

Abstract
Reactive microglia have been suggested to play a role in the Alzheimer's disease (AD) process, and previous studies have shown that expression of CD45, a membrane-bound protein-tyrosine phosphatase (PTP), is elevated in microglia in AD brain compared with controls. To investigate the possible role of CD45 in microglial responsiveness to β-amyloid (Aβ) peptides, we first co-treated primary cultured microglia with a tyrosine phosphatase inhibitor [potassium bisperoxo (1,10-phenanthroline) oxovanadate (phen), 5 μm] and freshly solubilized Aβ peptides (1000 nm). Data show synergistic induction of microglial activation as evidenced by tumor necrosis factor α (TNF-α) production and nitric oxide (NO) release, both of which we show to be dependent on activation of p44/42 mitogen-activated protein kinase (MAPK). Furthermore, co-treatment with phen and Aβ peptides results in microglia-induced neuronal cell injury. Stimulation of microglial CD45 by anti-CD45 antibody markedly inhibits these effects via inhibition of p44/42 MAPK, suggesting that CD45 is a negative regulator of microglial activation. Accordingly, primary cultured microglia from CD45-deficient mice demonstrate hyper-responsiveness to Aβ, as evidenced by TNF-α release, NO production, and neuronal injury after stimulation with Aβ peptides. As a validation of these findings in vivo, brains from a transgenic mouse model of AD [transgenic Swedish APP-overexpressing (Tg APPsw) mice] deficient for CD45 demonstrate markedly increased production of TNF-α compared with Tg APPsw mice. Taken together, these results suggest that therapeutic agents that stimulate the CD45 PTP signaling pathway may be effective in suppressing microglial activation associated with AD.
Keywords: Alzheimer's disease, β-amyloid, microlgia, neurons, mitogen-activated protein kinase, CD45, protein-tyrosine phosphatase, tyrosine phospatase inhibitor, TNF-α, nitric oxide
It has been suggested that activated microglia play a key role in the inflammatory processes of neurodegenerative diseases such as Alzheimer's disease (AD), because reactive microglia secrete cytokines, including tumor necrosis factor α (TNF-α) and interleukin-1β, which promote neurodegeneration (Meda et al., 1995; Rogers et al., 1996; Barger and Harmon, 1997). However, current anti-inflammatory therapeutics directed against AD, such as nonsteroidal anti-inflammatory drugs (NSAIDs), only partially suppress microglial activation (Mackenzie and Munoz, 1998) and, therefore, may not provide the greatest therapeutic benefits for AD. This suggestion is supported by clinical evidence, in which elderly persons using NSAIDs demonstrate only an ∼20% reduction in risk for AD (Beard et al., 1998), and AD patients using NSAIDs have only partial amelioration of disease symptoms (Rich et al., 1995). Thus, a more viable therapeutic strategy may be combination of NSAIDs with specific inhibitors of microglial activation.
Most of our knowledge concerning the molecular mediators of microglial activation comes from studies involving peripheral lymphocytes. For example, the CD40–CD40L signaling pathway is involved in both T-cell and microglial cell activation (Yang and Wilson 1996; Maxwell et al., 1999; Tan et al., 1999a), yet although blockade of this pathway has proved an efficient means of opposing T-cell activation (Grewal et al., 1996; Stuber et al., 1996), interruption of this pathway is largely unexplored as a means of opposing microglial activation. We have recently shown that ligation of microglial CD40 synergistically enhances activation of these cells by a low dose of freshly solubilized β-amyloid (Aβ; Tan et al., 1999b), indicating that the CD40–CD40L interaction is critically involved in microglial activation induced by Aβ. In searching for novel cell surface molecules that may play a role in opposing microglial activation, we focused on CD45, a functional transmembrane protein-tyrosine phosphatase (PTP), which, when cross-linked, has been shown to play a critical role in negative regulation of T and B lymphocyte activation (Justement, 1996). CD45 is particularly interesting, because microglia express it in the frontal cortex and hippocampus of normal aging individuals, and this expression level is markedly increased in these brain regions in AD cases (Masliah et al., 1991; Licastro et al., 1998). Furthermore, in an animal model of neurodegeneration, upregulation of phosphotyrosine signal associated with activated microglia was found in and around the degenerating brain region (Karp et al., 1994). These data led us to investigate the possible involvement of CD45 PTP signaling as a putative regulator of microglial activation.
MATERIALS AND METHODS
Materials. Monoclonal antibodies (purified rat anti-mouse CD45 and purified rat IgG2b control antibodies and FITC-conjugated rat anti-mouse CD45 and FITC-conjugated rat IgG2b control antibodies) were purchased from PharMingen (San Diego, CA). To test whether CD45 cross-linkage could result in stimulation of CD45 PTP activity, we measured free inorganic phosphate (Pi) in microglial cell lysates treated in the presence or absence of anti-CD45 antibody and found significantly higher levels of Pi in microglial cell lysates treated with CD45 antibody compared with untreated cells (data not shown). Antibodies for phospho-p44/42 mitogen-activated protein kinase (MAPK) (Thr-202/Tyr-204) and total p44/42 MAPK were obtained from New England Biolabs (Beverly, MA). Aβ1-40 and Aβ1-42peptides and control peptide (Aβ40-1) were obtained from QCB (Hopkinton, MA) and were freshly solubilized in distilled H2O immediately before use. To determine the oligomeric state of Aβ in our assays, Aβ was immunoprecipitated from cell supernatants after incubation with microglia and/or neurons, and Western blot analysis was performed at time points of 12, 24, and 48 hr. Data revealed that both Aβ1-40 and Aβ1-42, irrespective of the time points assayed, existed as a ladder of SDS-stable oligomers, with a predominant species of ∼32 kDa. Human CD45 recombinant protein (specific activity, 20,000 U/mg of protein) and PD98059 were obtained from Calbiochem (La Jolla, CA), as well as the phosphatase inhibitors including potassium bisperoxo (1,10-phenanthroline) oxovanadate (phen), sodium orthovanadate, and okadaic acid. Each of these was dissolved in DMSO before adding to complete cell culture medium, and DMSO alone was used as a solvent control, which did not differ from the untreated controls presented. Bacterial lipopolysaccharide (LPS) was purchased from Sigma (St. Louis, MO) and dissolved in complete cell culture medium. Anti-mouse HRP-conjugated IgG secondary antibody and Western blotting luminol reagent were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Immun-Blot polyvinylidene difluoride (PVDF) membranes were purchased from Bio-Rad (Hercules, CA). Anti-mouse TNF-α polyclonal antibody was obtained from R & D systems (Minneapolis, MN).
Murine primary cell culture. Breeding pairs of BALB/c and CD45-deficient (C57BL/6OlaHsd-Ptprc) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in the animal facility at the University of South Florida Health Science Center. Transgenic Swedish APP-overexpressing (Tg APPsw) mice are the 2576 line back-crossed to C57B6/SJL as previously described (Hsiao et al., 1995, 1996). Murine primary culture microglia were isolated from mouse cerebral cortices and were grown in RPMI 1640 medium supplemented with 5% fetal calf serum, 2 mm glutamine, 100 U/ml penicillin, 0.1 μg/ml streptomycin, and 0.05 mm2-mercaptoethanol according to previously described methods (Chao et al., 1992). Briefly, cerebral cortices from newborn mice (1–2 d old) were isolated under sterile conditions and were kept at 4°C before mechanical dissociation. Cells were plated in 75 cm2 flasks, and complete medium was added. Primary cultures were kept for 14 d so that only glial cells remained, and microglia were isolated by shaking flasks at 200 rpm in a Lab-Line incubator–shaker. More than 98% of these glial cells stained positive for Mac-1 (CD11b/CD18; Boehringer Mannheim, Indianapolis, IN). Mouse primary culture neuronal cells were prepared as previously described (Chao et al., 1992). Briefly, cerebral cortices were isolated from BALB/c mouse embryos, between 15 and 17 d in utero, and cortices were mechanically dissociated in trypsin (0.25%) after incubation for 15 min at 37°C. Cells were collected after centrifugation at 1200 rpm, resuspended in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal calf serum, 10% horse serum, uridine (33.6 mg/ml; Sigma) and fluorodeoxyuridine (13.6 mg/ml; Sigma), and plated in 24-well tissue culture plates at 2.5 × 105 cells per well after collagen coating the plates. Where microglia were isolated from CD45-deficient mice, to verify CD45 deficiency status, fluorescence-activated cell sorter analysis was performed as previously described (Tan et al., 1999a), and CD45 was undetectable in these cells (data not shown).
TNF-α ELISA and nitric oxide release assay. Primary cultured microglial cells were plated in 24-well tissue-culture plates (Costar, Cambridge, MA) at 5 × 104cells per well and stimulated for 12 hr with phen (5 μm), Aβ peptides (1000 nm), phen and Aβ peptides in the presence or absence of anti-CD45 antibody (1:200) or PD98059 (5 μm) pretreatment for 1 hr, or appropriate controls. Cell-free supernatants were collected and assayed by a TNF-α ELISA kit (Genzyme, Cambridge, MA) or a nitric oxide (NO) assay kit (Calbiochem) in strict accordance with the manufacturer's instructions. The Bio-Rad (Hercules, CA) protein assay was performed to measure total cellular protein from each of the cell groups under consideration just before quantification of cytokine release by ELISA or NO secretion by NO assay.
Lactate dehydrogenase release assay in neuronal and microglial co-cultures. After 5 d in vitro, neuronal cells were passed in preparation for subsequent experiments. More than 96% of these cells stained positive for neurofilament L (using rabbit anti-human neurofilament L antibody, 1:300; Serotec Ltd., Kidlington, Oxon, UK; data not shown), a marker of differentiated neuronal cells. Neuronal cells were seeded in 24-well tissue culture plates at 1 × 105 cells per well for 48 hr and used as target cells for lactate dehydrogenase (LDH) release assay. In this experimental paradigm, neuronal cells or neuronal–microglial co-cultures (microglia, 5 × 104 cells per well, a 2:1 ratio of neurons to microglia) were treated with phen (5 μm), Aβ1-40 (1000 nm), Aβ1-42 (1000 nm), control peptide (1000 nm), anti-CD45 antibody (1:200), phen and Aβ peptides, anti-CD45, phen, and Aβ peptides, or appropriate controls. An LDH release assay (Promega, Madison, WI) was performed as described (Tan et al., 1999a) after either 36 or 48 hr of treatment in neuronal cultures, microglial cultures or neuronal–microglial co-cultures. Total LDH release represents maximal lysis of target cells with 5% Triton X-100.
Immunocytochemistry in neuronal and microglial co-cultures.For morphological examination and immunocytochemistry, primary cultured neuronal cells were isolated as described above, passed onto glass coverslips in six-well tissue culture plates at 2 × 105 cells per well, and then co-cultured with various groups of microglia at 1 × 105 cells per well (yielding a 2:1 ratio of neurons to microglia). Co-cultures then went untreated (control) or were treated with phen (5 μm), Aβ1-42 (1000 nm), or both for 48 hr. After this treatment period, morphological examination and immunocytochemistry were performed. Double immunocytochemical staining was performed with a combination of an indirect method (using an HRP-conjugated DAB chromogen system) and the Dako (Carpinteria, CA) EnVision system (with an alkaline phosphatase-conjugated fast red chromogen system), using anti-mouse CD11b antibody (clone M1/70, 1:50 dilution; Chemicon, Temecula, CA) as a microglia marker and anti-mouse neuorfilament L antibody (neurofilament 68 clone NR4, 1:300 dilution; Sigma) as a neuronal marker. Morphological changes in co-cultured primary neurons and microglia were recorded by bright-field microscopic examination.
Western immunoblotting. Murine primary culture micrgolia were plated in six-well tissue culture plates at a density of 8 × 105 cells per well. These cells were incubated for 30 min with or without phen (5 μm) and Aβ peptides (1000 nm) in the presence or absence of anti-CD45 or control antibodies (1:200 dilution for each) or PD98059 (5 μm) pretreatment for 1 hr, or appropriate controls. Immediately after culturing, microglia were washed in ice-cold PBS three times, scraped into ice-cold PBS, and lysed in an ice-cold lysis buffer containing 20 mm Tris, pH 7.5, 150 mmNaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mmβ-glycerolphosphate, 1 mmNa3VO4, 1 μg/ml leupeptin, and 1 mm PMSF. After incubation for 30 min on ice, samples were centrifuged at high speed for 15 min, and supernatants were collected. Total protein content was estimated using the Bio-Rad protein assay. An aliquot corresponding to 50 μg of total protein of each sample was separated by SDS-PAGE and transferred electrophoretically to Immun-Blot PVDF membranes. Nonspecific antibody binding was blocked with 5% nonfat dry milk for 1 hr at room temperature in Tris-buffered saline (20 mm Tris and 500 mm NaCl, pH 7.5). Membranes where first hybridized with a phospho-specific p44/42 MAPK antibody, stripped with β-mercaptoethanol stripping solution (62.5 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol), and then reprobed with an antibody that recognizes total p44/42 MAPK. Alternatively, membranes with identical samples were probed either with a phospho-specific p44/42 MAPK antibody or with an antibody that recognizes total p44/42 MAPK. Immunoblotting was performed with a primary antibody followed by an anti-mouse HRP-conjugated IgG secondary antibody as a tracer. The luminol reagent was used to develop the blots. Densitometric analysis was performed for all blots using the Flour-S MultiImager with Quantity One software (Bio-Rad).
For TNF-α Western blot, brains from 6-month-old transgenic mice were isolated under sterile conditions on ice and placed in ice-cold lysis buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mmβ-glycerolphosphate, 1 mmNa3VO4, 1 μg/ml leupeptin, and 1 mm PMSF. Brains were then sonicated on ice for ∼3 min, let stand for 15 min at 4°C, and centrifuged at 15,000 rpm for 15 min. Supernatants were then collected for protein assay and Western immunoblotting as described above.
Immune complex kinase assay. Primary culture microgial cells were seeded in six-well tissue culture plates at 8 × 105 per well. Thirty minutes after co-treatment with phen and Aβ peptides in the presence or absence of anti-CD45 antibody or appropriate controls, microglial cells were lysed in ice-cold lysis buffer (as described above). Total cellular protein was quantified with the Bio-Rad protein assay, and an aliquot of 100 μg of protein for each treatment condition was separated by SDS-PAGE. p44/42 MAPK activity was determined using the p44/42 MAP Kinase assay kit (New England Biolabs) in strict accordance with the manufacturer's instructions. The phosphorylated form of the Elk1 p44/42 MAPK fusion protein was visualized by Western immunoblotting (as described above) using a specific antibody for phosphorylated Elk1 supplied with the kit.
Statistical analysis. Data were analyzed using ANOVA followed by post hoc comparisons of means by Bonferroni's or Dunnett's T3 method, for which Levene's test for homogeneity of variances was used to determine the appropriate method of post hoc comparison. In instances of single-mean comparisons,t test for independent samples was used to assess significance. α levels were set at 0.05 for each analysis. All analyses were performed using SPSS for Windows release 9.0.
RESULTS
Co-treatment with phen and Aβ peptides results in synergistic microglial acitvation
It has been shown that a tyrosine phosphorylation cascade plays an important role in Aβ-induced microglial activation (McDonald et al., 1998; Combs et al., 1999). To test whether promotion of tyrosine phosphorylation could affect Aβ-induced microglial activation, we co-incubated primary cultured microglial cells with phen, a specific tyrosine phosphatase inhibitor, and Aβ peptides for 12 hr. Microglial activation was measured by TNF-α and NO production and neuronal cell injury in co-culture experiments. Data showed that phen synergistically enhanced Aβ-stimulated microglial activation (Fig.1). To further confirm that phen and Aβ activated microglia through inhibiting the PTP signaling pathway, we co-cultured microglia with Aβ and either sodium orthovanadate (50 μm, another PTP inhibitor) or okadaic acid (50 nm, a protein phosphatase 2A inhibitor) and measured NO and TNF-α release. Although sodium orthovanadate treatment in conjunction with Aβ produced results similar to those of phen and Aβ peptide co-treatment (data not shown), NO and TNF-α were not detectable in the media of okadaic acid- and Aβ-co-treated microglia (data not shown). This result led us to focus on stimulating microglial PTP activity to oppose Aβ-induced activation of these cells.
Fig. 1.
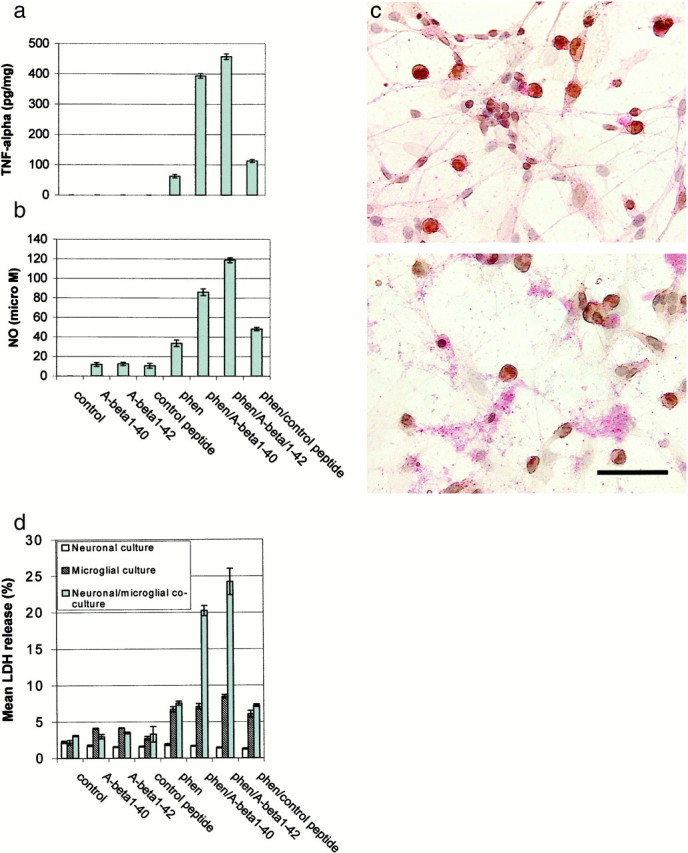
Co-treatment with phen and Aβ peptides synergistically affects microglial activation. Microglial cells were treated as indicated for 12 hr or co-cultured with primary neuronal cells (microglia/neurons, 1:2) under the indicated treatment conditions for 48 hr. Control peptide is Aβ40-1. Microgial activation was measured by TNF-α production (mean ± 1 SEM, picograms per milligram of total protein) in cultured media by TNF-α ELISA (a), NO release (mean ± 1 SEM, micromolar concentration per milligram of total protein) in cultured media by NO assay (b), neuronal cell injury by immunocytochemistry and microscopic examination (c), and neuronal cell injury by LDH assay [mean LDH (percent) release ± 1 SEM] (d). Data shown in a and b are representative of five independent experiments, and data in c andd are representative of two independent experiments. Forc, co-cultures of microglia and neurons are shown before (top panel) and after (bottom panel) co-treatment with phen and Aβ1-42. CD11b-positive cells (brown) are microglia, whereas neurofilament L-staining cells (red) are neurons. Scale bar, 50 μm (calculated for each panel). Neuronal, but not microglial, degeneration is apparent only after co-treatment and is not detectable after treatment with Aβ1-42 or phen alone. Fora, b, and the neuronal–microglial co-culture conditions in d, ANOVA revealed significant main effects of Aβ1-40 (p < 0.001), Aβ1-42 (p < 0.001), and phen (p < 0.001). There were also significant interactions between Aβ1-40 or Aβ1-42 and phen (p < 0.001). One-way ANOVA revealed significant between-group differences (p < 0.001), and post hoctesting revealed significant differences between phen/Aβ1-40 or phen/Aβ1-42 when compared with phen/control peptide (p < 0.001).
CD45 cross-linking significantly inhibits microglial activation induced by phen and Aβ peptides
It has been reported that CD45, a protein-tyrosine phosphatase that is constitutively expressed on microglia (Karp et al., 1994), is markedly increased on microglia from AD frontal cortices (Masliah et al., 1991; Licastro et al., 1998). To examine the putative role of CD45 in microglial activation, we treated primary cultured microglial cells with monoclonal anti-CD45 antibody before stimulation with phen and Aβ peptides. Microglial activation, as evidenced by TNF-α and NO release after co-treatment of microglia with phen and Aβ peptides, was significantly inhibited by cross-linking CD45 (Fig.2a,b). Hyperstimulation of microglia commonly results in bystander cell injury, and we went on to evaluate whether cross-linking of CD45 might protect neuronal cells against injury from activated microglia (resulting from phen and Aβ peptide co-treatment). When activated microglia were co-cultured with primary cultured neuronal cells in the presence of anti-CD45 antibody, we observed that neuronal cells were significantly protected against injury induced by reactive microglia (Fig. 2c). In addition, to determine whether directly increasing CD45 activity could block microglial activation resulting from co-treatment with phen and Aβ, we added CD45 recombinant protein (20 U/ml) to activated microglia and measured NO and TNF-α release. Results showed that release of NO and TNF-α were markedly decreased after addition of CD45 recombinant protein compared with the appropriate control (denatured CD45 protein; data not shown), further substantiating the role of CD45 in negative regulation of microglial activation.
Fig. 2.
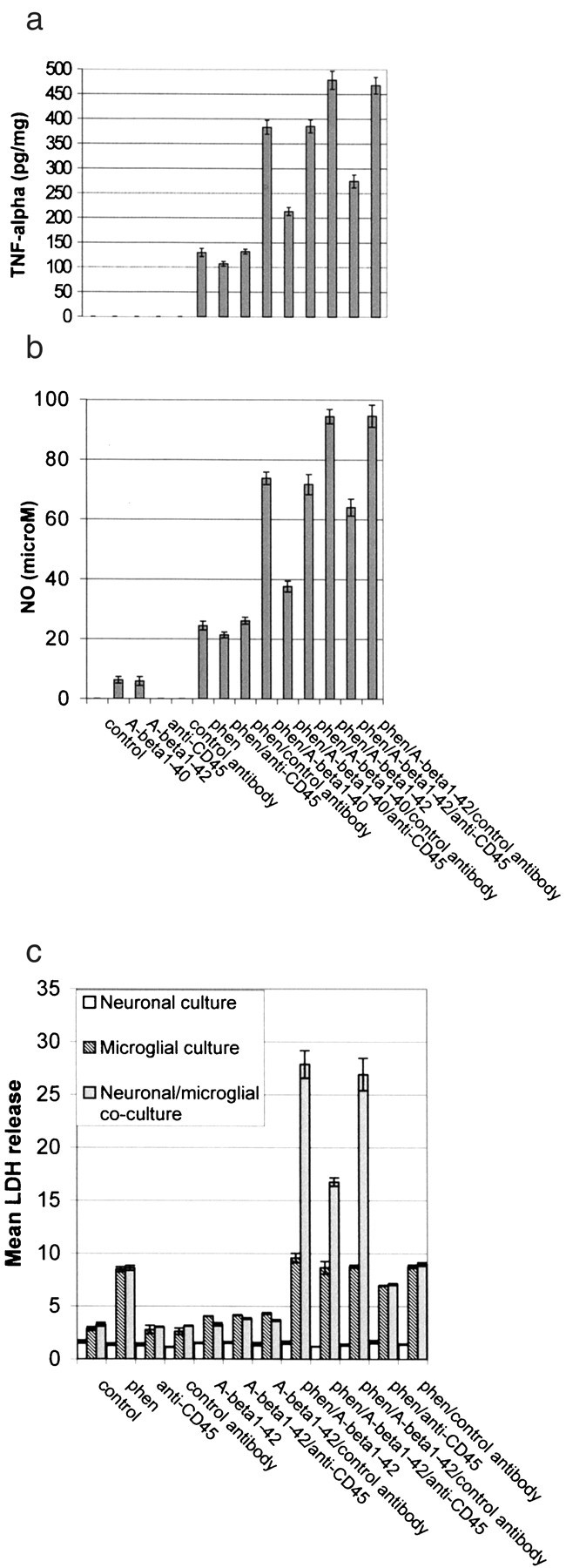
CD45 cross-linking markedly inhibits phen and Aβ peptide-induced microglial activation. Microglial cells were treated as indicated for 12 hr or co-cultured with primary cultured neuronal cells (microglia/neurons, 1:2) under the same treatment conditions for 48 hr. Control antibody is rat IgG2b. Microglial activation was determined by TNF-α production (mean ± 1 SEM, picograms per milligram of total protein) in cultured media (a), NO release (mean ± 1 SEM, micromolar concentration per milligram of total protein) in cultured media (b), and neuronal cell injury [mean LDH (percent) release ± 1 SEM] in co-culture experiments (c). Data shown in a andb are representative of five independent experiments, and data in c are representative of two independent experiments. For a and b, one-way ANOVA revealed significant between-group differences (p < 0.001), and post hoctesting revealed significant differences between phen/Aβ1–40/anti-CD45 and phen/Aβ1–40/control antibody (p < 0.01) or phen/Aβ1–42/anti CD45 and phen/Aβ1–42/control antibody (p < 0.02). For the neuronal–microglial co-culture conditions in c, one-way ANOVA revealed significant between-group differences (p < 0.001), and post hoc testing revealed a significant difference between phen/Aβ1–42/anti-CD45 and phen/Aβ1–42/control antibody (p < 0.02).
Cross-linking of CD45 suppresses microglial activation through a p44/42 MAPK-dependent pathway
Previous studies have shown that activation of mitogen-activated protein kinase kinase (MEK1/2) and downstream p44/42 MAPK is involved in TNF-α production in macrophages, monocytes, and microglia after activation of these cells with a variety of stimuli, including LPS and CD40 ligand (Hambleton et al., 1995; Suttles et al., 1999; Tan et al., 1999c). These data led us to ask whether the observed effect of CD45 cross-linking on opposing microglial activation might be mediated via activation of the MAPK module. Thus, we analyzed p44/42 MAPK phosphorylation status and activity in microglial cell lysates after co-treatment with phen and Aβ peptides or appropriate control conditions. Results showed that p44/42 MAPK phosphorylation and activity were both synergistically induced within 30 min after co-treatment with phen and Aβ1-40 or Aβ1-42 peptides (Fig.3a,b). Furthermore, we observed that treatment of microglia with PD98059, a selective inhibitor of MEK1/2, results in significant reduction of phen- and Aβ-mediated phosphorylation and activity of p44/42 MAPK (Fig.3c,d). To determine whether activation of p44/42 MAPK was responsible for TNF-α and NO production after co-treatment of microglia with phen and Aβ peptides, we treated microglia with PD98059 before stimulation with phen and Aβ peptides. Production of TNF-α and NO were markedly decreased compared with appropriate controls within 12 hr after treatment with PD98059 and phen and Aβ peptides (Fig. 3e,f). These data suggest that activation of p44/42 MAPK is crucial for microglial TNF-α and NO production after co-treatment of microglia with phen and Aβ peptides.
Fig. 3.

Co-treatment of microglia with phen and Aβ peptides activates p44/42 MAPK, resulting in microglial activation. Microglial treatment conditions are indicated and are further described in Materials and Methods. Control peptide is Aβ40-1. Cell lysates were analyzed by Western immunoblotting using specific antibodies that recognize phosphorylated or total p44/42 MAPK.a,b, Phosphorylation and activity of p44/42 MAPK after co-treatment with phen and Aβ peptides.c,d, Inhibition of this effect by PD98059 (a specific MEK1/2 inhibitor). Histograms below the immunoblots represent the mean band density ratio ± 1 SEM (phospho-p44/42 MAPK/total p44/42 MAPK) and band density in optical density units (phospho-Elk1), respectively (n = 3 for each condition presented). Microglial activation is evidenced by mean TNF-α release ± 1 SEM (picograms per milligram of total protein) (e) and mean NO release ± 1 SEM (micromolar concentration per milligrams of total protein) (f) in cultured media by ELISA or by NO release assay, respectively (n = 3 for each condition presented). Fora and b, ANOVA revealed significant main effects of phen, Aβ1-40, and Aβ1-42(p < 0.001), and there were significant interactive terms between phen and either Aβ1-40 or Aβ1-42 (p < 0.001). One-way ANOVA revealed significant between-group differences (p < 0.001), and post hoctesting showed significant differences between Aβ1-40 and phen/Aβ1-40 (p < 0.001) and between Aβ1-42 and phen/Aβ1-42(p < 0.001). Forc–f, one-way ANOVA revealed significant between-group differences (p < 0.001), and post hoc testing showed significant differences between phen/Aβ1-40 and phen/Aβ1-40/PD98059 (p < 0.01) and between phen/Aβ1-42 and phen/Aβ1-42/PD98059 (p < 0.01).
Having shown that cross-linking of CD45 opposed microglial activation, we wished to determine whether reduced p44/42 MAPK activity could be responsible for this effect. To investigate this possibility, microglial cells were co-incubated with anti-CD45 antibody, phen, and Aβ peptides. Cell lysates were then analyzed for phosphorylated forms of p44/42 MAPK by Western immunoblotting. Results showed that cross-linking of CD45 significantly inhibited phosphorylation of p44/42 MAPK induced by phen and Aβ peptide co-treatment compared with controls (Fig. 4a). To determine whether this effect could result in decreased MAPK activity, an immune complex kinase assay was performed. Results showed that cross-linking of CD45 markedly reduced p44/42 MAPK activity in phen- and Aβ peptide-co-treated cells (Fig. 4b), demonstrating the functionality of CD45 cross-linking on p44/42 MAPK activity.
Fig. 4.
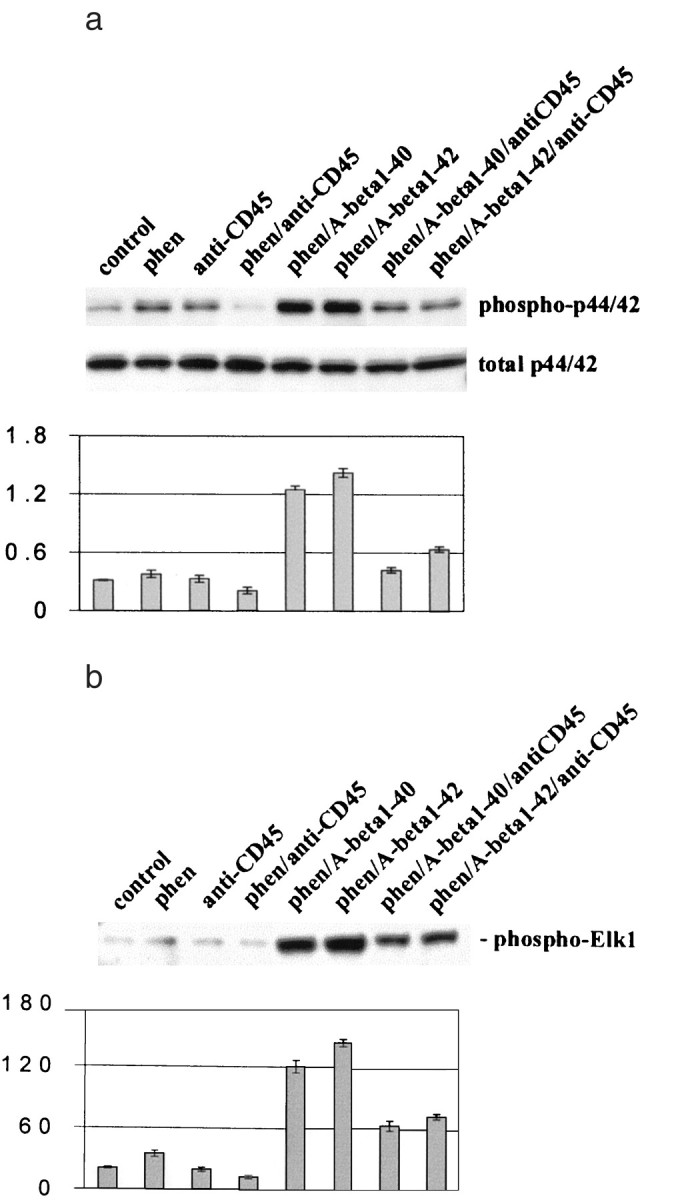
Cross-linking of microglial CD45 markedly suppresses p44/42 MAPK activation resulting from phen and Aβ peptide co-treatment. Microglial treatment conditions are indicated and are further described in Materials and Methods. Cell lysates were analyzed by Western immunoblotting using specific antibodies that recognize phosphorylated or total p44/42 MAPK (a) or the p44/42 MAPK fusion protein Elk-1 (b) by immune complex kinase assay. Histograms below immunoblots represent the mean band density ratio ± 1 SEM (phospho-p44/42 MAPK/total p44/42 MAPK) (a) or the mean band density ± 1 SEM in optical density units (b), with n = 3 for each condition presented. For a and b, ANOVA revealed significant main effects of Aβ1-40 and Aβ1-42 (p < 0.001), and there was statistical interaction between either Aβ1-40 or Aβ1-42 and phen (p < 0.001). One-way ANOVA revealed significant between-group differences (p < 0.001), and post hoctesting showed significant differences between phen/Aβ1-40 and phen/Aβ1-40/anti-CD45 (p < 0.001) and between phen/Aβ1-42 and phen/Aβ1-42/anti-CD45 (p < 0.001).
Stimulation of CD45-deficient microglia with Aβ peptides directly results in microglial activation
To further substantiate the role of CD45 in Aβ-mediated microglial activation, microglia were obtained from CD45-deficient or wild-type mice and treated with either Aβ or control peptide for 12 hr. Data showed that Aβ treatment greatly resulted in microglial activation, which was quantified by TNF-α and NO release. Results shown in Figure 5, aand b, indicate marked activation of CD45-deficient microglia compared with wild-type microglia after stimulation with Aβ peptides. Because we have consistently shown that microglial activation resulting from phen and Aβ peptide treatment first resulted in p44/42 MAPK phosphorylation and activation followed by TNF-α release and NO production, we assessed whether treatment of CD45-deficient microglia with Aβ peptides could result in increased p44/42 MAPK phosphorylation and activation at the 30 min time point. Results showed that Aβ treatment of CD45-deficient microglia greatly increased p44/42 MAPK phosphorylation and activation compared with CD45 wild-type microglia (data not shown). Furthermore, to determine whether CD45-deficient microglia in this scenario could cause neuronal cell injury, primary cultured cortical neurons and microglia were co-cultured, and morphological examination (data not shown) and LDH assay were performed. Data showed that cortical neurons were markedly injured by Aβ-treated, CD45-deficient microglia compared with wild-type cells (Fig. 5c). When taken together, these data show that CD45 is a negative regulator of Aβ-induced microglial activation.
Fig. 5.
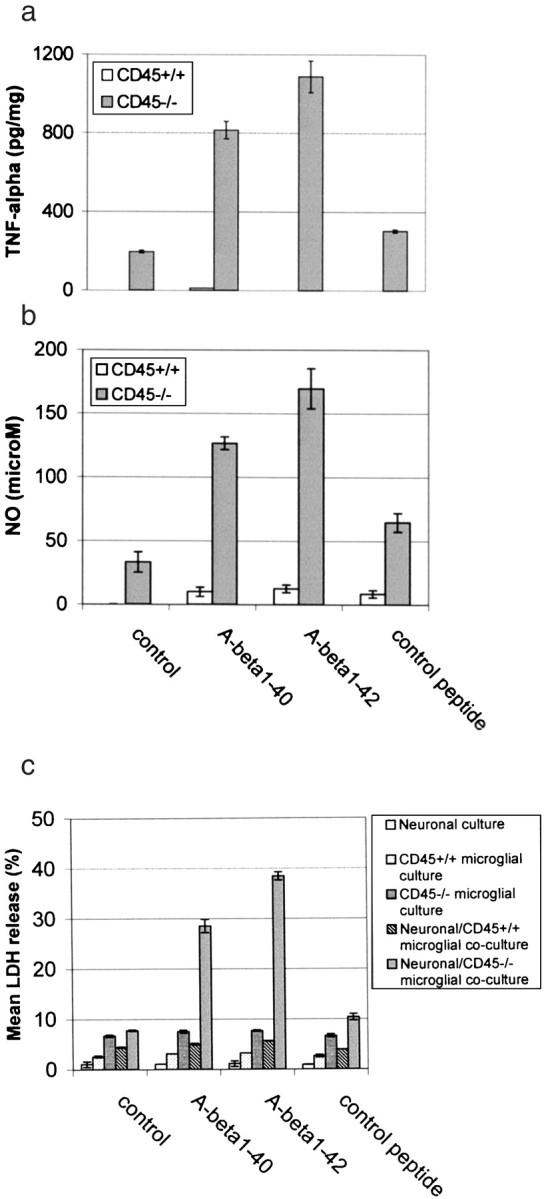
Stimulation of CD45-deficient microglia with Aβ peptides results in microglial activation. Primary cultured wild-type or CD45-deficient microglial cells were treated as indicated for 12 hr or co-cultured with primary cultured neuronal cells under the same treatment conditions for 36 hr. Control peptide is Aβ40-1. Microglial activation is evidenced by TNF-α production (mean ± 1 SEM; n = 3 for each condition presented) (a), NO assay (mean ± 1 SEM; n = 3 for each treatment condition) (b), and neuronal cell injury in co-culture experiments [mean LDH (percent) release ± 1 SEM;n = 3 for each condition presented] (c). For CD45-deficient microglia ina and b, one-way ANOVA revealed significant between-group differences (p < 0.001), and post hoc testing showed significant differences between control peptide and either Aβ1-40(p < 0.001) or Aβ1-42(p ≤ 0.001). For neuronal–CD45−/− microglial co-culture experiments, one-way ANOVA revealed significant between-group differences (p < 0.001), andpost hoc testing showed significant differences between control peptide and either Aβ1-40(p < 0.001) or Aβ1-42(p < 0.001).
Tg APPsw mice deficient for CD45 demonstrate marked TNF-α induction
To evaluate the possibility that CD45 might be a negative regulator of Aβ-mediated microglial activation in vivo, we crossed Tg APPsw mice with mice deficient for CD45 and measured TNF-α production in the brains of these animals. Results showed a marked increase in TNF-α protein in brain homogenates from these mice compared with Tg APPsw mice (Fig.6). We were only able to detect CD45 on microglia in Tg APPsw and control mice (data not shown), and CD45 has not been reported to be expressed by other CNS cells. Thus, these data suggest that microglial CD45 negatively regulates Aβ-induced microglial activation as evidenced by TNF-α production in vivo.
Fig. 6.

TNF-α production in vivo is markedly increased in Tg APPsw mice deficient for CD45. Brains from 6-month-old transgenic mice were isolated and prepared for TNF-α Western immunoblotting as described in Materials and Methods. TNF-α protein immunoblots (top panel) and mean band density ratios to actin ± 1 SEM (bottom panel; n = 3 for each condition) are shown. ANOVA revealed significant main effects of CD45 deficiency (CD45 def.; p < 0.001) and Tg APPswstatus (p < 0.001), as well as a significant interactive term between them (p< 0.01). One-way ANOVA revealed significant between-group differences (p < 0.001), and post hoctesting showed significant differences between control and Tg APPsw (p < 0.01) or CD45 def. (p < 0.05) and between either Tg APPsw or CD45 def. and the crossed mice (p < 0.001).
DISCUSSION
Microglial activation has been implicated as pathogenic in a variety of neurodegenerative diseases such as multiple sclerosis, AIDS dementia, and AD, raising the possibility that therapeutic strategies aimed at opposing microglial activation may be beneficial in treating such diseases. However, current attempts at reducing neuroinflammation mediated via microglial activation have only been partially efficacious (Rich et al., 1995), possibly because of the fact that such strategies are more general inhibitors of inflammation than specific inhibitors of microglial-associated neuroinflammation. For example, Mackenzie and Munoz (1998) examined postmortem brain tissue from AD patients who underwent NSAID treatment and control individuals who did not use NSAIDs (Mackenzie et al., 1998). These authors found that, although there were no significant differences in the mean numbers of senile plaques, senile plaque subtypes (diffuse or neuritic), or neurofibrillary pathology between cases and controls, the numbers of activated microglia were significantly decreased in NSAID-treated AD patients compared with controls. These data suggest that NSAIDs are prophylactic for AD partly by virtue of their opposition of microglial activation. Following from this idea, pharmacotherapeutics specifically aimed at blocking microglial activation may well be more efficient at ameliorating microglial-associated neuropathology in AD.
In this study, we focused on identifying a specific cell surface receptor target, which, when activated, could inhibit microglial activation far upstream of intracellular proinflammatory mediators such as the MAPK pathway. Our rationale for such investigation was that, if we could inhibit microglial activation very early on, the amplification of the inflammatory response associated with activation of proinflammatory intracellular signal transduction cascades could be abated. Our data show that microglia can be activated after treatment with Aβ peptides and the PTP inhibitor phen. This result led us to investigate stimulation of the membrane-bound PTP CD45 as a putative negative regulator of microglial activation. Data showed that cross-linking CD45 markedly reduced microglial activation resulting from Aβ and phen co-treatment. Furthermore, we observed decreased activation of p44/42 MAPK under these conditions, suggesting that CD45 cross-linking stimulates the CD45-associated PTP pathway, and that stimulation of this pathway negatively controls p44/42 MAPK activation. In accordance with this, co-treatment of Aβ- and phen-activated microglia with PD98059, an inhibitor of MEK1/2 (the upstream activator of p44/42 MAPK), resulted in statistically interactive blockade of microglial activation. We found that microglia deficient for CD45 could be directly activated by Aβ peptides in vitro, and brains from Tg APPsw mice deficient for CD45 demonstrated markedly increased TNF-α levels compared with Tg APPsw or CD45-deficient mouse brains. These results suggest that stimulation of CD45 is a viable approach for downregulating Aβ-induced microglial activation.
It has previously been shown that CD45 and the TNF receptor superfamily member CD40 can antagonize each another, because stimulation of CD45 opposes CD40-induced Ig class switching of human B cells to the IgE isotype (Loh et al., 1995). The mechanism underlying CD45–CD40 antagonism involves dephosphorylation and phosphorylation of tyrosine residues on their respective target signaling proteins, because CD45 is a PTP, and ligation of CD40 results in protein-tyrosine phosphorylation (Lazaar et al., 1998; Friedman and Greene, 1999). We have recently shown that ligation of CD40 leads to activation of microglia, as evidenced by TNF-α release and bystander-induced neurotoxicity (Tan et al., 1999a). We presently show that inhibition of microglial CD45 leads to activation of these cells, whereas stimulation of microglial CD45 opposes this effect. In the presence of low doses of freshly solubilized Aβ, synergistic enhancement of microglial activation is observed in either the CD40 ligation (Tan et al., 1999b) or the CD45 inhibition paradigm. When taken together, these observations suggest that, in microglia, CD40 and CD45 may have antagonistic effects on activation of these cells, whereby CD40 promotes and CD45 opposes it. Additionally, the possibility arises that Aβ is able to positively affect microglial activation via disruption of CD45–CD40 homeostasis.
We initially showed that co-treatment with the PTP inhibitor phen and Aβ peptides resulted in microglial activation as evidenced by increased NO and TNF-α release (Fig. 1a,b). However, the question arose of whether this effect was dependent on PTP inhibition as opposed to inhibition of other phosphatases. Thus, we co-treated wild-type primary culture microglia with Aβ and either sodium orthovanadate, another PTP inhibitor, or okadaic acid, an inhibitor of protein phosphatase 2A, and measured NO and TNF-α release. We observed that sodium orthovanadate treatment in conjunction with Aβ produced results similar to those of phen and Aβ peptide co-treatment. However, NO and TNF-α were not detectable in the media of okadaic acid- and Aβ-co-treated microglia. These data suggest that treatment of microglia with specific inhibitors of PTPs, as opposed to general phosphatase inhibitors, along with Aβ triggers microglial activation, further substantiating the specific effect of PTP stimulation via CD45 in opposing microglial activation induced by phen and Aβ peptides.
Past studies have shown that, in general, phosphatase activity decreases with aging and even more so in AD across various cell types (Gong et al., 1995; Pei et al., 1998). Specifically, it has been shown that peripheral T lymphocytes isolated from AD patients demonstrate decreased amounts of the CD45R isoform compared with age-matched nondemented control subjects (Ikeda et al., 1991). To examine whether increasing CD45 activity could block microglial activation resulting from co-treatment with phen and Aβ, we activated wild-type microglia with phen and Aβ, added CD45 recombinant protein (20 U/ml) to these cells, and measured NO and TNF-α release. We observed marked reduction of NO and TNF-α after addition of CD45 recombinant protein to activated microglia compared with appropriate controls. Interestingly, treatment of activated microglia with CD45 recombinant protein resulted in blockade of NO and TNF-α release to an extent similar to that resulting from cross-linking CD45, further substantiating that CD45 cross-linking stimulates the CD45 PTP pathway.
Our data thus far had focused on CD45-mediated downregulation of microglial activation induced by co-treatment with phen and Aβ. These data raised the question of whether CD45 may reduce microglial activation induced by other stimuli, such as LPS. To address this possibility, we incubated microglia with LPS (1 ng/ml) and anti-CD45 antibody. Data showed that CD45 cross-linking markedly attenuated microglial activation as evidenced by NO and TNF-α release (Fig.7). These data raise the possibility that stimulation of the CD45 pathway negatively controls microglial activation induced by various proinflammatory stimuli and suggest that pharmacotherapeutics targeting stimulation of CD45 may be beneficial in suppressing microglial activation, which is a pathogenic component of a variety of neurodegenerative diseases.
Fig. 7.

CD45 cross-linking markedly inhibits production of TNF-α and NO in LPS-treated microglia. Microglial cells were treated as indicated for 12 hr. TNF-α production (mean ± 1 SEM, picograms per milligram of total protein) and NO release (mean ± 1 SEM, micromolar concentration per milligram of total protein) were determined by TNF-α ELISA and nitric oxide assay, respectively. Data shown are representative of six independent experiments. One-way ANOVA revealed significant between-group differences (p < 0.001), and post hoctesting revealed a significant difference between LPS and LPS/anti-CD45 antibody (p < 0.001).
Footnotes
We are grateful to Diane and Robert Roskamp for their generous support, which helped make this work possible. We thank Jodi Kroeger for assistance in flow cytometric acquisition and analysis and Kiyoko Yokota for assistance in maintaining CD45-deficient mice.
Correspondence should be addressed to Dr. Jun Tan, Roskamp Institute, University of South Florida, 3515 East Fletcher Avenue, Tampa, FL 33613. E-mail: jtan@coml.med.usf.edu.
REFERENCES
- 1.Barger SW, Harmon AD. Microgial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 2.Beard CM, Waring SC, O'Brien PC, Kurland LT, Kokmen E. Nonsteroidal anti-inflammatory drug use and Alzheimer's disease: a case-control study in Rochester, Minnesota, 1980 through 1984. Mayo Clin Proc. 1998;73:951–955. doi: 10.4065/73.10.951. [DOI] [PubMed] [Google Scholar]
- 3.Chao CC, Hu S, Molitor TW, Shaskan ED, Peterson PK. Activated microglia mediate neuronal cell injury via nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 4.Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman WJ, Greene A. Neurotrophin signaling via Trks and p75. Exp Cell Res. 1999;253:131–142. doi: 10.1006/excr.1999.4705. [DOI] [PubMed] [Google Scholar]
- 6.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal K, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 7.Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA, Flavell RA., Jr Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273:1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- 8.Hambleton J, McMahon M, DeFranco AL. Activation of Raf-1 and mitogen-activated protein kinase in murine macrophages partially mimics lipopolysaccharide-inducing events. J Exp Med. 1995;182:147–154. doi: 10.1084/jem.182.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 10.Hsiao KK, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda T, Yamamoto K, Takahashi K, Yamada M. Immune system-associated antigens on the surface of peripheral blood lymphocytes in patients with Alzheimer's disease. Acta Psychiatr Scand. 1991;83:444–448. doi: 10.1111/j.1600-0447.1991.tb05573.x. [DOI] [PubMed] [Google Scholar]
- 12.Justement LB. The role of CD45 in signal transduction. Adv Immunol. 1996;66:1–65. doi: 10.1016/s0065-2776(08)60595-7. [DOI] [PubMed] [Google Scholar]
- 13.Karp HL, Tillotson ML, Sorita J, Reich C, Wood JG. Microglial tyrosine phosphorylation systems in normal and degenerating brain. Glia. 1994;11:284–290. doi: 10.1002/glia.440110310. [DOI] [PubMed] [Google Scholar]
- 14.Lazaar AL, Amrani Y, Hsu J, Panettieri RA, Fanslow WC, Jr, Albelda SM, Pure E. CD40-mediated signal transduction in human airway smooth muscle. J Immunol. 1998;161:3120–3127. [PubMed] [Google Scholar]
- 15.Licastro F, Mallory M, Hansen LA, Masliah E. Increased level of alpha-1-antichymotrypin in brains of patients with Alzheimer's disease correlate with activated astrocytes and are affected by APOE 4 genotype. J Neuoimmunol. 1998;88:105–110. doi: 10.1016/s0165-5728(98)00096-4. [DOI] [PubMed] [Google Scholar]
- 16.Loh RK, Jabara HH, Ren CL, Fu SM, Vercelli D, Geha RS. Role of protein tyrosine kinases and phosphatases in isotype switching: crosslinking CD45 to CD40 inhibits IgE isotype switching in human B cells. Immunol Lett. 1995;45:99–106. doi: 10.1016/0165-2478(94)00233-h. [DOI] [PubMed] [Google Scholar]
- 17.Mackenzie IR, Munoz DG. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology. 1998;50:986–990. doi: 10.1212/wnl.50.4.986. [DOI] [PubMed] [Google Scholar]
- 18.Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer's disease. Acta Neuropathol (Berl) 1991;83:12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- 19.Maxwell JR, Campbell JD, Kim CH, Vella AT. CD40 activation boosts T cell immunity in vivo by enhancing T cell clonal expansion and delaying peripheral T cell deletion. J Immunol. 1999;162:2024–2034. [PubMed] [Google Scholar]
- 20.McDonald DR, Bamberger ME, Combs CK, Landreth GE. beta-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 22.Pei JJ, Gong CX, Iqbal K, Grundke-Iqbal I, Wu QL, Winblad B, Cowburn RF. Subcellular distribution of protein phosphatases and abnormally phosphorylated tau in the temporal cortex from Alzheimer's disease and control brains. J Neural Transm. 1998;105:69–83. doi: 10.1007/s007020050039. [DOI] [PubMed] [Google Scholar]
- 23.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 24.Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P. Inflammation and Alzheimer's disease pathogenesis. Neurobiol Aging. 1996;17:681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- 25.Suttles J, Milhorn DM, Miller RW, Poe JC, Wahl LM, Stout RD. CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (il-4) and il-10 anti-inflammatory action. J Biol Chem. 1999;26:5835–5842. doi: 10.1074/jbc.274.9.5835. [DOI] [PubMed] [Google Scholar]
- 26.Stuber E, Strober W, Neurath M. Blocking the CD40-CD40L interaction in vivo specifically prevents the priming of T helper 1 cells through the inhibition of interleukin 12 secretion. J Exp Med. 1996;183:693–698. doi: 10.1084/jem.183.2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan J, Town T, Paris D, Placzek A, Parker T, Crawford F, Yu H, Humphrey J, Mullan M. Activation of microglial cells by the CD40 pathway: relevance to multiple sclerosis. J Neuroimmunol. 1999a;97:77–85. doi: 10.1016/s0165-5728(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 28.Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after β-amyloid stimulation. Science. 1999b;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 29.Tan J, Town T, Saxe M, Paris D, Wu Y, Mullan M. Ligation of microglial CD40 results in p44/42 mitogen-activated protein kinase-dependent TNF-α production that is opposed by TGF-β1 and IL-10. J Immunol. 1999c;163:6614–6621. [PubMed] [Google Scholar]
- 30.Yang Y, Wilson JM. CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science. 1996;273:1862–1864. doi: 10.1126/science.273.5283.1862. [DOI] [PubMed] [Google Scholar]