inhibition (∼40%) than that observed with a saturating dose of adenosine (∼20%). This finding in itself suggests the recruitment of an additional pool of G-proteins. However, it does indicate whether the increased inhibition is attributable to the activation of additional, perhaps reserve, Go/Gi or the recruitment of distinct G-protein types.
To test for channel inhibition via non-Go/GiG-proteins, we tested GTPγS after PTX treatment. Although the degree of GTPγS-dependent calcium current inhibition was reduced, a significant level persisted. However, an alternative interpretation for this PTX-insensitive fraction is that GTPγS overcomes the action of the toxin on Go/Gi and that calcium channel inhibition still involves these G-proteins. Early studies by Gilman (Katada et al., 1984) on purified G-proteins in vitro concluded that PTX treatment markedly impedes the activation of Gi by GTPγS. However, a later report by Huff and Neer (1986) contradicted this finding, reporting that GTPγS can overcome the inhibitory action of PTX. The anomaly in these two reports is readily attributable to differences in the assay conditions: the former study exposed the G-proteins to GTPγS for only 2 min at 30°C and, hence, tested only for short-term effects. However, the study byHuff and Neer (1986) incubated GTPγS with the G-proteins for 30 min at 30°C followed by overnight at 4°C, testing for completion of its action. Although no subsequent study has attempted to reconcile these findings directly, they can be explained if GTPγS can relieve PTX block but its latency or kinetics are greatly slowed. Indeed, such a slowing was demonstrated directly in an earlier study (Jakobs et al., 1984) in which activation of G-protein by GTPγS was assayed by the inhibition of adenylate cyclase. This study found that the onset of G-protein activation was markedly slowed after PTX, from ≤1 min in control cell fractions (the minimum time tested) to ∼5 min after toxin treatment. Thus, all previous reports are consistent with the conclusion that PTX slows activation of G-protein by GTPγS by several minutes. Thus, the inhibition of the presynaptic calcium current by GTPγS after PTX block within 5–10 msec, as noted here, strongly suggests the involvement of a Go/Gi-independent G-protein pathway.
Few studies have examined the spectrum of G-protein types in presynaptic nerve terminals and which of these might be involved in calcium channel modulation. We have examined the diversity and location of G-proteins at the chick calyx nerve terminal by immunocytochemistry and high-resolution imaging. All primary antibodies used in this study were characterized in detail, testing each against recombinant G-proteins to confirm specificity as well as against rat and chick neural tissue to confirm cross-reactivity. These studies indicated that Go, Gi (Fig. 4), Gq or G11 (Fig. 6), and G12 or G13 (Fig. 7) were located in the nerve terminal and at the transmitter release site regions. Gs was located primarily outside the nerve terminal and synaptic cleft and exhibited little staining on the surface membrane (Fig. 8), suggesting that it is less likely to act as a primary modulator of the calcium channels. Gzwas not observed on the surface membrane but instead colocalized with the neurofilaments (Fig. 9). The functional significance of this localization is unclear, but it makes it unlikely that this G-protein modulates the calcium channels at the transmitter release site. Thus, our study identifies Gq, G11, G12, and G13 as possible PTX-insensitive G-proteins capable of presynaptic calcium channel modulation at this terminal.
If, as suggested by our results, Go or Gi are the only G-proteins mediating calcium channel inhibition via neurotransmitter receptors at this nerve terminal, what then is the role of the PTX-insensitive pathway? An obvious possibility is that these G-proteins are linked to an as yet untested membrane receptor pathway that we have not explored. Alternatively, the potent action of GTPγS induces inhibition by G-proteins that are not normally involved in calcium channel modulation. However, if this was the case one might expect a much slower inhibition via the PTX-insensitive G-proteins, whereas a very rapid effect, more consistent with a membrane-delimited pathway, was noted (Fig. 5B). One exciting possibility is that mechanisms other than that initiated by metabotropic receptors can modulate the release site-associated calcium channels. Possibilities might include inhibition associated with the transmitter release steps, such as the loading or unloading of vesicles into the transmitter release site, and interactions between the presynaptic and postsynaptic cells mediated by extracellular matrix proteins.
Footnotes
This work was supported by Canadian Institute for Health Research Award FRN 38091. We are indebted to gifts of antibodies from Drs. J. K. Northup (National Institute on Deafness and Other Communication Disorders) and J. S. Gutkind (National Institute of Dental Research) and to suggestions from Drs. J. K. Northup (National Institutes of Health) and J. Mitchell (Department of Pharmacology, University of Toronto).
R.M. and X.Z. contributed equally to this work.
Correspondence should be addressed to Dr. Elis F. Stanley, Head, Cellular and Molecular Biology Division, Toronto Western Research Institute, MP14–320, 399 Bathurst Street, Toronto, Ontario M5T 2S8, Canada. E-mail: estanley@uhnres.utoronto.ca.
Dr. Stanley's present address: Cellular and Molecular Biology Division, Toronto Western Research Institute Toronto, Canada.
Dr. Mirotznik's present address: National Institutes of Health, National Institute of Neurological Disorders and Stroke, Building 36, Room 5B21, Bethesda, MD 20892-1408.
Dr. Zheng's present address: National Institutes of Health, National Institute of Neurological Disorders and Stroke, Building 10/5N250, 10 Center Drive, MSB-1408, Bethesda, MD 20892-1408.
REFERENCES
- 1.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature (Lond) 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 2.Diversé-Pierluissi M, Dunlap K. Distinct, convergent second messenger pathways modulate neuronal calcium currents. Neuron. 1993;10:753–760. doi: 10.1016/0896-6273(93)90175-q. [DOI] [PubMed] [Google Scholar]
- 3.Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G-proteins. J Physiol (Lond) 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolphin AC, Wooten JF, Scott RH, Trehtham DR. Photoactivation of intracellular guanosine triphosphate analogues reduces the amplitude and slows the kinetics of voltage-activated calcium channel currents in sensory neurons. Pflügers Arch. 1988;411:628–636. doi: 10.1007/BF00580858. [DOI] [PubMed] [Google Scholar]
- 5.Elmslie K. LHRH and GTP-gamma-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- 6.Filippov AK, Webb TE, Barnard EA, Brown DA. P2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents. J Neurosci. 1998;18:5170–5179. doi: 10.1523/JNEUROSCI.18-14-05170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grassi F, Lux H-D. Voltage-dependent GABA-induced currents in chick sensory neurons. Neurosci Lett. 1989;105:113–119. doi: 10.1016/0304-3940(89)90021-9. [DOI] [PubMed] [Google Scholar]
- 8.Haydon PG, Henderson E, Stanley EF. Localization of individual calcium channels at the release face of a presynaptic nerve terminal. Neuron. 1994;13:1275–1280. doi: 10.1016/0896-6273(94)90414-6. [DOI] [PubMed] [Google Scholar]
- 9.Hille B. Modulation of ion channels by G-protein coupled receptors. Trends Neurosci. 1994;17:531–535. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 10.Hille B, Beech DJ, Bernheim L, Mathie A, Shapiro MS, Wollmuth LP. Multiple G-protein-coupled pathways inhibit N-type Ca channels of neurons. Life Sci. 1995;56:989–992. doi: 10.1016/0024-3205(95)00038-8. [DOI] [PubMed] [Google Scholar]
- 11.Huff RM, Neer EJ. Subunit interactions of native and ADP-ribosylated α39 and α41, two guanine nucleotide-binding proteins from bovine cerebral cortex. J Biol Chem. 1986;261:1105–1110. [PubMed] [Google Scholar]
- 12.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G-protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 13.Jakobs KH, Aktories K, Schultz G. Mechanism of pertussis toxin action on the adenylate cyclase system. Eur J Biochem. 1984;140:177–181. doi: 10.1111/j.1432-1033.1984.tb08083.x. [DOI] [PubMed] [Google Scholar]
- 14.Jeong SW, Ikeda SR. G-protein alpha subunit G alpha z couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron. 1998;21:1201–1212. doi: 10.1016/s0896-6273(00)80636-4. [DOI] [PubMed] [Google Scholar]
- 15.Juhaszova M, Church PJ, Blaustein MP, Stanley EF. Location of calcium transporters at presynaptic terminals. Eur J Neurosci. 2000;12:839–846. doi: 10.1046/j.1460-9568.2000.00974.x. [DOI] [PubMed] [Google Scholar]
- 16.Katada T, Northup JK, Bokoch GM, Gilman AG. The inhibitory guanine nucleotide-binding regulatory component of adenylate cyclase. Subunit dissociation and guanine nucleotide-dependent hormonal inhibition. J Biol Chem. 1984;259:3578–3585. [PubMed] [Google Scholar]
- 17.Morikawa H, Fukuda K, Mima H, Shoda T, Kato S, Mori K. Nociceptin receptor-mediated Ca2+ channel inhibition and its desensitization in NG108–15 cells. Eur J Pharmacol. 1998;351:247–252. doi: 10.1016/s0014-2999(98)00306-9. [DOI] [PubMed] [Google Scholar]
- 18.Park D, Dunlap K. Dynamic regulation of calcium influx by G-proteins, action potential waveform, and neuronal firing frequency. J Neurosci. 1998;18:6757–6766. doi: 10.1523/JNEUROSCI.18-17-06757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanley EF. Single calcium channels on a cholinegic presynaptic nerve terminal. Neuron. 1991;7:587–591. doi: 10.1016/0896-6273(91)90371-6. [DOI] [PubMed] [Google Scholar]
- 20.Stanley EF. Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron. 1993;11:1007–1011. doi: 10.1016/0896-6273(93)90214-c. [DOI] [PubMed] [Google Scholar]
- 21.Stanley EF, Drachman DB. Denervation and the time course of resting membrane potential in skeletal muscle in vivo. Exp Neurol. 1980;69:253–259. doi: 10.1016/0014-4886(80)90209-5. [DOI] [PubMed] [Google Scholar]
- 22.Stanley EF, Goping G. Characterization of a calcium current in a vertebrate cholinergic presynaptic nerve terminal. J Neurosci. 1991;11:985–993. doi: 10.1523/JNEUROSCI.11-04-00985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein modulation of presynaptic calcium channels. Nature (Lond) 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- 24.Toth PT, Shekter LR, Ma GH, Philipson LH, Miller RJ. Selective G-protein regulation of neuronal calcium channels. J Neurosci. 1996;16:4617–4624. doi: 10.1523/JNEUROSCI.16-15-04617.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilk-Blaszczak MA, Singer WD, Gutowski S, Sternweis PC, Belardetti F. The G-protein G13 mediates inhibition of voltage-dependent calcium current by bradykinin. Neuron. 1994;13:1215–1224. doi: 10.1016/0896-6273(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 26.Yawo H, Chuhma N. Preferential inhibition of omega-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature (Lond) 1993;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]
- 27.Yawo H, Momiyama A. Re-evaluation of calcium currents in pre- and postsynaptic neurones of the chick ciliary ganglion. J Physiol (Lond) 1993;460:153–172. doi: 10.1113/jphysiol.1993.sp019464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, Ikeda SR. VIP inhibits N-type Ca2+ channels of sympathetic neurons via a pertussis toxin-insensitive but cholera toxin-sensitive pathway. Neuron. 1994;13:657–669. doi: 10.1016/0896-6273(94)90033-7. [DOI] [PubMed] [Google Scholar]