

Abstract
Plasmin is converted from its zymogen plasminogen by tissue type or urokinase type plasminogen activator (PA) and degrades many components of the extracellular matrix (ECM). To explore the possibility that the PA–plasmin system regulates synaptic plasticity, we investigated the effect of plasmin on degradation of ECM and synaptic plasticity by using organotypic hippocampal cultures. High-frequency stimulation produced long-term potentiation (LTP) in control slices, whereas the potentiation was induced but not maintained in slices pretreated with 100 nm plasmin for 6 hr. The baseline synaptic responses were not affected by pretreatment with plasmin. The impairment of LTP maintenance was not observed in slices pretreated with 100 nm plasmin for 6 hr, washed, and then cultured for 24–48 hr in the absence of plasmin. To identify substrates of plasmin, the expression of three major components of ECM, laminin, fibronectin, and type IV collagen, was investigated by immunofluorescence imaging. The three ECM components were widely distributed in the hippocampus, and only laminin was degraded by plasmin pretreatment. The expression level of laminin returned to normal levels when the slices were cultured for 24–48 hr after washout of plasmin. Furthermore, preincubation with anti-laminin antibodies prevented both the degradation of laminin and the impairment of LTP maintenance by plasmin. These results suggest that the laminin-mediated cell–ECM interaction may be necessary for the maintenance of LTP.
Keywords: plasmin, long-term potentiation, hippocampus, laminin, extracellular matrix, synaptic plasticity, organotypic culture
Long-term potentiation (LTP) in the hippocampus is one form of synaptic plasticity and is thought to be a cellular mechanism underlying learning and memory (Bliss and Collingridge, 1993). LTP is induced by high-frequency stimulation, and requires activation of NMDA type of glutamate receptors and consequent calcium entry into the postsynaptic spines, at least in the Schaffer collateral–CA1 pyramidal cell synapses (Nicoll and Malenka, 1995). Synaptic plasticity is recently considered to be related to the structural modification of postsynaptic regions (Geinisman, 1993; Buchs and Müller, 1996) and may be regulated by the interaction between cells and the extracellular matrix (ECM). Indeed, it has been reported that hippocampal LTP is regulated by several molecules involved in the cell–cell or cell–ECM interaction, i.e., integrin (Stäubli et al., 1990, 1998; Xiao et al., 1991; Bahr et al., 1997), cadherin (Tang et al., 1998), neural cell adhesion molecule, L1 (Lüthi et al., 1994; Rønn et al., 1995; Müller et al., 1996), andN-syndecan (Lauri et al., 1999).
Tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) convert the ubiquitous zymogen plasminogen to plasmin (Vassalli et al., 1991; Plow et al., 1995), which in turn functions to degrade ECM components (Werb, 1997). Although the PA–plasmin system is well known to be involved in the fibrinolysis of the blood clot, accumulating evi- dence suggests that this system also functions in the CNS. First, PAs are synthesized in most brain regions (Sappino et al., 1993). The localized expression of PAs in the spinal cord and migrating granule cells during neuronal development suggests that plasmin-mediated proteolysis facilitates neurite outgrowth and cell migration (Sumi et al., 1992; Ware et al., 1995). Second, tPA-deficient mice have been demonstrated to be resistant to excitotoxin-induced neuronal degeneration in the hippocampus and have an elevated threshold for seizure (Tsirka et al., 1995, 1997; Chen and Strickland, 1997), indicating that degradation of ECM by the PA–plasmin system is a critical event in the excitotoxic neuronal death. Third, tPA has been demonstrated also to be induced as an immediate-early gene accompanying seizure, kindling, or LTP, and might contribute to structural changes observed during activity-dependent synaptic plasticity (Qian et al., 1993). Fourth, mice overexpressing uPA showed impaired learning (Meiri et al., 1994). tPA-deficient mice showed a reduction in the maintenance of LTP (Frey et al., 1996; Huang et al., 1996), and mice overexpressing tPA showed an enhanced LTP (Madani et al., 1999). Furthermore, it has been reported recently that application of tPA during tetanic stimulation enhanced the late phase of LTP in rat hippocampal slices (Baranes et al., 1998). We have demonstrated previously that application of plasmin during tetanic stimulation facilitated the induction of LTP (Mizutani et al., 1996,1997), suggesting that plasmin promotes synaptic plasticity as an acute effect. However, it remained unknown how chronic treatment with plasmin affects LTP and whether the effect of plasmin on LTP is associated with ECM. Therefore, in the present study, we investigated the effect of chronic treatment with plasmin on LTP and the expression level of ECM by using organotypic hippocampal cultures, which is suitable for chronic application of drugs (Gähwiler et al., 1997).
MATERIALS AND METHODS
Chemicals and antibodies. Plasmin, α2-antiplasmin, fibronectin, rabbit anti-laminin antibody (antigen, laminin-1), fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG antibody, and peroxidase-conjugated anti-rabbit IgG antibody were purchased from Sigma (St. Louis, MO). Rabbit anti-fibronectin antibody was from Calbiochem-Novabiochem (La Jolla, CA). Rabbit anti-type IV collagen antibody was from LSL (Tokyo, Japan). Anti-mouse IgG antibody was from Amersham (Buckinghamshire, UK). Laminin (laminin-1) was from Upstate Biotechnology (New York, NY), and type IV collagen was from Biomedical Technologies (Stoughton, MA).
Organotypic cultures. Organotypic hippocampal cultures were prepared according to the interface method (Stoppini et al., 1991). Brains were rapidly removed from 8-d-old Wistar rats (SLC, Shizuoka, Japan), and 300-μm-thick horizontal entorhino-hippocampal slices were cut using a microslicer (DTK-1500; Dosaka EM, Kyoto, Japan). The slices were maintained in cold (4°C) and oxygenated (95% O2–5% CO2) Gey's balanced salt solution supplemented with 6.5 mg/ml glucose and placed on the transparent polytetrafluoroethylene membrane (Millicell-CM; Millipore, Bedford, MA) with 1.0 ml of culture medium consisting of 25% HBSS, 25% donor horse serum, and 50% minimum essential medium (MEM) (Life Technologies, Grand Island, NY) supplemented with 6.5 mg/ml glucose, 50 U/ml penicillin G potassium, and 100 μg/ml streptomycin sulfate. The slices were cultured at 37°C in a moist 5% CO2 atmosphere, and the medium was changed every 3.5 d.
Drug treatment. The hippocampal slices were used for experiments after being cultured for 10–15 d. The culture medium was replaced with serum-free culture medium (HBSS/MEM, 1:1) containing each drug or antibody.
Electrophysiological recordings. Part of the transparent membrane including the slice was cut out with a knife and transferred to a recording chamber in which the slice was continuously perfused with warmed (30°C) and oxygenated (95% O2–5% CO2) artificial CSF (ACSF) at a rate of 2.0 ml/min. ACSF had the following composition (in mm): NaCl 127, KCl 1.6, KH2PO4 1.24, MgSO4 1.3, CaCl2 2.4, NaHCO3 26, and glucose 10. To remove thoroughly the culture medium containing drugs, the slice was allowed to be perfused at least for 30 min before recording. The Schaffer collaterals were stimulated with a bipolar electrode, and the evoked field EPSP (fEPSP) was extracellularly recorded from the stratum radiatum of the CA1 region with a glass capillary microelectrode filled with 0.9% NaCl. A rectangular pulse of 50 μsec duration (20–40 μA) was delivered every 30 sec with an intensity that evoked a fEPSP of 50–60% of the maximum. LTP was induced by high-frequency stimulation (100 pulses at 100 Hz, twice at an interval of 30 sec). The rising slope of fEPSP was measured to evaluate changes in synaptic transmission.
Immunofluorescence imaging. Slices were washed in PBS at room temperature for 15 min, fixed with 4% paraformaldehyde and 4.5% sucrose in 0.1 m phosphate buffer at 4°C for 30 min, permeabilized with 0.3% Triton-X for 60 min, and stored in PBS containing 10% horse serum at 4°C overnight. Without washing, the slices were incubated with anti-laminin antibody (1:30 dilution), anti-fibronectin antibody (1:20 dilution), or anti-type IV collagen antibody (1:200 dilution) at 4°C for 6 hr, washed in PBS for 15 min, and then incubated with secondary FITC-conjugated anti-rabbit IgG antibody (1:1000 dilution) at 4°C for 3 hr. After washing in PBS for 15 min, immunofluorescence was imaged with a laser scanning confocal system (MRC-600; Bio-Rad, Hercules, CA) equipped with an inverted microscope (Nikon, Tokyo, Japan), an argon ion laser, and a host computer system. The tissue was illuminated with the excitation wave length of 488 nm, and FITC fluorescence images were obtained through a 515 nm bandpass filter using 4× or 60× objectives. A series of 10 μm optical sections was obtained, and all z-series accumulated images were analyzed. To quantify the intensity of FITC fluorescence, the pixel intensity values (0–255) of the images were calculated in each hippocampal area by creating three pixels (82.5 μm square) and expressed as the percentage of the value obtained from the pyramidal cell layer of CA1 in control slices, according to the method described previously (Nakagami et al., 1997).
Western blotting. Five hippocampal slices were solubilized in 0.3 ml of lysis buffer (2% Triton X-100, 0.5m NaCl, 10 mm Tris-HCl, 1 mm phenylmethyl sulfonylfluoride, and 10 mm EDTA, pH 7.4). The homogenates were incubated on ice for 30 min and centrifuged at 10,000 × g at 4°C for 20 min, and the supernatants were collected as samples. Purified laminin-1 was incubated with 1 μmplasmin for 3 hr in 150 mm NaCl and 20 mm Tris-HCl, pH 7.5, at 37°C for 3 hr. Samples were subjected to 6% SDS-PAGE under reducing conditions, followed by transfer onto a polyvinylidene difluoride membrane at 1 mA/cm2 for 45 min at room temperature. The membrane were incubated in PBS containing 0.5% Tween 20 (PBS-T) and 2% bovine serum albumin at room temperature for 1 hr and then with anti-laminin antibody (1:1000 dilution in PBS-T) at 4°C overnight. The membranes were washed in PBS-T for 30 min and further incubated with peroxidase-conjugated anti-rabbit IgG antibody (1:5000 dilution in PBS-T) at room temperature for 1 hr. Immunoreactive proteins were visualized using an enhanced chemiluminescence kit (NEN, Boston, MA).
Statistical analysis. All data are expressed as means ± SEM. Statistical significance was evaluated by ANOVA, followed by Tukey's test. Differences were considered significant ifp < 0.05.
RESULTS
Cultured hippocampal slices were pretreated with 100 nm plasmin for 6 hr. Extrinsic plasmin was washed out before recording of field potentials. Control slices were incubated with medium only for 6 hr. There was no apparent difference between control slices and plasmin-pretreated slices in the waveform of fEPSP (Fig. 1A) or in the size of maximal fEPSP (1.82 ± 0.21 mV, n = 11 vs 1.79 ± 0.27 mV, n = 10) or in paired-pulse facilitation induced by two consecutive stimulations with a 50 msec interval (the ratio of second fEPSP slope to first fEPSP slope; 143.4 ± 4.6%, n = 9 vs 151.5 ± 11.0%,n = 9). Moreover, propidium iodide staining and optical recording (Nakagami et al., 1997) demonstrated that the plasmin pretreatment did not induce any cell death and abnormal excitatory propagation (data not shown). In control slices, application of high-frequency stimulation (100 pulses at 100 Hz, twice at an interval of 30 sec) produced a robust potentiation of fEPSP, which lasted for 60 min (Fig. 1A,B). In plasmin-pretreated slices, the same stimulation produced a potentiation of fEPSP; however, the potentiation gradually declined and returned to the baseline level (Fig. 1A,B). Within-slice comparisons showed that the magnitude of potentiation 25–40 and 45–60 min after high-frequency stimulation was significantly smaller in plasmin-pretreated slices than in control slices (Table 1). To examine whether the effect of plasmin is reversible, the slices were pretreated with 100 nm plasmin for 6 hr, washed with fresh medium, and then cultured for 24–48 hr in the absence of plasmin. In these slices, LTP was normally induced by high-frequency stimulation (Fig. 1B). To confirm whether this action of plasmin was blocked by α2-antiplasmin, slices were pretreated with plasmin and α2-antiplasmin simultaneously. Figure 1C summarizes data of the following three groups: control slices, slices pretreated with 300 nm α2-antiplasmin alone, and slices pretreated with 100 nm plasmin and 300 nm α2-antiplasmin. This concentration of α2-antiplasmin was enough to block effects of plasmin in our previous reports (Mizutani et al., 1996, 1997). Pretreatment with α2-antiplasmin alone did not affect LTP, and α2-antiplasmin blocked the impairment of LTP maintenance by plasmin. The change of basal synaptic responses without high-frequency stimulation were also investigated (Fig. 1D). The baseline responses did not change over a 60 min period, indicating that the decline of LTP observed in plasmin-pretreated slices is not attributable to a decrease of synaptic response during the measurement.
Fig. 1.

The effect of pretreatment with plasmin on LTP in organotypic hippocampal cultures. A, Typical fEPSPs recorded in the stratum radiatum of the CA1 area by stimulating the Schaffer collaterals in control and plasmin-pretreated slices. The fEPSPs immediately before and 60 min after high-frequency stimulation (100 pulses at 100 Hz, twice at an interval of 30 sec) in control and plasmin-pretreated slices are superimposed at the right. Test stimulation was delivered at the time indicated byarrowheads. B, The time course of LTP in control slices (white circles; n = 6), slices pretreated with 100 nm plasmin for 6 hr (black circles; n = 6), and slices cultured for 24–48 hr after washout of plasmin (black squares; n = 5). High-frequency stimulation was applied at 0 min. LTP was plotted as the percentage of baseline fEPSP slope. C, The time course of LTP in control slices (white circles; n = 6), slices pretreated with 300 nm α2-antiplasmin alone for 6 hr (black diamonds; n = 4), and slices pretreated with 100 nm plasmin and 300 nmα2-antiplasmin for 6 hr (black triangles;n = 5). The same data from control slices are shown in B and C. D, Baseline fEPSPs without application of high-frequency stimulation in control (white circles; n = 4) and plasmin-pretreated slices (black circles;n = 4).
Table 1.
Summary of the effect of pretreatment with plasmin, α2-antiplasmin or both on the time course of LTP
| fEPSP slope (%) | |||
|---|---|---|---|
| 5–20 min | 25–40 min | 45–60 min | |
| Control | 197.9 ± 10.2 | 203.2 ± 11.9 | 207.9 ± 15.3 |
| 100 nm plasmin | 181.0 ± 4.3 | 160.9 ± 7.1* | 132.6 ± 6.6** |
| Washout | 172.2 ± 8.6 | 190.0 ± 9.3 | 185.9 ± 8.61-167 |
| 300 nm α2-antiplasmin | 201.5 ± 7.5 | 214.4 ± 6.8 | 202.4 ± 5.9 |
| 100 nmplasmin + 300 nm α2-antiplasmin | 204.9 ± 15.3 | 218.4 ± 10.61-167 | 183.3 ± 9.81-167 |
Experimental protocol was as in Figure 1. The average percentage of fEPSP slope 5–20, 25–40, or 45–60 min after high-frequency stimulation was calculated in each slice.
* p < 0.05, **p < 0.01 versus control slices;
F1-167: p < 0.01 versus plasmin-pretreated slices (n = 4–6).
We hypothesized that the maintenance of LTP requires some components of ECM, which are degraded by plasmin. The expression of three major ECM components, laminin, fibronectin, and type IV collagen, was analyzed by using immunofluorescence technique. Laminin was highly expressed in the pyramidal cell layer of the hippocampus and in the blood vessels of the hippocampus and the entorhinal cortex (Fig.2A). High expression of laminin was also observed in the medial site of the hippocampus, which possibly reflects the residue of dura mater. The expression level of laminin in the dentate gyrus was lower than that in CA1 and CA3 areas. When the anti-laminin antibodies were preabsorbed with laminin, there was no detectable fluorescence, confirming that this antibody specifically recognizes laminin (Fig. 2B). In the slices pretreated with plasmin, laminin was decreased in all hippocampal areas, including the blood vessels (Fig. 2C). However, in the slices cultured for 24–48 hr after washout of plasmin, laminin appeared again (Fig. 2D). The expression pattern of laminin was examined in more detail in the CA1 region. In control slices, laminin was expressed as “somatic form” at the site peripheral to the somatic membrane of the pyramidal cells (Fig.2E). In plasmin-pretreated slices, “punctate form” laminin was found exclusively (Fig. 2F). Fibronectin and type IV collagen were also expressed in all hippocampal areas (Fig.3A,B). Preabsorption of anti-fibronectin and anti-type IV collagen antibodies abolished the fluorescence completely (Fig.3C,D). However, neither fibronectin nor type IV collagen was affected by pretreatment with plasmin (Fig.3E,F). Quantitative data are shown in Figure 4. Laminin was significantly degraded by pretreatment with plasmin and reappeared in slices cultured after washout of plasmin (Fig. 4A). These disappearances and reappearances of hippocampal laminin were observed in all areas of the stratum oriens, the pyramidal cell layer, and the stratum radiatum. Similar results were obtained in CA3 and the dentate gyrus (data not shown). We also tested whether degradation of laminin by plasmin is blocked by α2-antiplasmin. Pretreatment with α2-antiplasmin alone did not show any effect on the expression of laminin but completely blocked the degradation of laminin by plasmin (Fig. 4A). Fibronectin (Fig. 4B) and type IV collagen (Fig.4C) were not degraded by pretreatment with plasmin. The time course of laminin degradation by plasmin is shown in Figure4D. The pretreatment with 100 nm plasmin for 6 hr was sufficient to degrade laminin.
Fig. 2.

The expression of laminin in organotypic hippocampal cultures. Immunofluorescence imaging of laminin in control slices (A), slices preabsorbed with laminin (B), slices pretreated with 100 nmplasmin for 6 hr (C), and slices cultured for 24–48 hr after washout of plasmin (D). Laminin was observed in control slices as the somatic form (E,white arrowheads) and in plasmin-pretreated slices as the punctate form (F, black arrowheads).Arrows in E and F indicate the blood vessels. SO, Stratum oriens;PL, pyramidal cell layer; SR, stratum radiatum. The immunofluorescence intensity is gray-coded as shown by the scale in F. Scale bars: A–D, 500 μm; E, F, 50 μm.
Fig. 3.

The expression of fibronectin (A,C, E) and type IV collagen (B, D, F) in organotypic hippocampal cultures. Immunofluorescence imaging of fibronectin and type IV collagen in control slices (A,B), slices preabsorbed with fibronectin or type IV collagen (C, D), and slices pretreated with 100 nm plasmin for 6 hr (E,F). The immunofluorescence intensity is gray-coded as shown by the scale in F. Scale bar, 500 μm.
Fig. 4.
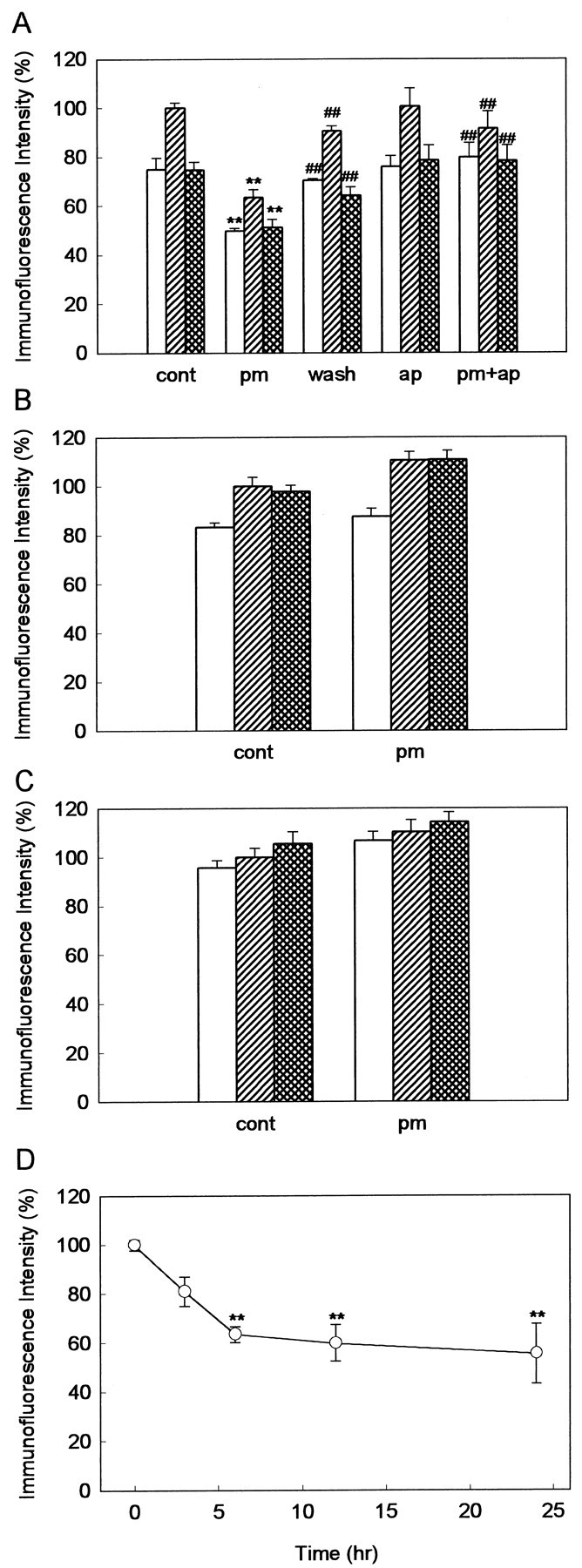
Quantitative analysis of the expression level of laminin (A), fibronectin (B), and type IV collagen (C) in the stratum oriens (open columns), the pyramidal cell layer (hatched columns), and the stratum radiatum (cross-hatched columns) of the CA1 area. cont, Control slices;pm, slices pretreated with plasmin; wash, slices cultured for 24–48 hr after washout of plasmin;ap, slices pretreated with α2-antiplasmin alone, pm+ap, slices pretreated with plasmin and α2-antiplasmin. There was no significant difference between the cont and apgroups. **p < 0.01 versus control slices; ##p < 0.01 versus plasmin-pretreated slices;n = 4–6. D, The time course of laminin degradation during plasmin treatment in the CA1 pyramidal cell layer. Results are expressed as the percentage of the value immediately before addition of plasmin (time 0). **p < 0.01 versus the value at time 0; n = 4–6.
Finally, we hypothesized that the impairment of LTP maintenance (Fig.1B) results from the degradation of laminin by plasmin. To prove this hypothesis, it is necessary to test whether the impairment of LTP maintenance is rescued by preventing the degradation of laminin. For this purpose, we used anti-laminin antibodies, which are expected to bind to laminin and protect laminin from the proteolytic action of plasmin. The slices were preincubated with the antibody (1:30 dilution) for 6 hr, pretreated with 100 nm plasmin for 6 hr in the presence of the antibody, and then washed by continuous perfusion with ACSF in a recording chamber for at least 30 min. Then, the slices were used for immunofluorescence imaging and electrophysiological recording. Indeed, immunofluorescence analysis showed that plasmin failed to degrade laminin in the presence of the anti-laminin antibody that we chose (Fig. 5A). Irrelevant anti-mouse IgG antibody did not affect the degradation of laminin by plasmin (Fig. 5A). The specificity of the anti-laminin antibody was also confirmed by Western blot analysis. Both the α1 chain (440 kDa) and β1/γ1 chain (220 kDa) of purified laminin-1 were degraded by plasmin (Fig. 5B, lanes 1, 2). As reported previously (Chen and Strickland, 1997), weak α1 chain and manifest β1/γ1 chain were detected in extracts of cultured hippocampal slices (Fig.5B, lane 3), and both were degraded by plasmin (Fig. 5B, lane 4). Preincubation with the anti-laminin antibody alone did not affect the expression level of laminin but protected laminin from the degradation by plasmin (Fig.5B, lanes 5, 6). Then, we tested whether the impairment of LTP maintenance by plasmin is rescued by preincubation with the anti-laminin antibody. To examine whether the antibodies are absent or remain in the slices during field potential recordings, some slices were fixed, incubated with FITC-conjugated anti-rabbit IgG antibody, and used for immunofluorescence analysis. The fluorescence intensity in the slices was ∼30% of that in slices without perfusion with ACSF (data not shown), indicating that part of the antibodies still remains during field potential recordings. However, the slices preincubated with the anti-laminin antibodies alone exhibited normal LTP, supporting that a small amount of the antibodies remaining in the slices has no effect on LTP (Fig. 5C). In the slices pretreated with plasmin in the presence of the anti-laminin antibody, LTP was normally induced and maintained (Fig.5C).
Fig. 5.
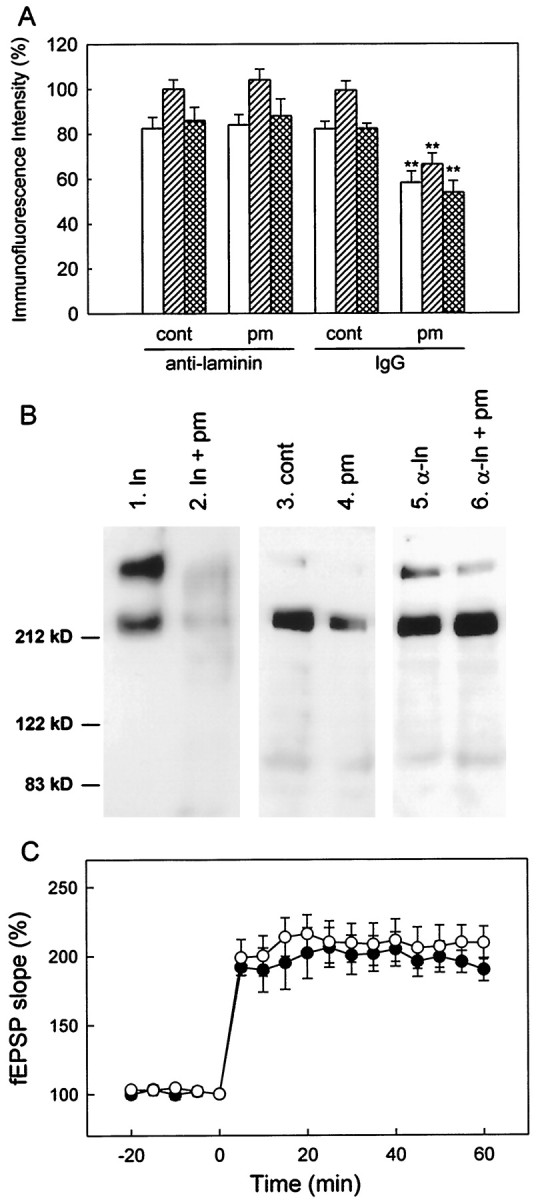
Influence of preincubation with anti-laminin antibodies on the effect of plasmin pretreatment. A, The expression of laminin in the stratum oriens (open columns), the pyramidal cell layer (hatched columns), and the stratum radiatum (cross-hatched columns) of the CA1 area. cont/anti-laminin, Control slices preincubated with anti-laminin antibodies for 6 hr;pm/anti-laminin, slices preincubated with anti-laminin antibodies for 6 hr and then pretreated with 100 nm plasmin for 6 hr; cont/IgG, control slices preincubated with irrelevant anti-mouse IgG antibodies for 6 hr; pm/IgG, slices preincubated with irrelevant anti-mouse IgG antibodies and then pretreated with 100 nm plasmin for 6 hr. **p < 0.01 versus cont/IgG;n = 6–8. B, Western blot analysis of laminin. Lane 1, Purified laminin (0.1 μg).Lane 2, Purified laminin (0.1 μg) incubated with 1 μm plasmin at 37°C for 3 hr. Lane 3, Control slices. Lane 4, Slices pretreated with plasmin.Lane 5, Control slices preincubated with anti-laminin anti body. Lane 6, Slices preincubated with anti-laminin antibody and then pretreated with plasmin.C, The time course of LTP in control slices preincubated with anti-laminin antibodies (white circles;n = 6) and slices preincubated with anti-laminin antibodies and then pretreated with 100 nm plasmin for 6 hr (black circles; n = 6).
DISCUSSION
In the present study, pretreatment with 100 nm plasmin for 6 hr impaired the maintenance of LTP in organotypic hippocampal cultures. Although plasmin has been reported to promote excitotoxic neuronal death in the hippocampus in vivo (Chen and Strickland, 1997), basal synaptic responses were stable in plasmin-pretreated slices, as well as in control slices, supporting that the decline of LTP is not caused by a cell damage possibly induced by plasmin pretreatment. There was no difference in the size or waveform of fEPSP between control and plasmin-pretreated slices, indicating that basal properties of synaptic excitation are not altered by the plasmin pretreatment. Although we have reported previously that GABA receptor-mediated inhibition is blocked by acute application of plasmin (Mizutani et al., 1996, 1997), it should be noted that exogenous plasmin was washed out before field potential recording in our present study. Spontaneous epileptiform potentials, which may appear accompanying a decrease in GABAergic inhibition, were not observed in plasmin-pretreated slices (data not shown). Paired-pulse facilitation, which is affected by changes in GABAergic inhibition (Nathan et al., 1990), was the same in control and plasmin-pretreated slices. In addition, NMDA receptor-dependent, short-term (<30 min) potentiation was normally induced in plasmin-pretreated slices. Therefore, the function of GABA or NMDA receptors is unlikely to be impaired by the plasmin pretreatment, although it remains possible that NMDA receptor function is slightly changed to the extent that it does not affect short-term potentiation. The effect of plasmin pretreatment appears to be specific for the mechanism involved in the maintenance of LTP. We considered ECM components as the possible target of plasmin. Immunofluorescence analysis demonstrated that laminin, but not fibronectin or type IV collagen, was degraded by this pretreatment with plasmin in organotypic hippocampal cultures. The result is consistent with a previous in vivo observation that laminin is expressed in the hippocampus and degraded by plasmin (Chen and Strickland, 1997). Furthermore, the expression level of laminin was correlated with the ability to maintain LTP. When the slices were allowed to recover after washout of plasmin, the expression level of laminin returned to normal level, and LTP was maintained. Simultaneous addition of α2-antiplasmin prevented both the degradation of laminin and the impairment of LTP by plasmin. Preincubation with anti-laminin antibodies protected laminin from the proteolysis and prevented the impairment of LTP maintenance. These results indicate that pretreatment with plasmin causes the degradation of laminin, which results in the impairment of LTP maintenance.
There are several reports studying the role of tPA in hippocampal LTP. tPA-deficient mice showed a reduction in the maintenance of LTP (Frey et al., 1996; Huang et al., 1996), and mice overexpressing tPA showed an enhanced LTP (Madani et al., 1999). The late phase of LTP was blocked by tPA inhibitors and enhanced by application of exogenous tPA (Baranes et al., 1998). These observations suggest that tPA is necessary for the maintenance of LTP. Because tPA is known to activate plasmin, the role of the PA–plasmin system in LTP appears to be opposite in their and our results. However, there are several differences between their and our studies. First, experimental protocol was completely different. Baranes et al. (1998) applied exogenous tPA from 10 min before to 10 min after tetanic stimulation, which was enough to enhance the late phase of LTP. In our studies, the slices were pretreated with plasmin for 6 hr and washed with ACSF before field potential recording. Exogenous plasmin was not present during tetanic stimulation in our studies. Because relatively longer time (2–5 hr) was required for exogenous plasmin to degrade laminin (Fig.4D), the rapid effect of tPA observed by Baranes et al. (1998) is unlikely to result from the degradation of laminin. Second, previous studies used acute hippocampal slices, whereas we used hippocampal slice cultures. Because the expression level of laminin was not significantly changed by the 6 hr treatment with 100 nm plasmin in acute hippocampal slices from adult rats (our unpublished data), we could not test the role of laminin using this preparation. Treatment with higher concentrations of plasmin for longer times may be necessary to degrade laminin in acute hippocampal slices, possibly because of the rigidity of ECM components or the slice thickness. More importantly, previous reports have investigated the role of tPA only, and the role for plasmin or laminin is unknown. Conceptually, overexpression or elimination of tPA should affect plasmin level. However, because plasmin production is regulated not only by tPA but also by other enzymes, including uPA, plasmin and laminin could be normal in tPA-deficient or overexpressing mice. To answer this question, it is necessary to determine the level of plasmin and laminin in these mice. Furthermore, it remains possible that tPA regulates LTP through the mechanism independent of plasmin or laminin. Indeed, tPA has been reported to induce microglial activation through a plasminogen-independent mechanism (Tsirka et al., 1997) or activate other substrates such as hepatocyte growth factor (Mars et al., 1993). Therefore, our results do not necessarily contradict previous reports. It is possible that tPA rapidly activates the mechanism involved in the maintenance of LTP, whereas long-term action of plasmin results in the impairment of LTP maintenance by degrading laminin.
Laminin is a glycoprotein composed of three (α, β, and γ) polypeptide chains, all of which are known to be susceptible to the proteolytic action of plasmin (Salonen et al., 1984; Paulsson et al., 1988). Indeed, Western blot analysis confirmed that α1 and β1/γ1 chains are expressed in our hippocampal cultures and both are degraded by plasmin (Fig. 5B). It has also been reported that laminin forms a self-assembled network around the cells (Timpl and Brown, 1994). Our immunofluorescence study revealed that laminin is present as the somatic form in control slices and is changed into the punctate form after pretreatment with plasmin. The change in the appearance of laminin may represent the degradation of laminin polymers to oligomers or smaller fragments. In addition, if laminin functions to regulate synaptic plasticity, it would be expected to be present at the synapses. In the CA1 area, laminin was expressed at a relatively high level in the pyramidal cell layer but also at a modest level in the stratum radiatum in which the Schaffer collaterals and CA1 pyramidal neurons make synaptic contact, and plasmin degraded laminin in the stratum radiatum as well as in the pyramidal cell layer. Electron microscopic examination is necessary to elucidate the role of laminin at the synapse.
Although the mechanism by which laminin contributes to the maintenance of LTP remains unknown, several possibilities can be argued. Because integrin, a cell-sulfate binding for laminin, has been proposed to be involved in the maintenance of LTP (Stäubli et al., 1990, 1998;Xiao et al., 1991; Bahr et al., 1997), laminin may regulate synaptic plasticity by interacting with integrin. Another possibility is a role of laminin in synaptic conformational stability. Laminin-11 has been reported to prevent glial entry into the synaptic cleft at the neuromuscular junction (Patton et al., 1998).
What is the physiological significance of regulation of LTP by plasmin and laminin? Many previous studies have shown that the PA–plasmin system plays a important role in cell migration and neurite extension by alternating the cell–ECM interaction (McGuire and Seeds, 1990; Shea and Beermann, 1992). In developing rat brain, the punctate form of laminin appears as early as embryonic day 10 and then disappears in the first week of postnatal life (Zhou, 1990). A similar form of laminin is also seen around vascular basement membranes, senile plaques, or reactive glia in the brain of patients with Alzheimer's disease (Luckenbill-Edds, 1997). Furthermore, it has been demonstrated recently that excitotoxic neuronal death is promoted by plasmin-catalyzed degradation of laminin (Chen and Strickland, 1997). Therefore, it is possible that a decrease of synaptic plasticity by plasmin-mediated laminin degradation occurs during development or under pathological conditions. Further investigations are underway to examine whether degradation of laminin by plasmin regulates synaptic plasticityin vivo.
In conclusion, we have demonstrated for the first time that the maintenance of LTP is impaired by degradation of laminin in organotypic hippocampal cultures. The laminin-mediated cell–ECM interaction may be necessary for the maintenance of LTP. If laminin is degraded by plasmin in physiological or pathological conditions, hippocampal synaptic plasticity would be decreased. Therefore, whether chronic treatment with plasmin for several days in vivo actually degrades laminin and affects synaptic plasticity should be elucidated. Further investigations on the role of plasmin and laminin in LTP will give more information on molecular mechanisms regulating synaptic plasticity.
Footnotes
This work was partially supported by Grant-in-Aid 7656 for Japan Society for the Promotion of Science (JSPS) Fellows from the Ministry of Education, Science, Sports, and Culture of Japan. Yasuhiro Nakagami is a Research Fellow of the JSPS.
Correspondence should be addressed to Dr. Norio Matsuki, Laboratory of Chemical Pharmacology, Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113–0033, Japan. E-mail: matsuki@mol.f.u-tokyo.ac.jp.
REFERENCES
- 1.Bahr BA, Stäubli U, Xiao P, Chun D, Ji Z-X, Esteban ET, Lynch G. Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. J Neurosci. 1997;17:1320–1329. doi: 10.1523/JNEUROSCI.17-04-01320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baranes D, Lederfein D, Huang Y-Y, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 4.Buchs P-A, Müller D. Induction of long-term potentiation is associated with major ultrastructural changes of activated synapses. Proc Natl Acad Sci USA. 1996;93:8040–8045. doi: 10.1073/pnas.93.15.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Z-L, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 6.Frey U, Müller M, Kuhl D. A different form of long-lasting potentiation revealed in tissue plasminogen activator mutant mice. J Neurosci. 1996;16:2057–2063. doi: 10.1523/JNEUROSCI.16-06-02057.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gähwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–477. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- 8.Geinisman Y. Perforated axospinous synapses with multiple, completely partitioned transmission zones: probable structural intermediates in synaptic plasticity. Hippocampus. 1993;3:417–434. doi: 10.1002/hipo.450030404. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y-Y, Bach ME, Lipp H-P, Zhuo M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind J-M, Mulligan R, Collen D, Carmeliet P. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci USA. 1996;93:8699–8704. doi: 10.1073/pnas.93.16.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauri SE, Kaukinen S, Kinnunen T, Ylinen A, Imai S, Kaila K, Taira T, Rauvala H. Regulatory role and molecular interaction of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J Neurosci. 1999;19:1226–1235. doi: 10.1523/JNEUROSCI.19-04-01226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luckenbill-Edds L. Laminin and the mechanism of neuronal outgrowth. Brain Res Rev. 1997;23:1–27. doi: 10.1016/s0165-0173(96)00013-6. [DOI] [PubMed] [Google Scholar]
- 12.Lüthi A, Laurent JP, Figurov A, Müller D, Schachner M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature. 1994;372:777–779. doi: 10.1038/372777a0. [DOI] [PubMed] [Google Scholar]
- 13.Madani R, Hulo S, Toni N, Madani H, Steimer T, Muller D, Vassali J-D. Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J. 1999;18:3007–3012. doi: 10.1093/emboj/18.11.3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mars W, Zarnegra R, Michalopoulos G. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 15.McGuire PG, Seeds NW. Degradation of underlying extracellular matrix by sensory neurons during neurite outgrowth. Neuron. 1990;4:633–642. doi: 10.1016/0896-6273(90)90121-u. [DOI] [PubMed] [Google Scholar]
- 16.Meiri N, Masos T, Rosenblum K, Miskin R, Dudai Y. Overexpression of urokinase-type plasminogen activator in transgenic mice is correlated with impaired learning. Proc Natl Acad Sci USA. 1994;91:3196–3200. doi: 10.1073/pnas.91.8.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizutani A, Saito H, Matsuki N. Possible involvement of plasmin in long-term potentiation of rat hippocampal slices. Brain Res. 1996;739:276–281. doi: 10.1016/s0006-8993(96)00834-7. [DOI] [PubMed] [Google Scholar]
- 18.Mizutani A, Tanaka T, Saito H, Matsuki N. Postsynaptic blockade of inhibitory postsynaptic currents by plasmin in CA1 pyramidal cells of rat hippocampus. Brain Res. 1997;761:93–96. doi: 10.1016/s0006-8993(97)00338-7. [DOI] [PubMed] [Google Scholar]
- 19.Müller D, Wang C, Skibo G, Toni N, Cremer H, Calaora V, Rougon G, Kiss JZ. PSA-NCAM is required for activity-induced synaptic plasticity. Neuron. 1996;17:413–422. doi: 10.1016/s0896-6273(00)80174-9. [DOI] [PubMed] [Google Scholar]
- 20.Nakagami Y, Saito H, Matsuki N. Basic fibroblast growth factor and brain-derived neurotrophic factor promote survival and neuronal circuit formation in organotypic hippocampal culture. Jpn J Pharmacol. 1997;75:319–326. doi: 10.1254/jjp.75.319. [DOI] [PubMed] [Google Scholar]
- 21.Nathan T, Jensen MS, Lambert JD. GABAB receptors play a major role in paired-pulse facilitation in area CA1 of the rat hippocampus. Brain Res. 1990;531:55–65. doi: 10.1016/0006-8993(90)90757-3. [DOI] [PubMed] [Google Scholar]
- 22.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 23.Patton BL, Chiu AY, Sanes JR. Synaptic laminin prevents glial entry into the synaptic cleft. Nature. 1998;393:698–701. doi: 10.1038/31502. [DOI] [PubMed] [Google Scholar]
- 24.Paulsson M, Saladin K, Landwehr R. Binding of Ca2+ influences susceptibility of laminin to proteolytic digestion and interactions between domain-specific laminin fragments. Eur J Biochem. 1988;177:477–481. doi: 10.1111/j.1432-1033.1988.tb14397.x. [DOI] [PubMed] [Google Scholar]
- 25.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL. The cell biology of the plasminogen system. FASEB J. 1995;9:939–945. doi: 10.1096/fasebj.9.10.7615163. [DOI] [PubMed] [Google Scholar]
- 26.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 27.Rønn LCB, Bock E, Linnemann D, Jahnsen H. NCAM-antibodies modulate induction of long-term potentiation in rat hippocampal CA1. Brain Res. 1995;677:145–151. doi: 10.1016/0006-8993(95)00147-i. [DOI] [PubMed] [Google Scholar]
- 28.Salonen E-M, Zitting A, Vaheri A. Laminin interacts with plasminogen and its tissue-type activator. FEBS Lett. 1984;172:29–32. doi: 10.1016/0014-5793(84)80866-2. [DOI] [PubMed] [Google Scholar]
- 29.Sappino AP, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli JD. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shea TB, Beermann ML. Regulation of neuronal migration and neuritogenesis by distinct surface proteases. Relative contribution of plasmin and a thrombin-like protease. FEBS Lett. 1992;307:190–194. doi: 10.1016/0014-5793(92)80765-9. [DOI] [PubMed] [Google Scholar]
- 31.Stäubli U, Vanderklish P, Lynch G. An inhibitor of integrin receptors blocks long-term potentiation. Behav Neural Biol. 1990;53:1–5. doi: 10.1016/0163-1047(90)90712-f. [DOI] [PubMed] [Google Scholar]
- 32.Stäubli U, Chun D, Lynch G. Time-dependent reversal of long-term potentiation by an integrin antagonist. J Neurosci. 1998;18:3460–3469. doi: 10.1523/JNEUROSCI.18-09-03460.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoppini L, Buchs P-A, Müller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 34.Sumi Y, Dent MA, Owen DE, Seeley PJ, Morris RJ. The expression of tissue and urokinase-type plasminogen activators in neural development suggests different modes of proteolytic involvement in neuronal growth. Development. 1992;116:625–637. doi: 10.1242/dev.116.3.625. [DOI] [PubMed] [Google Scholar]
- 35.Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- 36.Timpl R, Brown JC. The laminins. Matrix Biol. 1994;14:275–281. doi: 10.1016/0945-053x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 37.Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 38.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vassalli J-D, Sappino A-P, Belin D. The plasminogen activator/plasmin system. J Clin Invest. 1991;88:1067–1072. doi: 10.1172/JCI115405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ware JH, DiBenedetto AJ, Pittman RN. Localization of tissue plasminogen activator mRNA in the developing rat cerebellum and effects of inhibiting tissue plasminogen activator on granule cell migration. J Neurobiol. 1995;28:9–22. doi: 10.1002/neu.480280103. [DOI] [PubMed] [Google Scholar]
- 41.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 42.Xiao P, Bahr BA, Stäubli U, Vanderklish PW, Lynch G. Evidence that matrix recognition contributes to the stabilization but not induction of LTP. NeuroReport. 1991;2:461–464. doi: 10.1097/00001756-199108000-00013. [DOI] [PubMed] [Google Scholar]
- 43.Zhou FC. Four patterns of laminin-immunoreactive structure in developing rat brain. Dev Brain Res. 1990;55:191–201. doi: 10.1016/0165-3806(90)90200-i. [DOI] [PubMed] [Google Scholar]