

Abstract
We investigated a role for cAMP/protein kinase A (PKA) in light/glutamate (GLU)-stimulated state changes of the mammalian circadian clock in the suprachiasmatic nucleus (SCN). Nocturnal GLU treatment elevated [cAMP]; however, agonists of cAMP/PKA did not mimic the effects of light/GLU. Coincident activation of cAMP/PKA enhanced GLU-stimulated state changes in early night but blocked light/GLU-induced state changes in the late night, whereas inhibition of cAMP/PKA reversed these effects. These responses are distinct from those mediated by mitogen-activated protein kinase (MAPK). MAPK inhibitors attenuated both GLU-induced state changes. Although GLU induced mPer1 mRNA in both early and late night, inhibition of PKA blocked this event only in early night, suggesting that cellular mechanisms regulating mPer1 are gated by the suprachiasmatic circadian clock. These data support a diametric gating role for cAMP/PKA in light/GLU-induced SCN state changes: cAMP/PKA promotes the effects of light/GLU in early night, but opposes them in late night.
Keywords: suprachiasmatic nucleus, glutamate, signal transduction, mPer1, protein kinase A, MAP kinase, rat
The circadian clock is a complex biological structure in which a dynamic set of cellular state progressions generates near 24 hr rhythms at behavioral, physiological, cellular, and molecular levels. The oscillatory mechanism is a self-sustained transcriptional–translational feedback loop (for review, see King and Takahashi, 2000). In mammals, rhythmicity is modulated by the impingement of numerous neurochemical inputs, which provide information regarding the external and internal environments, on a central clock in the suprachiasmatic nucleus (SCN) (Moore and Lenn, 1972; Pickard, 1982; Johnson et al., 1988). These afferent signals use specific signal transduction pathways to interface with core clock elements to induce long-term state changes, or phase shifts, in the clock. Clock sensitivity to state-altering signals and activation of signaling pathways are gated by the SCN (Gillette, 2000). For example, cAMP activation alters circadian timing only during subjective day, whereas sensitivity to cGMP-dependent state changes are restricted to subjective night (for review, see Gillette and Tischkau, 1999). Thus, the accessibility of specific signaling pathways is key to regulation of timing.
Light signaling pathways intersect with the molecular clockworks via the mammalian period1 (mPer1) gene. Translation of mPer1 mRNA is required for light-induced phase resetting in the early night (Akiyama et al., 1999). Light elevatesmPer1 mRNA in both early and late night (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997; Takumi et al., 1998; Zylka et al., 1998). Light responsiveness of other clock genes differs. Levels of mPer2 increase robustly after light in the early night, but the response in the late night is not as clear (Albrecht et al., 1997; Takumi et al., 1998; Zylka et al., 1998).mTim mRNA levels are also augmented in response to light in the early night [Tischkau et al. (1999), but see King and Takahashi (2000)].
Dissection of elements required for SCN processing of light information points to commonalities and disparities between early and late night. Glutamate (GLU) is the primary neurotransmitter transmitting light signals to the SCN (Pickard, 1982; Johnson et al., 1988; Castel et al., 1993; De Vries et al., 1993; Ding et al., 1994; Shirakawa and Moore, 1994; Hannibal et al., 2000). Throughout the night, a phase-resetting light stimulus evokes release of GLU from the retinohypothalamic tract (RHT) and activation of multiple GLU receptor types, of which NMDA receptors are critical (Colwell and Menaker, 1992; Ding et al., 1994; Shibata et al., 1994; Shirakawa and Moore, 1994, Mintz et al., 1999). The consequent influx of Ca2+ activates nitric oxide synthase (NOS) to produce nitric oxide (NO) (Ding et al., 1994; Amir et al., 1995). After liberation of NO, the light signaling pathways diverge (Gillette, 2000). In early night, light/GLU-induced state change, which delays the clock's rhythm, requires activation of neuronal ryanodine receptors (RYRs) to release intracellular stores of Ca2+ (Ding et al., 1998). In late night, the light/GLU signal activates an RYR-independent, cGMP/protein kinase G (PKG)-dependent signal transduction cascade to initiate a phase advance (Weber et al., 1995; Mathur et al., 1996; Ding et al., 1998). Finally, light/GLU signaling in both early and late night induces phosphorylation of Ca2+/cAMP response element binding protein (CREB) and CRE-mediated transcriptional activation (Ginty et al., 1993; Ding et al., 1997;Obrietan et al., 1998, 1999).
Although activation of PKA can be downstream from NMDA receptor-triggered Ca2+ transients in other brain regions (Greengard et al., 1991; Bito et al., 1997), a role for cAMP in nocturnal light/GLU signaling SCN state changes has yet to be determined. Recently, pituitary adenylyl cyclase-activating polypeptide (PACAP) was demonstrated to modulate circadian state changes stimulated by light/GLU (Chen et al., 1999). These data suggest that activation of cAMP/PKA could contribute to light/GLU signal transduction. We hypothesized that a GLU-primed cAMP/PKA response system modulates light/GLU-induced state changes. We examined the effects of cAMP/PKA modulation of early and late night glutamatergic input to the SCN in terms of rodent behavioral and SCN electrical activity rhythms and on mPer1 mRNA.
MATERIALS AND METHODS
Animals and circadian time. Long–Evans rats (6–12 weeks old) were used for all in vitro experiments. This line has been inbred for >35 generations, surpassing the requirements for genetic homogeneity, which leads to low variation in physiological experiments. Rats were provided food and water ad libitumand entrained to a daily cycle of 12 hr light and 12 hr dark. Over the 2–3 d of experimentation in vitro, the SCN generates stable, near 24 hr oscillations in neuronal activity that do not deviate from the entrained 24 hr cycle (Prosser and Gillette, 1989). Therefore, in vitro clock time was determined from the lighting cycle in the donor colony. The time of lights-on was designated as circadian time (CT) 0; subjective day was CT 0–12. Subjective night (CT 12–24) corresponded to the dark portion of the donor's cycle.
Preparation and treatment of brain slices. Brain slices were prepared ≥2 hr before the onset of the dark phase of the light/dark cycle. A block of hypothalamic tissue was cut with a mechanical chopper into 500 μm coronal slices containing the SCN. Slices were studied for up to 3 d in vitro with continuous perifusion of Earle's Essential Balanced Salt Solution (EBSS, Sigma, St. Louis, MO), supplemented with 24.6 mm glucose, 26.2 mm NaHCO3, and 2.5 mg/l gentamicin, and saturated with 95% O2/5% CO2 at 37°C, pH 7.4. Neuronal activity, measured by single-unit recording, is low at night and peaks around midday (CT 7) (Gillette and Prosser, 1988). Therefore, measurement of time-of-peak provides an accurate assessment of circadian phase (Gillette et al., 1995).
For all experiments except scintillation proximity assay (SPA), SCN slices were treated in the brain slice chamber. Perifusion was stopped during treatment. GLU (10 mm, 10 min) was applied by microdrop (1 μl) to the SCN at the surface of the slice. All other treatments were applied 30 min before the time of GLU treatment by replacing the bath with EBSS containing the reagent to be tested. At the conclusion of treatment, slices were washed, the bath was replaced with fresh EBSS, and perifusion was resumed. For SPA experiments, SCN slices were trimmed to contain only the SCN and underlying optic chiasm. This reduced slice preparation retains SCN clock properties (Gillette and Reppert, 1987).
cAMP measurements. At either CT 14 or CT 20, reduced slices were transferred to microfuge tubes containing 100 μl of 10 mm GLU or vehicle control. Tubes containing reduced slices and treatment media were snap frozen in a dry ice/methanol bath at the conclusion of treatment. After ethanol extraction from samples, cAMP was assayed using the SPA (Amersham, Arlington Heights, IL) according to the protocol provided. Sample size was based on rigorous statistical testing of the hypothesis.
Single-unit recordings of SCN neuronal activity. The technique used to record single units extracellularly from the ensemble of SCN neurons has been described in detail previously and validated thoroughly (Prosser and Gillette, 1989). Briefly, under visual guidance, a glass microelectrode filled with 5 m NaCl was lowered into the SCN using a hydraulic microdrive until the signal from a single cell was encountered. Electrical signals from single units exceeding twice the background level were isolated, observed for stability, and counted for 4 min using LabView software. The electrode was advanced until a different cell was encountered and its activity was counted. After a complete pass through the brain slice, the electrode was repositioned within the SCN to sample throughout the entire nucleus over the course of the experiment. Firing rates of individual neurons recorded during a single experiment were grouped into 2 hr running averages using 15 min lags. The time-of-peak for each experiment was determined by visual inspection of a plot of 2 hr running averages for the symmetrically highest point. Certain experiments were performed with the experimenter blind to treatment conditions. Results of such experiments were identical to “non-blind” recordings.
The average peak electrical activity of the ensemble of SCN neurons, which reliably occurs near CT 7 in untreated brain slices maintainedin vitro, provides an accurate measure of clock time. A phase advance was defined as a peak that appeared before CT 7 on the days after treatment; the time-of-peak appeared later than CT 7 for phase delays. Thus, phase shifts were determined by comparing the mean time-of-peak from treatment groups with vehicle-treated controls.
In situ hybridization. After early- or late-night treatment, brain slices containing the SCN were fixed overnight in 4% paraformaldehyde. Slices were transferred to 0.1 m PBS with 20% sucrose and maintained at 4°C until sectioning. Ten micrometer sections were cut at −17°C on a cryostat. Hybridization was performed as previously described (Tischkau et al., 1999). The template for the mPer1 probe was kindly provided by Dr. U. Schibler (Université de Genève, Switzerland) (Balsalobre et al., 1998) and corresponded to nucleotides 660–780 of the published sequence (Sun et al., 1997). The probe was labeled with digoxygenin by in vitro transcription. A digoxygenin-labeled sense or antisense riboprobe for mPer1(5 ng/μl) was applied in hybridization buffer (4× SSC, 40% formamide, 10% dextran sulfate, 1× Denhardt's solution, 10 mm DTT, 1 mg/ml yeast tRNA, 1 mg/ml salmon sperm DNA) overnight at 42°C. Probe hybridization was visualized using an alkaline phosphatase-labeled anti-digoxygenin antibody (1:100, Roche). Analysis of mPer1-positive cells was made in a midcaudal section of the SCN by an individual blind to the experimental design and identity of the samples.
In vivo wheel-running experiments. Male Syrian hamsters (Mesocricetus auratus) were housed individually in cages equipped with running wheels (5.5 inch diameter). Animals were entrained to a 14 hr light/10 hr dark schedule for at least 21 d before release into constant darkness (DD). Wheel revolutions were counted by activation of reed switches (by a magnet attached to the wheel) and tallied using DATACOL 3 data acquisition software (Minimitter, Sun River, OR). Actograms were plotted using Ratman (Klemfuss and Clopton, 1993). Treatment times were calculated from the time of activity onset, designated CT 12. The magnitude of phase resetting was measured as the distance between regression lines drawn through data from at least 5 d immediately before treatment and 5 d after reestablishment of a stable circadian rhythm after treatment (Ding et al., 1998).
At least 7 d before release into DD, animals (70–110 gm at time of surgery) were stereotaxically implanted with guide cannulae directed into the third ventricle (Ding et al., 1994). Under an anesthesia mixture of ketamine (125 mg/kg), xylazine (20 mg/kg), and acepromazine maleate (2 mg/kg), guide cannulae (26 gauge; Plastics One, Roanoke, VA) were implanted 1.0 mm anterior to bregma at the midline (upper incisor bar at 0) to a depth of 2.8 mm inferior to the dural surface. Guide cannulae were secured with a jeweler's screw and cranioplastic cement (Plastics One). Stainless steel stylets (33 gauge; Small Parts, Miami, FL) were inserted into cannulae to maintain patency.
Animals were allowed to free-run under constant conditions for 10–14 d before the first intracerebroventricular injection. Injections were performed as described previously (Chen et al., 1999). Briefly, saline and KT5720 (5 μm) injections were performed in dim red light (∼1 lux). Injections (2 μl) were administered over a 1 min interval. When injections were accompanied by a light pulse, the light stimulus (15 min, 20 lux) (Weber et al., 1995) was initiated 15 min after the injection. Subsequent injections were set at 10–14 d intervals to allow recovery of a stable, free-running rhythm. Each animal received up to three treatments (saline + light, KT5720, KT5720 + light).
RESULTS
GLU stimulates cAMP accumulation in the SCN
To evaluate cAMP-mediated signal transduction in state changes, or phase shifts, that arise as a consequence of light/GLU stimulation, we first determined whether SCN cAMP levels change in response to GLU in early (CT 14) and late (CT 20) subjective night. CT 14 and CT 20 correspond to the circadian times most sensitive to phase resetting in response to GLU in the rat. A time course for the cAMP response at CT 14 established that SCN cAMP levels increased significantly 2 min after GLU treatment (p < 0.05), peaked at 3 min (p < 0.01), and returned to basal levels 10 min after treatment (Fig.1A,B). At CT 20, cAMP was also elevated significantly 3 min (p < 0.01) after GLU treatment. Data shown are the results of six independent experiments for each treatment.
Fig. 1.

cAMP measurements after GLU stimulation at CT 14 or CT 20. A, At CT 14, GLU elevated cAMP levels in a time-dependent manner. cAMP was elevated significantly 2 min (p < 0.01), 3 min (p < 0.01), and 5 min (p < 0.01) after GLU treatment, but returned to basal levels by 10 min after GLU treatment. Peak levels were observed 3 min after GLU stimulation. B, cAMP levels were elevated after GLU treatment at both CT 14 and CT 20. ** indicates statistical significance (p < 0.01) compared with controls (ANOVA with Student–Newman–Keuls).
cAMP potentiates GLU-induced phase delays but inhibits GLU-induced phase advances in vitro
To test the hypothesis that cAMP modulates GLU-induced state changes that are expressed as phase shifts of circadian rhythms, we examined the effects of GLU on SCN clock phase in the presence of the membrane-permeable cAMP analog, 8-Br-cAMP. Control slices exhibit a persistent, near 24 hr oscillation with a mean time-of-peak occurring at CT 6.80 ± 0.15 (Fig.2A,H) (n = 6). Application of 8-Br-cAMP at CT 14 (time-of-peak, CT 6.75 ± 0.25, n = 3) or CT 20 (time-of peak, CT 6.62 ± 0.15, n = 4) had no effect on the subsequent phase of the SCN neuronal activity rhythm (Fig. 2, B and E, respectively), confirming earlier observations (Prosser and Gillette, 1989). GLU application at CT 14 stimulated a characteristic (Ding et al., 1994) phase delay (time-of-peak, CT 9.9 ± 0.35, n = 5) of ∼3 hr (Fig. 2C,H). Interestingly, coapplication of 8-Br-cAMP potentiated the phase delay stimulated by GLU. The 4.29 ± 0.15 hr phase delay that ensued after treatment with 8-Br-cAMP + GLU at CT 14 (time-of-peak, CT 11.09 ± 0.13;n = 4) (Fig.2D,H) was significantly greater (p < 0.05) than the phase delay produced by GLU alone.
Fig. 2.

Exogenous cAMP modulates GLU-induced phase resetting of the SCN electrical activity rhythm in vitro. A, A control recording demonstrated that the electrical activity rhythm persists for 2 d in vitro, with a peak near CT 7 on both days. B, The membrane-permeable cAMP analog 8-Br-cAMP (200 μm) had no effect on the time-of-peak electrical activity at CT 14.C, At CT 14, GLU (10 mm) induced a 3 hr phase delay in the SCN electrical activity rhythm. D, 8-Br-cAMP significantly increased (p < 0.05, Student's t test) the magnitude of the GLU-induced phase delay from ∼3 to ∼4.5 hr. E, 8-Br-cAMP (200 μm) had no effect on the electrical activity rhythm when applied alone at CT 20. F, At CT 20, GLU (10 mm) advanced the electrical activity rhythm by ∼3 hr. G, 8-Br-cAMP (200 μm) blocked the GLU-stimulated phase advance. H, Summary of the phase shifting effects of GLU ± 8-Br-cAMP at CT 14 and CT 20. Bars indicate the mean ± SEM of three to six experiments.a, b, and c indicate values significantly different from each other and from controls (p < 0.05; Student's ttest).
At CT 20, GLU induced a phase advance of ∼3.5 hr (Fig.2F,H) (time-of-peak CT 3.29 ± 0.32, n = 4), corroborating previous studies (Ding et al., 1994). When 8-Br-cAMP was applied in combination with GLU at CT 20 (Fig. 2G,H), the time-of-peak neuronal activity occurred, as in controls, at CT 6.55 ± 0.17 (n = 4). Thus, 8-Br-cAMP blocked the GLU-induced phase advance at CT 20.
PKA inhibition blocks GLU-induced phase delays but potentiates GLU-induced phase advances in vitro
A ubiquitous consequence of cAMP elevation is activation of PKA. To determine whether PKA mediates the effects of GLU that generate phase-shifting state changes in the SCN throughout subjective night, we used KT5720, an isoquinoline inhibitor of PKA. Application of KT5720 alone at either CT 14 (time-of-peak, CT 6.75 ± 0.18,n = 4) or CT 20 (time-of-peak 6.66 ± 0.25,n = 4) had no effect on the time-of-peak electrical activity from the ensemble of SCN neurons (Figs.3, C, H andF, H, respectively). Coapplication of KT5720 completely blocked the GLU-induced phase delay of the SCN electrical activity rhythm at CT 14 (time-of-peak CT 6.98 ± 0.30,n = 3) (Fig.3D,H). Conversely, at CT 20, KT5720 potentiated the phase-advancing effects of GLU. KT5720 + GLU caused a 6.25 ± 0.42 hr (n = 4) (Fig.3G,H) phase advance, which is significantly larger (p < 0.01) than the phase advance induced by application of GLU alone at this time. Thus, although GLU stimulation increases cAMP levels throughout the night, the resultant PKA activation produces opposite consequences on phase resetting in response to glutamatergic stimuli in early versus late night.
Fig. 3.
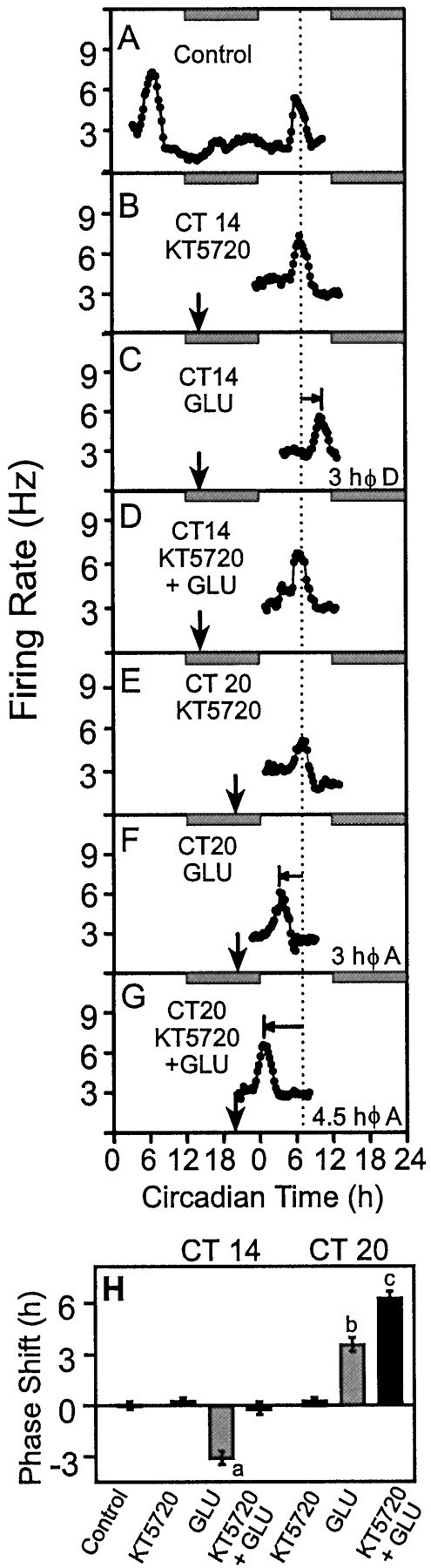
Inhibition of PKA, using KT5720, modulates GLU-induced phase resetting of the SCN electrical activity rhythmin vitro. A, The same control recording described in Figure 2. B, KT5720 (100 nm) had no effect on the time-of-peak electrical activity at CT 14.C, At CT 14, GLU (10 mm) induced a 3 hr phase delay in the SCN electrical activity rhythm. D, KT5720 fully blocked the GLU-induced phase delay. E, KT5720 (100 nm) had no effect on the electrical activity rhythm when applied alone at CT 20. F, At CT 20, GLU (10 mm) advanced the electrical activity rhythm by ∼3 hr.G, KT5720 enhanced the GLU-stimulated phase advance by 1.5 hr to a total of 4.5 hr. H, Summary of the phase shifting effects of GLU ± KT5720 at CT 14 and CT 20. Bars indicate the mean ± SEM of three to six experiments.a, b, and c indicate values significantly different from each other and from controls (p < 0.05; Student's ttest).
Mitogen-activated protein kinase inhibition partially blocks both GLU-induced phase delays and advances
GLU-induced signal transduction can also activate the mitogen-activated protein kinase (MAPK) cascade to initiate neuronal state changes (Bading and Greenberg, 1991; Rosen et al., 1994). cAMP-dependent activation of the MAPK pathway has been implicated in the phase-resetting effects of light/GLU in the SCN (Ginty et al., 1993; Obrietan et al., 1998). To determine whether the cAMP-dependent effects that we observed were caused by activation of MAPK, we inhibited the MAPK signal transduction cascade at two different sites in conjunction with GLU treatment at CT 14 and CT 20. Olomoucine (Abraham et al., 1995) and apigenin (Kuo and Yang, 1995) inhibit p44 MAPK. PD98059 inhibits MEK (MAP kinase kinase) (Kultz et al., 1998). Each inhibitor partially blocked the GLU-induced phase delay at CT 14 (Fig. 4) (n = 3 for each inhibitor). Likewise, but in contrast to the effects of KT5720 in tandem with GLU at CT 20, each MAPK inhibitor partially blocked the GLU-induced phase advance. Thus, our data support a role for MAPK as one of several elements essential to signal transduction events stimulated by GLU that initiate phase resetting at night. However, permissive effects of PKA on the MAPK pathway do not account for the effects we observe with KT5720. Importantly, inhibition of MAPK alone cannot explain the potentiation of the GLU-induced phase advance observed in the presence of KT5720.
Fig. 4.

Inhibition of the MAPK kinase cascade attenuates GLU-induced phase shifts at both CT 14 and CT 20. Bars indicate the mean ± SEM of three to six experiments. Inhibitors of p44 MAPK, apigenin (50 μm), and olomoucine (100 μm) diminish the amplitudes of the GLU-induced phase delay at CT 14 from ∼3 to ∼1.5 hr (n = 3, p < 0.05, Student's t test) and phase advance at CT 20 from ∼3.5 to ∼1.75 hr (n = 3, p< 0.05, Student's t test). PD98059 (50 μm), an inhibitor of MEK, also decreased the GLU-induced phase delay to ∼2 hr (n = 3, *p < 0.05, Student's t test) and phase advance to ∼1.5 hr (n = 3, *p < 0.05, Student's ttest).
PKA inhibition potentiates light-induced phase shiftsin vivo
The importance of activation of cAMP/PKA signal transduction in response to light/GLU in the SCN was further evaluated in vivo. A 20 lux light pulse at the time of maximal sensitivity to phase resetting by light caused a 1.24 ± 0.37 hr (n = 8) phase advance in the onset of the wheel-running rhythm in the hamster (Fig. 5). Injection of KT5720 at 30 min before a light pulse significantly potentiated light-induced phase advances (1.92 ± 0.25 hr, n = 10; p < 0.05) (Fig.5B,D). Injection of KT5720 alone had no effect on the phase of wheel-running activity (Fig.5C) (n = 8).
Fig. 5.

The PKA inhibitor, KT5720, potentiated light-induced phase advances of hamster wheel-running activity rhythmin vivo. A–C, Sequential daily activity records from three hamsters representing the effects of the various treatments. Each line represents 24 hr. Light exposure (20 lux, 15 min) at CT 18 (6 hr after activity onset, indicated bystar) after vehicle (2 μl) administration by intracerebroventricular injection induced a 1.24 ± 0.37 hr (n = 8) phase advance. KT5720 injection (2 μl) before light exposure significantly augmented the light-induced phase advance (1.92 ± 0.25 hr, n = 10;a, b indicate statistically significant differences, *p < 0.05, ANOVA with Student–Newman–Keuls). KT5720 alone did not induce significant phase shifts (0.08 ± 0.15 hr; n = 8).
PKA inhibition differentially alters GLU-induced mPer1mRNA accumulation in a state-dependent manner
To determine whether cAMP/PKA mediates the rapid, transient elevation of mPer1 mRNA required for both phase delays (Akiyama et al., 1999) and phase advances (S. A. Tischkau and M. U. Gillette, unpublished observations) in response to nocturnal light/GLU,in situ hybridizations were performed after treatment of SCN slices with GLU ± KT5720. Endogenous mPer1 mRNA was low at CT 14 and CT 20 (Fig.6A,D). Consistent with the effects of light in vivo (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997; Zylka et al., 1998), mPer1 mRNA was significantly elevated 60 min after GLU treatment at either CT 14 or CT 20 (Fig.6B,E) (n = 6). There was no significant difference in total content of GLU-stimulatedmPer1 at either time (Student's t test), but at CT 14, GLU-stimulated mPer1 was observed throughout the SCN, whereas GLU-stimulated mPer1 was restricted to the retinorecipient SCN at CT 20. KT5720 blocked the GLU-stimulated rise inmPer1 at CT 14 (Fig. 6C) (n = 6) but did not diminish the accumulation of mPer1 after GLU treatment at CT 20. In contrast to samples treated with GLU alone at CT 20, mPer1 was observed throughout the SCN in these samples (Fig. 6F) (n = 6). KT5720 had no effect on mPer1 levels when applied alone at either time (data not shown).
Fig. 6.

Effects of PKA inhibition on GLU-inducedmPer1 mRNA levels in the SCN at CT 14 and CT 20. A digoxygenin-labeled cRNA probe detected low levels ofmPer1 mRNA in control sections at CT 14 (A) and CT 20 (D). GLU significantly elevated mPer1 mRNA 60 min after GLU treatment at either CT 14 (B) or CT 20 (E). Inhibition of PKA blocked GLU-inducedmPer1 at CT 14 (C), but had no effect on GLU-induced mPer1 at CT 20 (F). G, For quantitation, positive cells were counted from one SCN in a single, midcaudal section by an experimenter blind to the treatment. Bars represent mean ± SEM for six independent experiments. ** represents statistically significant differences compared with control values at the same circadian time as determined by AVOVA (p < 0.01) with Student–Newman–Keuls post hocanalysis.
DISCUSSION
Overt SCN clock sensitivity to reagents that activate cAMP pathways (the neuromodulatory ligands PACAP and serotonin), stimulate cAMP accumulation (forskolin, cAMP phosphodiesterase inhibitors), or activate PKA (cAMP analogs) is restricted to subjective day; cAMP/PKA at night has no effect on clock phase (Prosser and Gillette, 1989;Medanic and Gillette, 1992; Shibata et al., 1992; Hannibal et al., 1997). Thus, daytime activation of cAMP signal transduction is typical of a primary signaling pathway. Direct activation of a downstream component, cAMP, evokes the same response as an extracellular signal, PACAP or serotonin, whereas inhibition of a downstream component, PKA, blocks the response to the extracellular signal.
This study reveals a different role for cAMP/PKA-dependent processes in modulation of nocturnal glutamatergic input to the SCN. PKA acts to gate long-term state changes invoked by GLU, because inhibition of cAMP/PKA alters the GLU effects, but cAMP/PKA has no effect independent of GLU. cAMP/PKA gating provides regulation by either blocking or enhancing signal flow through a primary pathway (Iyengar, 1996), as observed in other systems. Developmentally, PKA elevation blocks differentiation by the morphogenic Hedgehog signal inDrosophila (Blair, 1995; Jiang and Struhle, 1995, Li et al., 1995; Perrimon, 1995) and mice (Fan et al., 1995). cAMP blocks H-Ras-induced transformation of NIH-3T3 cells (Chen and Iyengar, 1994) and EGF-induced Raf-1 phosphorylation in rat1 fibroblasts (Wu et al., 1993), whereas cAMP alone is ineffective. In contrast, cAMP/PKA activation promotes synaptic potentiation by BDNF in Xenopusneuromuscular synapses (Boulanger and Poo, 1999) and the early phase of postsynaptic long-term potentiation (LTP) (Blitzer et al., 1995,1998), whereas exogenous cAMP has no effect.
Interestingly, both the positive and negative functions of the cAMP/PKA gate occur in the SCN (Fig.7A,B). Light/GLU activation of the cAMP/PKA gate in early night permits signal transduction through the primary pathway but blocks signal transmission through the same primary signaling pathway in late night. Thus, the clock itself determines the effects of cAMP/PKA activation, and as such, cAMP/PKA contributes contextual information regarding clock state at the time of light exposure.
Fig. 7.

A, A schematic summary of the effects of the cAMP/PKA gate on long-term changes in the SCN neuronal activity rhythm induced by light/GLU. 8-Br-cAMP is ineffective in altering circadian state when applied alone at night. However, raising cAMP enhances GLU-in-duced phase delays in early night but blocks GLU-induced phase advances in late night. In contrast, blocking PKA in the presence of GLU abolishes GLU-induced phase delays in early night but potentiates GLU-induced phase advances in late night. In contrast, inhibition of the MAPK kinase cascade attenuates both GLU-induced phase delays and phase advances by 50%. B, A model depicts interaction of the cAMP/PKA gating pathway with the primary GLU signaling pathway that confers state change in the SCN when activated at night. Nocturnal light activates an NMDA receptor-mediated pathway that leads to liberation of NO in both early and late night. After NO, the pathway leading to circadian phase delay in early night diverges from the pathway that evokes phase advance in late night. However, each pathway eventually leads to activation of mPer1 mRNA and ultimately alters circadian time. The cAMP/PKA gating pathway modulates activity of the main pathway in both early and late night. Activation of cAMP/PKA in the early night opens the gate, thereby permitting signal flow through the main pathway. In contrast, activation of cAMP/PKA in the late night hinders signal transfer through the primary pathway; the cAMP/PKA gate is closed. Thus, the phase of the circadian clock itself is the gatekeeper that determines the downstream consequences of cAMP/PKA activation. The mechanism for activation of cAMP/PKA remains speculative at this time as indicated by the question marks. NMDA receptor-mediated Ca2+ influx may both activate the primary pathway and the cAMP/PKA gate, as occurs during hippocampal LTP (Blitzer et al., 1995). Alternatively, NMDA receptor-mediated Ca2+ influx may prime the system for other extracellular signals, such as PACAP (Chen et al., 1999) or serotonin (Moriya et al., 1998), to activate adenylyl cyclase.
State changes initiated by GLU in both early and late night are mediated by NMDA receptor-mediated Ca2+influx that activates NOS (Ding et al., 1994). Subsequent signaling activates multiple pathways specific to clock state but encompasses parallel, divergent, and convergent elements that induce phase-delaying state change in early night, or phase-advancing state change in late night. Convergent elements may include the MEK/MAPK pathway and CREB. MAPK (Obrietan et al., 1998), CREB phosphorylation (Ginty et al., 1993; Ding et al., 1997; Obrietan et al., 1998), and CRE-mediated gene transcription (Obrietan et al., 1999) have been implicated in light/GLU-induced state changes in the SCN. Activation of MAPK, but not PKA, can substitute for light/GLU to induce CRE-mediated gene transcription in the SCN (Obrietan et al., 1999).
This study provides functional evidence that MAPK signaling contributes to the effects of light/GLU on clock phase in both early and late night. Inhibition of MAPK attenuates both GLU-induced phase shifts by ∼50% (Fig. 4). Although higher concentrations of the MAPK inhibitors were not tested, we predict that 50% is the maximal response. Like KT5720, each of the MAPK inhibitors was used at a concentration two times the IC50. More importantly, these same concentrations of olomoucine and PD98059 block depolarization-induced CREB and Erk phosphorylation in hippocampal slices (Impey et al., 1998). Moreover, MAPK inhibition reduces light-induced phosphorylation of CREB on serine-133 in the SCN by 50% (Obrietan et al., 1998). PKA-dependent nuclear translocation of Erk/MAPK can lead to CREB phosphorylation (Impey et al., 1998).
These data predict that CREB functions as a convergence point, or coincidence detector, within the primary light/GLU-signaling pathway. An unidentified pathway(s) must contribute the other 50% of the light/GLU effect. Although PKA-dependent activation of MAPK can partially account for the signals that cause phase delay, the fact that PKA inhibition increases the magnitude of the phase advance suggests activation of an additional, alternative pathway. Although further studies are necessary to determine whether direct activation of MAPK can substitute for GLU to induce phase resetting at night, the aggregate data place MAPK on the primary pathway leading to long-term state changes in response to light/GLU.
Recent reports support a link between light signaling through GLU, NO, MAPK, and CREB phosphorylation to CRE elements in the mPer1promoter (Obrietan, 2000; Tei et al., 2000). CREB is not likely a direct substrate for cAMP/PKA because nocturnal activation of cAMP/PKA alone in the SCN does not activate CRE-mediated gene transcription (Obrietan et al., 1999). This argues for placing CREB on the primary pathway, independent from the cAMP/PKA gating pathway. CRE elements in the promoter allow tentative placement of mPer1 on the primary pathway because the light/GLU stimulus that inducesmPer1 (Albrecht et al., 1997; Shearman et al., 1997;Shigeyoshi et al., 1997; Zylka et al., 1998; present study) also generates phosphorylated CREB (Ginty et al., 1993; Ding et al., 1997; Obrietan et al., 1998) and stimulates CRE-mediated transcription. Furthermore, antisense oligonucleotides against mPer1completely block both GLU-induced phase delays (Akiyama et al., 1999) and phase advances (Tischkau and Gillette, unpublished observations). Although we cannot exclude placement of mPer1 on the gating pathway in early night, the fact that inhibition of PKA does not inhibit mPer1 in late night eliminates it from a position on the cAMP/PKA gating pathway at that time. Thus, we favor placement ofmPer1 on the primary pathway, downstream of CREB, with intersection of the cAMP/PKA gate upstream of these convergent elements (Fig. 7).
Our results demonstrate that activation of the cAMP/PKA gate, a signaling consequence of adding GLU to the slice, attenuates activity of the primary GLU pathway in the late night. PKA inhibition releases this brake, potentiating GLU-induced phase resetting. However, PKA inhibition did not potentiate the induction of mPer1 in the presence of GLU. These differences may reflect interpretive limitations imposed by experimental design. mPer1 induction was evaluated 60 min after GLU-stimulation of rat SCN slices. In the mouse, light-stimulated mPer1 in vivo is a rapid, transient event that is initiated by 15–30 min in the retinorecipient SCN, peaks at 60 min, and spreads throughout the nucleus by 120 min (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997; Takumi et al., 1998; Zylka et al., 1998). Our data suggest that the kinetics ofmPer1 induction may differ between species and/or may depend on the experimental paradigm. Sixty minutes after light in early night, expression of mPer1 in the rat is not limited to the retinorecipient SCN but has spread throughout the nucleus (Fig.6B). In contrast, 60 min after light in late night,mPer1 is found only in the retinorecipient SCN (Fig. 6, compare B and E). However, consistent with the concept that PKA inhibition removes a brake on the GLU-signaling pathway, mPer1 was expressed throughout the entire SCN (Fig.6, compare F and E) when slices were co-treated with a PKA inhibitor and GLU in late night. PKA inhibition may enhance flow through the signaling pathway, altering the kinetics ofmPer1 induction in response to GLU. A thorough time course may reveal differences in total mPer1 that ultimately reflect the magnitude of the GLU-induce phase response.
Mechanisms for intersection of cAMP/PKA gating pathway with the primary light/GLU pathway are unknown. Because cAMP has opposite effects on the light/GLU-induced signal transduction in early versus late night, the intersection between the cAMP/PKA gating pathway and the primary pathway likely occurs at elements unique to each pathway. For example, it seems unlikely that cAMP/PKA would activate the NMDA receptor in early night and then inhibit this same molecule in late night. Therefore, it is not unreasonable to speculate that the intersection points between the cAMP/PKA gating pathway and the primary signaling pathways lie somewhere downstream of NO, the divergence point (Fig.7B). In the early night, cAMP/PKA must regulate the primary pathway upstream of mPer1, because inhibition of PKA blocksmPer1 induction, i.e., the signal is blocked before it reaches mPer1.
The mechanism for raising cAMP in response to GLU remains unresolved. Gating pathways (Jordan and Iyengar, 1998) can be constitutively active, as in cAMP gating of the Hedgehog signal during development (Blair, 1995; Fan et al., 1995; Jiang and Struhle, 1995, Li et al., 1995; Perrimon, 1995), activated by intracellular signals, as in cAMP gating of early hippocampal LTP (Blitzer et al., 1995), or activated by a second extracellular signal, as in cAMP gating of neuropeptide-induced survival of retinal ganglion cells (Meyer-Franke et al., 1995). In the SCN, cAMP is not constitutively elevated. Peak levels occur near the end of subjective day and near the end of subjective night; low levels of cAMP are observed throughout the period of sensitivity to light/GLU (Prosser and Gillette, 1991). GLU could activate both the primary pathway and the cAMP/PKA gate.
More likely, GLU activates the primary pathway and then permits activation of the cAMP pathway via a second extracellular signal. Both PACAP (Chen et al., 1999), an extracellular messenger that colocalizes with GLU in the RHT (Hannibal et al., 2000), and the 5-HT1A-specific agonist MKC-242 (Moriya et al., 1998) have the same modulatory effects as cAMP/PKA on light/GLU-induced state changes. Thus, any afferent signal capable of accessing the cAMP/PKA gate in the presence of light/GLU may play a modulatory role. The cAMP/PKA gating pathway may provide contextual information to allow integration of multiple signals that occur coincidentally with light/GLU. Moreover, cAMP/PKA gating may explain discrepancies between the magnitudes of light-induced phase shifts in vivo (as in Fig. 5), where the response reflects integration of information from the RHT, as well as other afferents, and GLU-induced phase shifts in vitro (as in Figs. 2-4), where the response is isolated to the SCN. Elucidating the sites of intersection between the cAMP/PKA gating pathway and the primary signaling pathways stimulated by GLU will provide insights into mechanisms for integrating multiple signals to generate adaptive behavioral state changes.
Footnotes
This research was supported by Public Health Service Grants NS22155, HL67007 (M.U.G), and NS10170 (S.A.T).
Correspondence should be addressed to Dr. Martha U. Gillette, Department of Cell and Structural Biology, University of Illinois, B107 CLSL, 601 S. Goodwin Avenue, Urbana, IL 61801. E-mail:mgillett@uiuc.edu.
REFERENCES
- 1.Abraham RT, Acquarone M, Andersen A, Asensi A, Belle R, Berger F, Bergouniuox C, Brunn G, Buquet-Fagot C, Fagot D. Cellular effects of olomoucine, an inhibitor of cyclin-dependent kinases. Biol Cell. 1995;83:105–120. doi: 10.1016/0248-4900(96)81298-6. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci. 1999;19:1115–1121. doi: 10.1523/JNEUROSCI.19-03-01115.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albrecht U, Sun ZS, Eichele EE, Lee CC. A differential response of two putative mammalian circadian regulators, mPer1 and mPer2, to light. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- 4.Amir S, Robinson B, Edelstein K. Distribution of NADPH-diaphorase staining and light-induced Fos expression in the rat suprachiasmatic nucleus region supports a role for nitric oxide in the circadian system. Neuroscience. 1995;69:545–555. doi: 10.1016/0306-4522(95)00252-e. [DOI] [PubMed] [Google Scholar]
- 5.Bading H, Greenberg ME. Stimulation of protein tyrosine kinase phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 6.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 7.Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- 8.Blair SS. Hedgehog digs up an old friend. Nature. 1995;373:656–657. doi: 10.1038/373656a0. [DOI] [PubMed] [Google Scholar]
- 9.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 10.Blitzer RD, Conner JH, Brown GP, Wong T, Shenoliker S, Iyengar R, Landau EM. Gating of CAMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- 11.Boulanger L, Poo M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- 12.Castel M, Belenky M, Cohen S, Ottersen OP, Storm-Matheson J. Glutamate-like immunoreactivity in retinal terminals of the mouse suprachiasmatic nucleus. Eur J Neurosci. 1993;5:368–381. doi: 10.1111/j.1460-9568.1993.tb00504.x. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Buchanan GF, Ding JM, Hannibal J, Gillette MU. Pituitary adenylyl cyclase-activating peptide: a pivotal modulator of glutamatergic regulation of the suprachiasmatic circadian clock. Proc Natl Acad Sci USA. 1999;96:13468–13473. doi: 10.1073/pnas.96.23.13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Iyengar R. Suppression of Ras-induced transformation of NIH 3T3 cells by activated GαS. Science. 1994;263:1278–1281. doi: 10.1126/science.8122111. [DOI] [PubMed] [Google Scholar]
- 15.Colwell CS, Menaker M. NMDA as well as non-NMDA receptor antagonists can prevent the phase-shifting effects of light on the circadian system of the golden hamster. J Biol Rhythms. 1992;7:125–136. doi: 10.1177/074873049200700204. [DOI] [PubMed] [Google Scholar]
- 16.De Vries MJ, Nunes Cardozo B, van der Want J, de Wolf A, Meijer JH. Glutamate immunoreactivity in terminals of the retinohypothalamic tract of the brown Norwegian rat. Brain Res. 1993;612:231–237. doi: 10.1016/0006-8993(93)91665-f. [DOI] [PubMed] [Google Scholar]
- 17.Ding JM, Chen D, Weber ET, Faiman LE, Rea MA, Gillette MU. Resetting the biological clock: mediation of nocturnal phase shifts by glutamate and NO. Science. 1994;266:1713–1717. doi: 10.1126/science.7527589. [DOI] [PubMed] [Google Scholar]
- 18.Ding JM, Hurst WJ, Faiman LE, Kuriashkina LR, Gillette MU. Resetting the biological clock: mediation of nocturnal CREB phosphorylation through light, glutamate and nitric oxide. J Neurosci. 1997;17:667–675. doi: 10.1523/JNEUROSCI.17-02-00667.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding JM, Buchanan GF, Tischkau SA, Chen D, Kuriashkina L, Faiman LE, Alster JM, McPherson PS, Campbell KP, Gillette MU. A neuronal ryanodine receptor mediates light-induced phase delays of the circadian clock. Nature. 1998;394:381–384. doi: 10.1038/28639. [DOI] [PubMed] [Google Scholar]
- 20.Fan C-M, Porter JA, Chiang DT, Beachy PA, Tessier-Lavigne M. Long range sclerotome induction by Sonic Hedgehog: direct role of the amino-terminal cleavage product and modulation by the cAMP signaling pathway. Cell. 1995;81:457–465. doi: 10.1016/0092-8674(95)90398-4. [DOI] [PubMed] [Google Scholar]
- 21. Gillette MU. Cellular regulators of circadian timing. The regulation of sleep Borbely AA, Hayaishi O, Sejnowski T, Altman J. 2000. Human Frontier Science Program 10th Anniversary Workshop, Strasbourg, FR, June, 1999.
- 22.Gillette MU, Prosser RA. Circadian rhythm of the rat suprachiasmatic brain slice is rapidly reset by daytime application of cAMP analogs. Brain Res. 1988;474:348–352. doi: 10.1016/0006-8993(88)90449-0. [DOI] [PubMed] [Google Scholar]
- 23.Gillette MU, Reppert SM. The hypothalamic suprachiasmatic nuclei: circadian patterns of vasopressin secretion and neuronal activity in vitro. Brain Res Bull. 1987;19:135–139. doi: 10.1016/0361-9230(87)90176-6. [DOI] [PubMed] [Google Scholar]
- 24.Gillette MU, Tischkau SA. Suprachiasmatic nucleus: the brain's circadian clock. Rec Prog Hormone Res. 1999;54:33–59. [PubMed] [Google Scholar]
- 25.Gillette MU, Medanic M, McArthur AJ, Liu C, Ding JM, Faiman LE, Weber ET, Tcheng TK, Gallman EA. Circadian clocks and their adjustment (Ciba Foundation Symposium 183) Wiley; Chichester, UK: 1995. Intrinsic neuronal rhythms in the suprachiasmatic nuclei and their adjustment. pp. 134–153. [DOI] [PubMed] [Google Scholar]
- 26.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 27.Greengard P, Jen J, Nairn AC, Stevens CF. Enhancement of the glutamate response by cAMP-dependent protein kinase in hippocampal neurons. Science. 1991;253:1135–1138. doi: 10.1126/science.1716001. [DOI] [PubMed] [Google Scholar]
- 28.Hannibal J, Ding JM, Chen D, Fahrenkrug J, Larsen PJ, Gillette MU, Mikkelsen JD. Pituitary adenylate cyclase-activating peptide (PACAP) in the retinohypothalamic tract: a potential daytime regulator of the biological clock. J Neurosci. 1997;17:2637–2644. doi: 10.1523/JNEUROSCI.17-07-02637.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hannibal J, Moller M, Ottersen OP, Fahrenkrug J. PACAP and glutamate are co-stored in the retinohypothalamic tract. J Comp Neurol. 2000;418:147–155. [PubMed] [Google Scholar]
- 30.Impey S, Obrietan K, Wang ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 31.Iyengar R. Gating by cyclic AMP: expanded role for an old signaling pathway. Science. 1996;271:461–463. doi: 10.1126/science.271.5248.461. [DOI] [PubMed] [Google Scholar]
- 32.Jiang J, Struhle G. Protein kinase and hedgehog signaling in Dros-ophila limb development. Cell. 1995;80:563–572. doi: 10.1016/0092-8674(95)90510-3. [DOI] [PubMed] [Google Scholar]
- 33.Johnson RF, Moore RY, Morin LP. Loss of entrainment and anatomical plasticity after lesions of the hamster retinohypothalamic tract. Brain Res. 1988;460:297–313. doi: 10.1016/0006-8993(88)90374-5. [DOI] [PubMed] [Google Scholar]
- 34.Jordan JD, Iyengar R. Modes of interactions between signaling pathways. Biochem Pharmacol. 1998;55:1347–1352. doi: 10.1016/s0006-2952(97)00620-5. [DOI] [PubMed] [Google Scholar]
- 35.King DP, Takahashi JS. Molecular genetics of circadian rhythms in mammals. Annu Rev Neurosci. 2000;23:713–742. doi: 10.1146/annurev.neuro.23.1.713. [DOI] [PubMed] [Google Scholar]
- 36.Klemfuss H, Clopton PL. Seeking tau: a comparison of six methods. J Interdiscip Cycle Res. 1993;24:1–16. [Google Scholar]
- 37.Kultz D, Madhany S, Burg MB. Induction of GADD45 and GADD153 by osmosensing stress-activated protein kinase. J Biol Chem. 1998;273:13645–13651. doi: 10.1074/jbc.273.22.13645. [DOI] [PubMed] [Google Scholar]
- 38.Kuo ML, Yang NC. Reversion of v-H-ras-transformed NIH 3T3 cells by apigenin through inhibiting mitogen activated protein kinase and its downstream oncogenes. Biochem Biophys Res Commun. 1995;212:767–775. doi: 10.1006/bbrc.1995.2035. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Ohlymeyer JT, Lane ME, Kalderon D. Function of protein kinase A in hedgehog signal transduction and Drosophila imaginal disc development. Cell. 1995;80:553–562. doi: 10.1016/0092-8674(95)90509-x. [DOI] [PubMed] [Google Scholar]
- 40.Mathur A, Golombek DA, Ralph MR. cGMP-dependent kinase inhibitors block light-induced phase advances of hamster circadian rhythms in vivo. Am J Physiol. 1996;270:R1031–R1036. doi: 10.1152/ajpregu.1996.270.5.R1031. [DOI] [PubMed] [Google Scholar]
- 41.Medanic M, Gillette MU. Serotonin regulates the phase of the rat suprachiasmatic circadian pacemaker in vitro only during the subjective day. J Physiol (Lond) 1992;450:629–642. doi: 10.1113/jphysiol.1992.sp019147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 43.Mintz EM, Marvel CL, Gillespie CF, Price KM, Albers HE. Activation of NMDA receptors in the suprachiasmatic nucleus produces light-like phase shifts of the circadian clock in vivo. J Neurosci. 1999;19:5124–5130. doi: 10.1523/JNEUROSCI.19-12-05124.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore RY, Lenn NJ. A retinohypothalamic projection in the rat. J Comp Neurol. 1972;146:1–14. doi: 10.1002/cne.901460102. [DOI] [PubMed] [Google Scholar]
- 45.Moriya T, Yoshinobu Y, Yokota S, Akiyama M, Shibata S. Potentiating action of MKC-242, a selective 5-HT1A receptor agonist, on the photic entrainment of the circadian activity rhythm in hamsters. Br J Pharmacol. 1998;125:1281–1287. doi: 10.1038/sj.bjp.0702176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obrietan K (2000) Circadian regulation of cAMP response element-mediated expression in the SCN. Meeting of the Society for Research on Biological Rhythms, Amelia Island, FL, May.
- 47.Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- 48.Obrietan K, Impey S, Smith D, Athos J, Storm DR. Circadian regulation of cAMP response element-mediated gene expression in the suprachiasmatic nuclei. J Biol Chem. 1999;274:17748–17756. doi: 10.1074/jbc.274.25.17748. [DOI] [PubMed] [Google Scholar]
- 49.Perrimon N. Hedgehog and beyond. Cell. 1995;80:517–520. doi: 10.1016/0092-8674(95)90503-0. [DOI] [PubMed] [Google Scholar]
- 50.Pickard GE. The afferent connections of the suprachiasmatic nucleus of the golden hamster with emphasis on the retinohypothalamic connection. J Comp Neurol. 1982;211:65–83. doi: 10.1002/cne.902110107. [DOI] [PubMed] [Google Scholar]
- 51.Prosser RA, Gillette MU. The mammalian circadian clock in the suprachiasmatic nuclei is reset in vitro by cAMP. J Neurosci. 1989;9:1073–1081. doi: 10.1523/JNEUROSCI.09-03-01073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prosser RA, Gillette MU. Cyclic changes in cAMP concentration and phosphodiesterase activity in a mammalian circadian clock studied in vitro. Brain Res. 1991;568:234–238. doi: 10.1016/0006-8993(91)91396-i. [DOI] [PubMed] [Google Scholar]
- 53.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 54.Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF, Reppert SM., Jr Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 55.Shibata S, Tsuneyoshi A, Hamada T, Tominaga K, Watanabe S. Phase-resetting effect of 8-OH-DPAT, a serotonin 1A receptor agonist, on the circadian rhythm of firing rate in the rat suprachiasmatic nuclei in vitro. Brain Res. 1992;582:353–356. doi: 10.1016/0006-8993(92)90156-4. [DOI] [PubMed] [Google Scholar]
- 56.Shibata S, Watanabe A, Hamada T, Tominaga K, Watanabe S. N-methyl-d-aspartate induces phase shifts in circadian rhythm of neuronal activity of rat SCN in vitro. Am J Physiol. 1994;267:R360–R364. doi: 10.1152/ajpregu.1994.267.2.R360. [DOI] [PubMed] [Google Scholar]
- 57.Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 58.Shirakawa T, Moore RY. Glutamate shifts the phase of the circadian neuronal firing rhythm in the rat suprachiasmatic nucleus in vitro. Neurosci Lett. 1994;178:47–50. doi: 10.1016/0304-3940(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 59.Sun ZS, Albrecht U, Zhuchenko O, Bailey J, Eichele G, Lee CC. RIGUI, a putative mammalian ortholog of the Drosophila period gene. Cell. 1997;19:1003–1011. doi: 10.1016/s0092-8674(00)80366-9. [DOI] [PubMed] [Google Scholar]
- 60.Takumi T, Matsubara C, Shigeyoshi Y, Taguchi K, Yagita K, Maebayashi Y, Sakikida Y, Okumura K, Takashima N, Okamura H. A new mammalian period gene predominantly expressed in the suprachiasmatic nucleus. Genes Cells. 1998;3:167–176. doi: 10.1046/j.1365-2443.1998.00178.x. [DOI] [PubMed] [Google Scholar]
- 61.Tei H, Numano R, Sakaki Y, Yamazaki S, Menaker M (2000) Transgenic analysis of a mammalian clock gene,Per1. Meeting of Society for Research on Biological Rhythms Abstr 132.
- 62. Tischkau SA, Barnes JA, Lin F-J, Myers EM, Barnes JW, Meyer-Bernstein EL, Hurst WJ, Burgoon PW, Chen D, Sehgal A, Gillette MU. Oscillation and light induction of timeless mRNA in the mammalian circadian clock. J Neurosci 19 1999. RC15 (1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weber ET, Gannon RL, Rea MA. cGMP-dependent protein kinase inhibitor blocks light-induced phase advances of hamster circadian rhythms in vivo. Neurosci Lett. 1995;197:227–230. doi: 10.1016/0304-3940(95)11961-u. [DOI] [PubMed] [Google Scholar]
- 64.Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, Sturgill TW. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science. 1993;262:1065–1069. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- 65.Zylka MJ, Shearman LP, Weaver DR, Reppert SM. Three period homologs in mammals: differential light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of the brain. Neuron. 1998;20:1103–1110. doi: 10.1016/s0896-6273(00)80492-4. [DOI] [PubMed] [Google Scholar]