

Abstract
In mesial temporal lobe epilepsy (mTLE), the predominant form of epilepsy in adults, and in animal models of the disease, there is a conspicuous loss of the intracellular Ca2+-binding protein calbindin-D28k (CB) from granule cells (GCs) of the dentate gyrus. The role of this protein in nerve cell function is controversial, but here we provide evidence for its role in controlling Ca2+ influx into human neurons. In patients with Ammon's horn sclerosis (AHS), the loss of CB from GCs markedly increased the Ca2+-dependent inactivation of voltage-dependent Ca2+ currents (ICa), thereby diminishing Ca2+ influx during repetitive neuronal firing. Introducing purified CB into GCs restored Ca2+current inactivation to levels observed in cells with normal CB content harvested from mTLE patients without AHS. Our data are consistent with the possibility of neuroprotection secondary to the CB loss. By limiting Ca2+ influx through an enhanced Ca2+-dependent inactivation of voltage-dependent Ca2+ channels during prolonged neuronal discharges, the loss of CB may contribute to the resistance of surviving human granule cells in AHS.
Keywords: Ammon's horn sclerosis, mesial temporal lobe epilepsy, granule cells, voltage-dependent Ca2+ currents, calcium channels, calbindin-D28k
Ammon's horn sclerosis (AHS), present in approximately two-thirds of patients with mesial temporal lobe epilepsy (mTLE) (Blümcke et al., 1999), is characterized by a devastating loss of hippocampal neurons, comprising the hilus, and the CA1, CA3, and CA4 regions, whereas dentate gyrus granule cells (GCs) are conspicuously more resistant to the damage (Margerison and Corsellis, 1966; Blümcke et al., 1999). This selective loss of some, and sparing of other, neurons remains puzzling. Work on mTLE and animal models of limbic epilepsy has suggested that the neuronal damage may result from excessive and sustained epileptic seizures (Sagar and Oxbury, 1987; Meldrum, 1993; Wasterlain, 1997; Jackson et al., 1998). If frequent and prolonged seizures can kill nerve cells, then a likely mechanism for the ensuing neuronal loss is the activity-dependent accumulation of cytoplasmic Ca2+ entering the cells through voltage- and ligand-gated Ca2+-permeable channels, or being released from intracellular stores (Miller, 1991; Ghosh and Greenberg, 1995;Berridge, 1998). Indeed, neurons tightly control the cytoplasmic concentration of free Ca2+ because it regulates critical cellular events ranging from signal transduction cascades and induction of gene transcription to neuronal death (Ghosh and Greenberg, 1995; Berridge, 1998).
Numerous cellular changes have been considered to be associated with AHS (Blümcke et al., 1999). Specific molecular events such as altered GABAA receptor subunits (Brooks-Kayal et al., 1998) or the loss of the neuronal Ca2+-binding protein calbindin-D28k (CB) (Baimbridge and Miller, 1984) occur even before the generalization of seizures. Of the known cellular changes, the loss of CB in the surviving dentate gyrus granule cells of human mTLE patients (Maglóczky et al., 1997), also present in several animal models of the disease (Miller and Baimbridge, 1983;Baimbridge and Miller, 1984; Baimbridge et al., 1985; Shetty and Turner, 1995; Yang et al., 1997), gives the closest clue to an altered Ca2+ homeostasis in these mTLE-affected neurons. The role of CB in nerve cells is not well understood (Baimbridge et al., 1992). Many reports (Sloviter, 1989; Lledo et al., 1992; Cheng et al., 1994) consider its presence in neurons to convey resistance against excessive Ca2+-dependent neuronal damage. A contrasting view, based on recent findings in CB knock-out animals (CB−/−) (Klapstein et al., 1998) is that its absence may actually alleviate Ca2+-dependent neuronal damage. The discovery of a Ca2+-dependent promoter for CB (Arnold and Heintz, 1997), and the loss of CB from granule cells in several animal models of TLE (Miller and Baimbridge, 1983;Baimbridge and Miller, 1984; Köhr et al., 1991; Shetty and Turner, 1995; Yang et al., 1997) raises the possibility that levels of CB may be regulated in a Ca2+-dependent manner subsequent to changes in neuronal excitability. In the kindling model, the CB loss has been suggested to serve a neuroprotective role, by limiting the amount of Ca2+ entry during prolonged action potential firing (Köhr and Mody, 1991). We directly addressed the regulation of Ca2+ entry into human hippocampal neurons that survived AHS associated with mTLE. A reduced Ca2+ entry, compared to that found in cells with no CB loss obtained from nonsclerotic hippocampi, would be consistent with the loss of CB and the ensuing diminished Ca2+ entry having saved the granule cells from devastation in AHS. The availability of recombinant CB made possible its infusion into the very neurons that lost it during the course of the disease, thus directly addressing its role in the control of neuronal Ca2+ entry. We have exploited the unique opportunities provided by our epilepsy surgery programs (Engel, 1993; Schwartzkroin, 1993) to perform concomitant morphological and physiological analyses on human hippocampi resected from mTLE patients.
MATERIALS AND METHODS
Patient subjects. All patients had therapy-refractory mTLE and were under a full antiepileptic drug regimen at the time of surgery (Engel, 1993). Clinical data were not significantly different between the AHS and the lesion group (Table1). Hippocampal control specimens were obtained at autopsy from two individuals without any history of neurological or psychiatric disease (cause of death: respiratory insufficiency caused by severe pneumonia and cardiogenic shock caused by myocardial infarction, respectively). Informed consent was obtained from all patients for additional histopathological and electrophysiological evaluation. All procedures were approved by the respective ethics committees of the University of Bonn Medical Center and the UCLA Human Subject Protection (protocol HSPC no. 93-11-642-11) and conform to standards set by the Declaration of Helsinki (1989).
Table 1.
Clinical data on epileptic patients
| Code | Age and gender | Clinical history in addition to TLE | Duration | Seizure type | Diagnosis | No. of cells |
|---|---|---|---|---|---|---|
| A1 | 50 f | Perinatal hypoxia | 48 | SGS, CPS | AHS | 4 |
| A2 | 19 f | Complicated febrile seizure | 14 | SPS, CPS, SGS | AHS | 1 |
| A3 | 23 f | — | 23 | SPS, CPS | AHS | 3 |
| A4 | 42 m | Febrile seizures, meningitis | 35 | SPS, CPS | AHS | 3 |
| A5 | 19 f | Migraine, febrile seizures, mild hemiparesis | 5 | CPS, SGS | AHS | 4 |
| A6 | 32 f | Febrile seizures | 32 | SPS, CPS, SGS | AHS | 4 |
| A7 | 38 f | — | 5 | CPS, SGS | AHS | 1 |
| A8 | 42 m | IDDM, diabetic PNP, paranoid psychosis | 34 | CPS, SGS | AHS | 1 |
| A9 | 41 m | — | 11 | SPS, CPS, SGS | AHS | 6 |
| A10 | 38 m | Meningitis with febrile seizure | 38 | SPS, CPS, SGS | AHS | 1 |
| A11 | 32 m | Febrile seizure | 24 | CPS, SGS | AHS | 4 |
| A12 | 32 f | — | 25 | CPS, SGS | AHS | 2 |
| A13 | 22 f | Tuberculous meningitis | 14 | SPS, CPS, SGS | Postencephalitic scars | 3 |
| A14 | 42 f | — | 8 | CPS | AHS | 5 |
| A15 | 28 m | Multiple seizure disorders | 9 | CPS | Mild HS, cryptogenic | 3 |
| A16 | 52 f | Schizophrenia, psychosis | 52 | CPS | AHS | 5 |
| A17 | 49 f | — | 43 | CPS | AHS | 2 |
| L1 | 33 m | Pilocytic Astrocy-toma (WHO grade 1) | 25 | CPS, GMS | Multiple GNH | 1 |
| L2 | 37 m | Contusio cerebri (17 yrs) | 19 | CPS | Multiple glial scars | 1 |
| L3 | 10 m | One-sided spastic hemiparesis | 2 | CPS, SGS | Polymicro-gyria | 1 |
| L4 | 33 f | Partly resected angioma left temporal (22 y) | 11 | CPS | Arterio-venous malformation | 3 |
| L5 | 32 f | Proencephalic cyst, iron deficiency anaemia, hemiparesis | 29 | CPS, SGS | Glial scars | 2 |
| L6 | 42 m | Perinatal hypoxia | 27 | SPS, CPS, SGS | GNH | 2 |
| L7 | 25 f | — | 14 | SPS, CPS, SGS | Ganglio-glioma | 1 |
| L8 | 49 f | — | 24 | SPS, CPS | Dysembryoblastic neuroepithelial tumor | 3 |
m, Male; f, female; IDDM, insulin-dependent diabetes mellitus; PNP, polyneuropathy; SPS, simple partial seizures; CPS, complex partial seizures; SGS, secondary generalized seizures; AHS, Ammon's horn sclerosis; GNH, glio-neuronal hamartia; HS, hippocampal sclerosis; TLE, temporal lobe epilepsy.
All subjects were adults and suffered from medically intractable epilepsy. Patients were grouped into two classes according to histopathological diagnosis of either Ammon's horn sclerosis (A) or a lesion (L) in the temporal lobe but not in the hippocampus proper.
Immunohistochemistry and quantification of CB-immunoreactive GCs. Immunohistochemistry for CB was performed on 4-μm-thick paraffin-embedded sections of the hippocampi obtained from the patients whose cells were used for patch-clamp recordings. In addition, hippocampal control specimens were obtained at autopsy from two individuals without any history of neurological or psychiatric disease. All specimens were processed in a single batch according to standard methods. Specimens were stained with a primary antibody (1:1000; anti-calbindin-D28K; Swant, Bellinzona, Switzerland) overnight at 4°C. Binding of the primary antibody was detected by the avidin—biotin–complex peroxidase method (ABC Elite; Vector Laboratories, Burlingame, CA), using 3,3′-diaminobenzidine (ICN Biochemicals, Cleveland, OH) as a chromogen. Sections were lightly counterstained with hematoxylin, dehydrated, and mounted. Control experiments with omission of primary antibodies, as well as substitution of the primary antibody by equivalent dilutions of nonimmune rabbit IgG serum (Dako, Glostrup, Denmark), showed no immunoreaction product. For quantitative analysis of CB immunoreactivity, sections were scanned at a magnification of 1000× using a Vanox microscope (Olympus, Tokyo, Japan), a CCD video camera (Sony, Tokyo, Japan), and the IP Lab imaging analysis software (Signal Analytics Corporation, Vienna, Austria) on a MacIntosh computer (7100/66; Apple). Optical densities (OD) were recorded as values on a gray scale ranging from 0 (black) to 255 (white) for 10 randomly selected GC bodies, each within three defined regions of interest (superior blade, inferior blade, and apex of the dentate gyrus) per specimen. ODs of the alveus devoid of immunoreaction product served as reference values. Within each specimen, ODs determined for the GC bodies were subtracted from the reference value.
Preparation of acutely isolated dentate granule cells.Isolated hippocampal granule cells were prepared as described previously (Beck et al., 1997a,b, 1999; Nägerl and Mody, 1998). Human hippocampal specimens were placed in ice-cold artificial CSF (ACSF) immediately after surgical removal. Coronal slices, 400-μm-thick, were prepared from the corpus of the hippocampus with a vibratome and transferred to a storage chamber with warmed (32°C) ACSF. After an equilibration period of 60 min, enzymatic digestion was performed for 25 min with 2–3 mg/ml pronase (protease type XIV; Sigma, St. Louis, MO). The dentate gyrus was dissected under a binocular microscope (Zeiss, Oberkochen, Germany) and triturated in 2 ml of trituration solution with fire-polished glass pipettes. The cell suspension was then placed in a Petri dish for subsequent patch-clamp recordings. Only neurons with ovoid somata and a single dendrite reminiscent of granule cell morphology in situ were included in this study. The isolated cells were superfused with an extracellular solution containing (in mm): tetraethylammonium chloride (TEA) 140, 4-aminopyridine (4-AP) 5, CaCl2 5, glucose 10, and HEPES 10, and tetrodotoxin (TTX) 1 μm (chemicals obtained from Sigma and Fluka, Buchs, Switzerland). The osmolarity was adjusted to 283 mOsm with sucrose.
Patch-clamp whole-cell recording. Patch pipettes were made from borosilicate glass capillaries (outer diameter, 1.5 mm; inner diameter, 1 mm; Science Products, Hofheim, Germany or Garner Glass) with a resistance of 2–3 MΩ. The pipettes were filled with an intracellular solution containing (in mm): Cs-methanesulfonate 80, TEA 20, MgCl2 5, HEPES 10, ATP 10 and GTP 0.2, pH 7.4, CsOH. The osmolarity was adjusted to 275–280 mOsm with sucrose in all intracellular solutions. In some experiments, CB (500–1300 ng/μl) was included in the intracellular solution. For this purpose, lyophilized CB (Swant; Dr. K. G. Baimbridge, University of British Columbia) was reconstituted with bidistilled water, followed by buffer replacement with intracellular solution using Bio-Spin Chromatography Columns (Bio-Gel P-6; Bio-Rad, Munich, Germany) according to the supplier's recommendation. Homodimers of CB were not present in the intracellular solution, as demonstrated by subjecting 3 μg of CB (Swant) reconstituted in bidistilled water as well as 3 μg of CB purified by a Bio-Spin Chromatography Column (Bio-Rad) to 10% SDS-PAGE. Visualization of proteins on gels by Coomassie blue showed a single band at ∼28 kDa (data not shown). For the recordings including CB in the intracellular solution, the tip of the electrode was dipped briefly (1 sec) into CB-free intracellular solution and back-filled with CB-containing solution. Recordings performed with different intracellular solutions were interleaved in individual patients to minimize effects caused by interpatient variability. Tight-seal whole-cell recordings were obtained at room temperature (21–24°C) (Beck et al., 1997a,b, 1999; Nägerl and Mody, 1998). Membrane currents were recorded using the EPC9 patch-clamp amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany) and the TIDA acquisition and analysis program (HEKA Elektronik). To generate AP waveforms for voltage-clamp commands, a single action potential was recorded from GCs using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) in the fast current-clamp mode and the WinWCP Strathclyde Electrophysiology Software version 2.1 (courtesy of Dr. J. Dempster, University of Strathclyde). It was Bessel-filtered (8-pole) at 5 kHz, digitized at 20 kHz, and subsequently concatenated to produce a 1 sec 108 Hz AP train. The train was digital-to-analog-converted with an update rate of 10 kHz (PCI-MIO E16–4; National Instruments, Austin, TX) and applied as a voltage command. To minimize capacitive artifacts, all current traces were linear leak-subtracted according to the P/4 protocol (Bezanilla and Armstrong, 1977) or using the EPC9 leak subtraction protocol. Series resistance was compensated by 60–80%. The maximal residual voltage error estimated by multiplying the maximal Ca2+ current amplitude with the effective series resistance after compensation did not exceed 3 mV. A liquid junction potential of −9.7 mV was calculated between the intracellular and extracellular solution with the generalized Henderson liquid junction potential equation, as described (Barry and Lynch, 1991). This value was subtracted fromVh. All results were expressed as mean values ± SEM. Differences were proven with a Mann–WhitneyU Wilcoxon rank test, with the significance level set to 0.05 and denoted with asterisks in the figures.
RESULTS
Distribution of CB in the hippocampi of mTLE patients
We have obtained hippocampal specimens for electrophysiological and morphological investigations from mTLE patients. The patients were divided into two distinct groups (see Table 1 for clinical patient data). The first group comprised patients with a histopathological diagnosis of AHS characterized by severe neuronal loss in the CA1, CA3, and CA4 subfields and relative sparing of CA2 and the dentate gyrus (AHS group; Margerison and Corsellis, 1966). In this group, most cells in the granule cell layer were devoid of CB immunoreactivity (Fig.1C). The second group consisted of patients without AHS but with lesions in the temporal lobe (lesion group) that did not involve the hippocampus proper. In this group, GCs show a pattern of CB immunoreactivity comparable to that of control biopsy specimens stained under identical conditions (Fig.1A,B). The cumulative probability plot (Fig.1D) of the OD of the immunoreaction product from single GC bodies quantitatively illustrates this dichotomy in hippocampal CB content. The average density of the CB immunoreactivity over the cells was: AHS, 4.1 ± 1.2; lesion, 41.1 ± 9.3; and control, 56.5 ± 6.1.
Fig. 1.
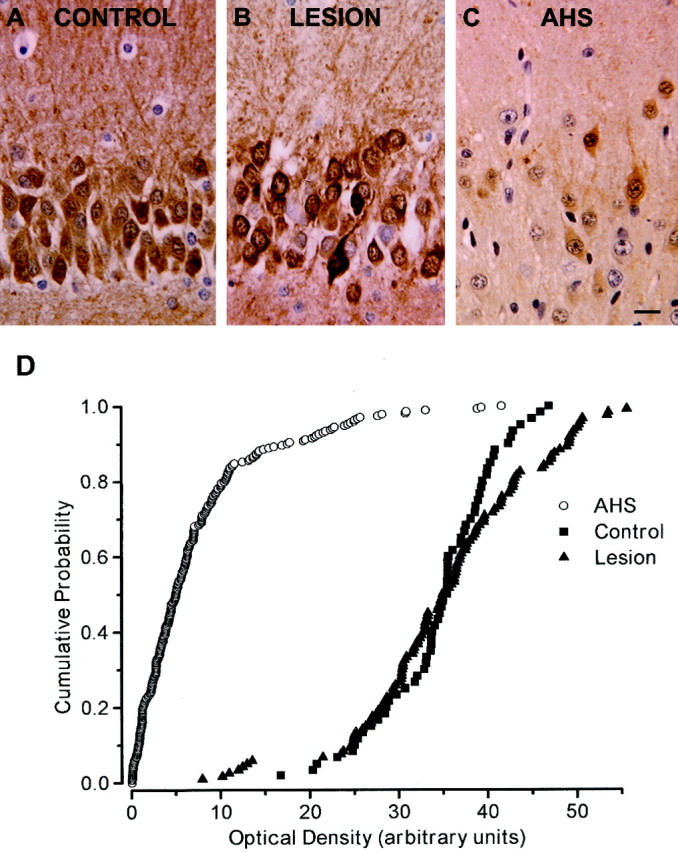
Immunohistochemistry for CB in dentate granule cells. All three photographs represent the granule cell layer with the granule cell bodies (stained dark for CB in A andB). Above the granule cells, the inner molecular layer is visible, whereas below the granule cell layer, part of the polymorphic cell layer of the dentate gyrus can be seen.A, In autopsy control hippocampi, the majority of dentate granule cells were intensely stained for CB. B,Specimens obtained from mTLE patients of the lesion group showed pronounced immunoreactivity in most dentate granule cells, corresponding well to the immunoreactivity pattern seen in the autopsy specimens. C, In contrast, AHS specimens displayed very faint or no CB immunoreactivity in the majority of dentate granule cells. Only a few neurons were immunopositive for CB. Sections are counterstained with hematoxylin. Scale bar, 10 μm. D,Cumulative probability plots of OD in hippocampal sections from autopsy control (filled triangles), lesion (filled circles), and AHS (open circles) patients as a measure for CB immunoreactivity. Each symbol represents the referenced OD for a single GC body.
Increased inactivation of ICa in human neurons lacking CB
Hippocampal specimens were obtained at surgery, and GCs were isolated according to standard protocols (Beck et al., 1996, 1997a,1999; Nägerl and Mody, 1998). In parallel, the specimens were also classified based on CB immunohistochemistry. As a quantitative measure for the Ca2+-dependent inactivation, we recorded voltage-dependent Ca2+ currents in the whole-cell configuration (Fig. 2A) using double-pulse experiments. A U-shaped relationship between the command voltage of the conditioning prepulse (P1; Fig. 2B, inset) and theICa amplitude evoked by a test pulse (P2) delivered 50 msec later is indicative of a Ca2+-dependentICa inactivation process. This amplitude directly measures the level of inactivation produced by previous Ca2+ entry (Eckert and Chad, 1984). Relating the peak amplitude ofICa evoked by single voltage pulses to the rate of its decay represents an alternative measure for the Ca2+ dependency ofICa inactivation, which we have used previously to characterize the enhancedICa inactivation in GCs obtained from AHS mTLE patients (Nägerl and Mody, 1998; Beck et al., 1999). According to the double-pulse experiments, Ca2+-dependent inactivation is considerably more pronounced in the AHS group (Fig.2A), as measured by the ratios of the test pulseICa amplitude and the peakICa evoked by P2. The minimal ratios were observed at +10 mV P1 voltage for the AHS group (0.59 ± 0.08,n = 25) and at +20 mV for the lesion group (0.81 ± 0.07, n = 14, p < 0.005; Fig.2B). The decay time constants of theICa in the three experimental conditions were (in msec): AHS, 52.7 ± 5.5; lesion, 172.6 ± 94; and AHS + CB, 146.2 ± 73.6.
Fig. 2.

Ca2+-dependentICa inactivation as measured by double-pulse experiments. A, Representative traces illustrating the inactivation of Ca2+ currents evoked by P2 after previous influx of Ca2+ during a 100 msec prepulse (P1, marked byasterisk) in GCs isolated from a patient with AHS and a patient with lesion-associated epilepsy. Note the more rapid Ca2+ current inactivation during P1 in patients with AHS. B, Peak current amplitudes during the test pulse (P2) were normalized to their maximal amplitudes, averaged separately for the lesion group (filled circles; n = 14) and the AHS group (open circles; n = 25), and plotted versus the command voltage of the 100 msec prepulse. The bottom panel shows the normalized amplitudes of the Ca2+ current during the conditioning prepulse P1.
We used nifedipine to estimate the contribution of L-type Ca2+ channels to the whole-cellICa inactivation. Bath application of nifedipine blocked up to 45.1 ± 3.7% (n = 4) of the ICa amplitude and diminished inactivation in AHS GCs from a ratio of 0.59 ± 0.08 (n = 25) to 0.88 ± 0.12 (n = 4) (data not shown). This indicates that most of the observed inactivation can be ascribed to L-type Ca2+ channels and that >75% of the L-type current inactivates after a 100 msec depolarization.
ICa inactivation after infusion of purified CB into AHS neurons devoid of CB
Next, we tested whether introduction of purified CB into granule cells of patients with AHS restores the Ca2+-dependent inactivation to levels seen in the lesion group. Indeed, inclusion of 500 ng/μl (∼20 μm) of purified CB into the intracellular solution (Fig.3A, bottom panel) substantially reduced Ca2+-dependent inactivation measured by double-pulse protocols compared to recordings without CB obtained in a GC from the same patient (Fig. 3B, top panel). Figure 3C demonstrates the effect of CB on ICa in a single cell as CB gradually diffuses into the cell after patch rupture. Recordings with (n = 6) and without (n = 7) CB were alternated to ensure that the effects of CB were not caused by interpatient variability. When the normalized ratios of Ca2+ current amplitudes during P2 were averaged, the presence of purified CB in the patch pipette considerably reduced the inactivation (0.75 ± 0.05, n = 6) compared to the corresponding recordings without CB (0.57 ± 0.05, n = 7; Fig.3D). This effect of CB was comparable to those obtained with including the synthetic Ca2+ buffer BAPTA (5 mm; inactivation ratio 0.85 ± 0.07,n = 5) into the patch pipette.
Fig. 3.

The effect of 500 ng/μl (∼20 μm) CB infused into GCs lacking CB that were obtained from patients with AHS. A, Representative traces illustrating Ca2+-dependent inactivation of Ca2+ currents in GCs from patients with AHS with and without purified CB included in the intracellular solution. Note that the Ca2+ current inactivation becomes slower after the intracellular dialysis of CB. B,Ca2+-dependent inactivation was measured as in Figure 2B. Averages of three neurons treated with CB (gray circles) and four neurons without CB (open circles) from one individual patient are shown.C, Time course of the effect of intracellular dialysis of CB into one GC on ICa inactivation at 5, 11, and 25 min after patch rupture. D, Bar graph showing the minimal ratios as calculated in Figure 2B for the four experimental conditions: AHS (n = 32), lesion (n = 14), AHS + CB (n = 6), and AHS + BAPTA (5 mm, n = 5). Recordings with different intracellular solutions were interleaved to minimize interpatient variability. Asterisks indicate significant differences (Mann–Whitney U Wilcoxon rank test, p < 0.05).
CB controls the Ca2+ entry induced by a train of action potentials
Using trains of action potential (AP) waveforms in the whole-cell voltage-clamp mode, we mimicked physiological Ca2+ entry during high-frequency neuronal firing to probe possible functional consequences of the CB-dependent Ca2+ current inactivation in mTLE GCs. Intracellular CB perfusion via the patch pipette into GCs from AHS patients gradually slowed the rate of decay of the Ca2+ entry evoked by AP trains. To quantify the AP-dependent Ca2+ entry, theICa associated with the last and the first AP in the train were integrated to calculate the total charge. The respective charge ratios were taken as a measure of the effect of CB on spike train-induced Ca2+ entry. As shown in Figure 4B, the charge ratio of a 1 sec 108 Hz train is significantly increased to 0.50 ± 0.02 (n = 4; p < 0.005) in AHS GCs after CB infusion (1300 ng/μl at 50 μm) from 0.37 ± 0.02 (n = 4) in CB-deficient GCs (Fig. 4A), whereas the total charge associated with the first AP of the train did not differ under the two conditions (0.191 ± 0.006 mC/F with CB, 0.195 ± 0.036 mC/F without CB, total charge normalized to cell capacitance). The infusion of the synthetic Ca2+chelator BAPTA (5 mm) instead of CB also increased the charge ratio to 0.52 ± 0.03 (n = 5;p < 0.005 compared to AHS GCs), an effect indistinguishable from that of CB.
Fig. 4.

Ca2+ entry during action potential trains is reduced in the absence of CB. A,Representative Ca2+ currents evoked by a 1 sec train of action potential waveforms at 108 Hz are shown. In CB-deficient GCs the total amount of Ca2+ entry per AP waveform decreases from the first (AP1) to the last AP (AP100) by ∼75% (seeinset). B, Reintroduction of 1300 ng/μl (∼50 μm) purified CB into GCs via the patch pipette prevents the decrease of total Ca2+ entry during an AP train. The difference in integrated charge between the last AP and the first is only ∼50% (see inset).
DISCUSSION
We have shown that by controlling the Ca2+-dependent inactivation of the high-voltage-activated L-type Ca2+current, CB can regulate the amount of Ca2+ entry into human epileptic CNS neurons. Two mTLE patient groups identified on the basis of hippocampal cell loss also diverged with regard to the CB content of dentate gyrus granule cells. These two patient groups provided us with the opportunity to record from human neurons with reduced and normal CB levels. The group with severe hippocampal cell loss (AHS) showed a marked reduction in granule cell CB levels (Maglóczky et al., 1997), an enhanced Ca2+ current inactivation (Nägerl and Mody, 1998; Beck et al., 1999), and a reduced Ca2+ entry after repetitive firing. Granule cells in nonsclerotic hippocampi (lesion group) displayed normal levels of CB and moderate Ca2+ current inactivation. A similar correlation of CB loss in GCs and altered Ca2+ current inactivation is found in an experimental animal model of TLE (kindling; Köhr and Mody, 1991;Köhr et al., 1991). We have now demonstrated the involvement of CB in altered Ca2+ current inactivation, because the reduced Ca2+ entry in AHS granule cells could be reversed by directly introducing CB into the neurons.
The Ca2+-dependent inactivation of voltage-gated Ca2+ channels is a key factor in limiting neuronal Ca2+ entry. If CB is to play an active role in the control of this process, it must either interact directly with the channel or it has to reach sufficiently high levels around the point Ca2+of entry to effectively buffer the entering Ca2+ ions. Interestingly, the Ca2+-binding protein calmodulin was recently shown to be the Ca2+ sensor responsible for inactivation (Lee et al., 1999; Levitan, 1999; Qin et al., 1999; Zuhlke et al., 1999). Because both CB and calmodulin bind Ca2+ to EF hand structures with possibly similar kinetics and affinities, it is conceivable that the estimated cytoplasmic CB concentrations of ∼100 μm (Baimbridge et al., 1992) are normally sufficient to disrupt the negative Ca2+ feedback on the channel. However, the relatively low concentrations of infused CB sufficient in our experiments to restore the inactivation of Ca2+ are consistent with a close physical association or even an interaction between CB and the Ca2+ channels. Another EF-hand Ca2+-binding protein, sorcin, has been shown to tightly associate with the α1 subunits of the cardiac and sarcoplasmic L-type Ca2+ channels (Meyers et al., 1998). At this time, there is no evidence for a similar association between CB any Ca2+ channel subunit, but recent evidence indicates a considerable fraction of CB to be found in the plasma membrane (Winsky and Kuznicki, 1995).
Regardless of the manner how CB controls the inactivation of Ca2+ entry through voltage-gated Ca2+ channels, the critical question is whether the loss of CB can lead to neuroprotection. Albeit counterintuitive, such an idea is consistent with the enhanced neuroprotection after ischemic insults in CB−/− mice (Klapstein et al., 1998). In addition to the Ca2+dependent inactivation of Ca2+ channels, some of the cellular mechanisms involved in neuroprotection in the CB−/− mice may include enhanced activation of Ca2+ dependent K+ channels and changes in synaptic facilitation. Nevertheless, most Ca2+entry into neurons takes place when voltage-gated Ca2+ channels open during APs. Repetitive AP firing such as that occurring during seizures may thus exacerbate Ca2+ entry unless there is a Ca2+-dependent negative feedback. Our experiments using action potential waveforms have shown that even at high frequencies, the first few action potentials in the train will produce comparable Ca2+ entry regardless of the presence or absence of CB. But in CB-deficient cells, the reduced Ca2+ entry during action potentials at the end of the train may be viewed as a use-dependent block of Ca2+ influx, turning to be gradually more pronounced as the duration of neuronal firing gets longer or its frequency becomes higher. If vulnerable neurons in AHS die directly from an excessive Ca2+ entry caused by prolonged high-frequency activity during seizures, then the protective mechanism against large Ca2+loads provided by the loss of CB may indeed be responsible for the preservation of granule cells. Conversely, one would predict that granule cells that did not lose CB would be more prone to cell death in AHS patients. Yet, Maglóczky et al. (1997) did not report any differences at the EM level between CB-deficient and CB-containing GCs. Nevertheless, the timing or mechanism of cell death in CB-positive neurons in AHS may preclude the identification of the neurodegenerative process in ultrastructural studies.
Presently, it is unknown what mechanisms control the decrease of CB levels in AHS but not in lesion-associated epilepsy. In neurons, the synthesis of CB is not vitamin D-dependent like in the periphery (Baimbridge et al., 1992), but the CB gene has a Ca2+-dependent promoter (Arnold and Heintz, 1997) that may regulate CB synthesis, depending on the amount of neuronal activity. Interestingly, the only study examining the levels of CB mRNA when protein levels were clearly reduced in kindled animals found no change in the amount of message (Sonnenberg et al., 1991). This lack of change in CB mRNA in kindling raises the possibility of an enhanced activity-dependent degradation of CB.
Our findings also show that in spite of the protection of GCs against neuronal injury in AHS, the loss of granule cell CB did not prevent mTLE. In the future, the impact of the CB loss from GCs on the function of the dentate gyrus will have to be considered in the context of the entire hippocampal network. Nevertheless, for the first time in adult human neurons, we have directly demonstrated the role of CB in regulating Ca2+ influx in surviving cells of the sclerotic hippocampus. Thus, our findings demonstrate a strong link between cellular CB levels and the intracellular Ca2+ homeostasis, providing further insight into the relationship between cellular CB content and neuronal vulnerability.
Footnotes
This study was supported by an American Epilepsy Foundation predoctoral fellowship to U.V.N., National Institutes of Health/ National Institute of Neurological Disorders and Stroke (NINDS) Grant NS 36141 and the Coelho Endowment to I.M., and grants from BONFOR 111/2, SFB 400, and DFG EL 122/7–1 to H.B. and C.E.E. The human tissue was kindly provided by Dr. Itzak Fried (Division of Neurosurgery, UCLA School of Medicine) and Dr. Masako Isokawa (Brain Research Institute, UCLA School of Medicine), members of the NINDS Program Project “A Clinical Research Program for the Partial Epilepsies” (Dr. Jerome Engel Jr, Program Director, Department of Neurology, UCLA School of Medicine) and Drs. Schramm, Zentner, and van Roost (Department of Epileptology, University of Bonn Medical Center). We thank Dr. K. G. Baimbridge (Department of Physiology, University of British Columbia) for supplying recombinant human CB.
Correspondence should be addressed to Istvan Mody, Department of Neurology RNRC 3-155, UCLA School of Medicine, 710 Westwood Plaza, Los Angeles, CA 90095-1769. E-mail: mody@ucla.edu.
REFERENCES
- 1.Arnold DB, Heintz N. A calcium responsive element that regulates expression of two calcium binding proteins in Purkinje cells. Proc Natl Acad Sci USA. 1997;94:8842–8847. doi: 10.1073/pnas.94.16.8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–308. doi: 10.1016/0166-2236(92)90081-i. [DOI] [PubMed] [Google Scholar]
- 3.Baimbridge KG, Miller JJ. Hippocampal calcium-binding protein during commissural kindling-induced epileptogenesis: progressive decline and effects of anticonvulsants. Brain Res. 1984;324:85–90. doi: 10.1016/0006-8993(84)90624-3. [DOI] [PubMed] [Google Scholar]
- 4.Baimbridge KG, Mody I, Miller JJ. Reduction of rat hippocampal calcium-binding protein following commissural, amygdala, septal, perforant path, and olfactory bulb kindling. Epilepsia. 1985;26:460–465. doi: 10.1111/j.1528-1157.1985.tb05681.x. [DOI] [PubMed] [Google Scholar]
- 5.Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. J Membr Biol. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- 6.Beck H, Blümcke I, Kral T, Clusmann H, Schramm J, Wiestler OD, Heinemann U, Elger CE. Properties of a delayed rectifier potassium current in dentate granule cells isolated from the hippocampus of patients with chronic temporal lobe epilepsy. Epilepsia. 1996;37:892–901. doi: 10.1111/j.1528-1157.1996.tb00043.x. [DOI] [PubMed] [Google Scholar]
- 7.Beck H, Steffens R, Heinemann U, Elger CE. Properties of voltage-activated Ca2+ currents in acutely isolated human hippocampal granule cells. J Neurophysiol. 1997a;77:1526–1537. doi: 10.1152/jn.1997.77.3.1526. [DOI] [PubMed] [Google Scholar]
- 8.Beck H, Clusmann H, Kral T, Schramm J, Heinemann U, Elger CE. Potassium currents in acutely isolated human hippocampal dentate granule cells. J Physiol (Lond) 1997b;498:73–85. doi: 10.1113/jphysiol.1997.sp021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck H, Steffens R, Heinemann U, Elger CE. Ca2+-dependent inactivation of high-threshold Ca2+ currents in hippocampal granule cells of patients with chronic temporal lobe epilepsy. J Neurophysiol. 1999;82:946–954. doi: 10.1152/jn.1999.82.2.946. [DOI] [PubMed] [Google Scholar]
- 10.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 11.Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blümcke I, Beck H, Lie AA, Wiestler OD. Molecular neuropathology of human mesial temporal lobe epilepsy. Epilepsy Res. 1999;36:205–223. doi: 10.1016/s0920-1211(99)00052-2. [DOI] [PubMed] [Google Scholar]
- 13.Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- 14.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 15.Eckert R, Chad JE. Inactivation of Ca channels. Prog Biophys Mol Biol. 1984;44:215–267. doi: 10.1016/0079-6107(84)90009-9. [DOI] [PubMed] [Google Scholar]
- 16.Engel J., Jr . Surgical treatment of the epilepsies. Raven; New York: 1993. [Google Scholar]
- 17.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 18.Jackson GD, McIntosh AM, Briellmann RS, Berkovic SF. Hippocampal sclerosis studied in identical twins. Neurology. 1998;51:78–84. doi: 10.1212/wnl.51.1.78. [DOI] [PubMed] [Google Scholar]
- 19.Klapstein GJ, Vietla S, Lieberman DN, Gray PA, Airaksinen MS, Thoenen H, Meyer M, Mody I. Calbindin-D28k fails to protect hippocampal neurons against ischemia in spite of its cytoplasmic calcium buffering properties: evidence from calbindin-D28k knockout mice. Neuroscience. 1998;85:361–373. doi: 10.1016/s0306-4522(97)00632-5. [DOI] [PubMed] [Google Scholar]
- 20.Köhr G, Mody I. Endogenous intracellular calcium buffering and the activation/inactivation of HVA calcium currents in rat dentate gyrus granule cells. J Gen Physiol. 1991;98:941–967. doi: 10.1085/jgp.98.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köhr G, Lambert CE, Mody I. Calbindin-D28K (CaBP) levels and calcium currents in acutely dissociated epileptic neurons. Exp Brain Res. 1991;85:543–551. doi: 10.1007/BF00231738. [DOI] [PubMed] [Google Scholar]
- 22.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 23.Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- 24.Lledo PM, Somasundaram B, Morton AJ, Emson PC, Mason WT. Stable transfection of calbindin-D28k into the GH3 cell line alters calcium currents and intracellular calcium homeostasis. Neuron. 1992;9:943–954. doi: 10.1016/0896-6273(92)90246-a. [DOI] [PubMed] [Google Scholar]
- 25.Maglóczky Z, Halász P, Vajda J, Czirják S, Freund TF. Loss of calbindin-D28k immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76:377–385. doi: 10.1016/s0306-4522(96)00440-x. [DOI] [PubMed] [Google Scholar]
- 26.Margerison JH, Corsellis JA. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- 27.Meldrum BS. Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 1993;3:405–412. doi: 10.1111/j.1750-3639.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- 28.Meyers MB, Puri TS, Chien AJ, Gao T, Hsu PH, Hosey MM, Fishman GI. Sorcin associates with the pore-forming subunit of voltage-dependent L-type Ca2+ channels. J Biol Chem. 1998;273:18930–18935. doi: 10.1074/jbc.273.30.18930. [DOI] [PubMed] [Google Scholar]
- 29.Miller JJ, Baimbridge KG. Biochemical and immunohistochemical correlates of kindling- induced epilepsy: role of calcium binding protein. Brain Res. 1983;278:322–326. doi: 10.1016/0006-8993(83)90264-0. [DOI] [PubMed] [Google Scholar]
- 30.Miller RJ. The control of neuronal Ca2+ homeostasis. Prog Neurobiol. 1991;37:255–285. doi: 10.1016/0301-0082(91)90028-y. [DOI] [PubMed] [Google Scholar]
- 31.Nägerl UV, Mody I. Calcium-dependent inactivation of high-threshold calcium currents in human dentate gyrus granule cells. J Physiol (Lond) 1998;509:39–45. doi: 10.1111/j.1469-7793.1998.039bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagar HJ, Oxbury JM. Hippocampal neuron loss in temporal lobe epilepsy: correlation with early childhood convulsions. Ann Neurol. 1987;22:334–340. doi: 10.1002/ana.410220309. [DOI] [PubMed] [Google Scholar]
- 34.Schwartzkroin PA. Basic research in the setting of an epilepsy surgery center. In: Engel J Jr, editor. Surgical treatment of the epilepsies. Raven; New York: 1993. pp. 755–773. [Google Scholar]
- 35.Shetty AK, Turner DA. Intracerebroventricular kainic acid administration in adult rat alters hippocampal calbindin and non-phosphorylated neurofilament expression. J Comp Neurol. 1995;363:581–599. doi: 10.1002/cne.903630406. [DOI] [PubMed] [Google Scholar]
- 36.Sloviter RS. Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol. 1989;280:183–196. doi: 10.1002/cne.902800203. [DOI] [PubMed] [Google Scholar]
- 37.Sonnenberg JL, Frantz GD, Lee S, Heick A, Chu C, Tobin AJ, Christakos S. Calcium binding protein (calbindin-D28k) and glutamate decarboxylase gene expression after kindling induced seizures. Brain Res Mol Brain Res. 1991;9:179–190. doi: 10.1016/0169-328x(91)90001-e. [DOI] [PubMed] [Google Scholar]
- 38.Wasterlain CG. Recurrent seizures in the developing brain are harmful. Epilepsia. 1997;38:728–734. doi: 10.1111/j.1528-1157.1997.tb01244.x. [DOI] [PubMed] [Google Scholar]
- 39.Winsky L, Kuznicki J. Distribution of calretinin, calbindin D28k, and parvalbumin in subcellular fractions of rat cerebellum: effects of calcium. J Neurochem. 1995;65:381–388. doi: 10.1046/j.1471-4159.1995.65010381.x. [DOI] [PubMed] [Google Scholar]
- 40.Yang Q, Wang S, Hamberger A, Celio MR, Haglid KG. Delayed decrease of calbindin immunoreactivity in the granule cell- mossy fibers after kainic acid-induced seizures. Brain Res Bull. 1997;43:551–559. doi: 10.1016/s0361-9230(97)00006-3. [DOI] [PubMed] [Google Scholar]
- 41.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]